


Abstract
The organic anion transporters 1 and 3 (OAT1 and OAT3) and organic cation transporter 2 (OCT2) are important for renal tubular drug secretion. In contrast, evidence for OAT2 expression in the human kidney is limited, and its role in renal drug transport is unknown. Both mRNA (real-time polymerase chain reaction) and protein (Western blotting) for OAT2 were detected in renal cortex from eight donors, and interindividual variability in protein levels was 3-fold. OAT2 protein in the renal cortex was localized (by immunohistochemistry) to the basolateral domain of tubules, as were OAT1 and OAT3. The absolute abundance of OAT2 mRNA was similar to that of OAT1 mRNA and 3-fold higher than that of OCT2 mRNA but 10-fold lower than that of OAT3 mRNA. A previous observation that OAT2 transports cGMP led us to examine whether acyclovir, ganciclovir, and penciclovir are OAT2 substrates; they are guanine-containing antivirals that undergo active tubular secretion. Transport of the antivirals into human embryonic kidney cells was stimulated 10- to 20-fold by expression of OAT2, but there was little to no transport of the antivirals by OAT1, OAT3, or OCT2. The Km values for acyclovir, ganciclovir, and penciclovir transport were 94, 264, and 277 μM, respectively, and transport efficiencies were relatively high (6–24 μl · min−1 · mg protein−1). This study provides definitive evidence for the expression of OAT2 in the human kidney and is the first to demonstrate that OAT2, compared with OAT1, OAT3, or OCT2, has a preference for antiviral drugs mainly eliminated in the urine via active secretion.
Introduction
Active proximal tubular secretion represents an important pathway in excreting from the body a diverse array of xenobiotics, including many environmental toxins and clinically relevant therapeutic agents. The organic anion transporters 1 and 3 (OAT1 and OAT3) and organic cation transporter 2 (OCT2) are expressed in basolateral membranes of renal proximal tubule cells and represent the initial step in tubular secretion of many of these xenobiotics (Wright and Dantzler, 2004). Attention has recently been placed on these transporters because evidence suggests they are clinically relevant (Giacomini et al., 2010). For example, OAT1, OAT3, and OCT2 influence the pharmacokinetics of numerous drugs, and there are cases in which diversity in the genes encoding these transporters cause altered drug pharmacokinetics (e.g., OCT2 and metformin) (Giacomini et al., 2010). In addition, drug interactions and adverse drug reactions have been suggested to occur at the level of these transporters (Giacomini et al., 2010). In contrast, there is only limited evidence for the expression of OAT2 protein in the human kidney (Enomoto et al., 2002), and its role in renal tubular handling of drugs and toxins is unknown.
OAT2 functions as a Na+-independent exchanger and shares many substrates in common with OAT1 and OAT3, such as para-aminohippurate, estrone-3-sulfate, and glutarate (Kobayashi et al., 2005). Two splice variants of OAT2 have been described, with variant 2 (OAT2-548aa, GenBank accession number NM_153320) containing an additional two amino acids in its sequence compared with variant 1 (OAT2-546aa, GenBank accession number NM_006672) (Cropp et al., 2008). Using real-time PCR it was shown that mRNA levels of the two variants were similar to each other in the tissues examined, which included the kidney and liver (Cropp et al., 2008). When expressed in human embryonic kidney cells, OAT2-546aa trafficked to the plasma membrane and was functional, whereas OAT2-548aa was retained in an intracellular compartment (Cropp et al., 2008). The OAT2-546aa variant was found to transport a number of purine and pyrimidine nucleobases, as well as nucleosides and nucleotides, with a preference for the guanine-containing second messenger cGMP (Cropp et al., 2008).
Valacyclovir, valganciclovir, and famciclovir are orally administered prodrugs that undergo extensive first-pass metabolism to form the active drugs acyclovir, ganciclovir, and penciclovir, respectively. Acyclovir and penciclovir are used in the treatment of herpes virus, whereas ganciclovir is used to treat cytomegalovirus infections. These antivirals are predominately eliminated in the urine in their active form, and their total body clearance is highly dependent on renal function (Krasny et al., 1982; Sommadossi et al., 1988; Gill and Wood, 1996). In addition, their renal excretion seems to occur, in part, as a result of active tubular secretion, i.e., their renal clearance exceeds the glomerular filtration rate (de Miranda et al., 1982; Fletcher et al., 1986; Fowles et al., 1992). For example, the renal clearance of penciclovir approaches renal plasma flow, indicating that this drug undergoes robust active tubular secretion (Fowles et al., 1992). Given that these antivirals undergo active tubular secretion and are structurally similar to cGMP (guanine-containing) (Fig. 1), we hypothesized that they would be substrates of OAT2-546aa and that the transporter may be important for their renal excretion.

Chemical structures of cyclic guanosine monophosphate, acyclovir, ganciclovir, and penciclovir.
The present study addressed several important questions regarding OAT2 expression in the human kidney and its potential role in proximal tubular secretion of acyclovir, ganciclovir, and penciclovir. First, is OAT2 detectable in the human renal cortex from multiple donors, where is it expressed, and is there interindividual variability in its expression level? How does the level of OAT2 mRNA in the human kidney compare with transcript levels of renal uptake transporters known to be clinically relevant, namely OAT1, OAT3, and OCT2? Finally, does OAT2-546aa transport acyclovir, ganciclovir, and penciclovir, and is there a potential for this transport activity to be relevant to the tubular secretion of these drugs in vivo?
Materials and Methods
Chemicals.
[3H]Acyclovir (13.6 Ci/mmol), [3H]ganciclovir (3.4 Ci/mmol), and [3H]pencilcovir (10.6 Ci/mmol) were from Moravek Biochemicals (Brea, CA). [3H]1-Methyl-4-phenylpyridinium (MPP) (80 Ci/mmol), [3H]estrone-3-sulfate (ES) (50 Ci/mmol), [3H]cGMP (14.3 Ci/mmol), and [3H]para-aminohippuric acid (PAH) (5 Ci/mmol) were from American Radiolabeled Chemicals (St. Louis, MO). Nonradiolabeled acyclovir, penciclovir, ganciclovir, PAH, ES, MPP, and nitrobenzylthioinosine were from Sigma-Aldrich (St. Louis, MO). Platinum High Fidelity DNA polymerase, Native pfu DNA polymerase, phleomycin (Zeocin), hygromycin, Flp recombinase expression plasmid (pOG44), phosphate-buffered saline (PBS), Hanks' balanced salt solution, Dulbecco's modified Eagle's growth medium (DMEM), HEK293 Flp-In cells, and synthetic oligonucleotides for cloning were from Invitrogen (Carlsbad, CA). The mammalian transfection reagent FuGENE 6 was from Roche Diagnostics Corporation (Indianapolis, IN). The QIAquick DNA gel extraction kit and QIAprep DNA purification kit were from QIAGEN (Valencia, CA). TaqMan real-time PCR gene expression assays for GAPDH (Hs02758991_g1), OAT2 (Hs00198527_m1), OCT2 (Hs00231269_m1), ENT1 (Hs01085706_m1), ENT2 (Hs00155426_m1), ENT3 (Hs00217911_m1), ENT4 (Hs00542001_m1), and SGLT2 (Hs00894642_m1) were from Applied Biosystems (Foster City, CA). Oligonucleotide primer sets and FAM-labeled TaqMan MGB probes for OAT1 and OAT3, TaqMan Universal PCR Master Mix, and the MagMAX-96 Total RNA isolation kit were also from Applied Biosystems. The primary antibodies (affinity-purified) against human OAT1, OAT2, and OAT3 were obtained from a commercial source (CosmoBio, Tokyo, Japan). Details concerning the generation and characterization of these antibodies can be found in the following publications: Hosoyamada et al. (1999), Cha et al. (2001), and Enomoto et al., 2002. The primary antibody against the human multidrug and toxin extrusion transporter 1 (MATE1) was a generous gift from Dr. Yosinori Moriyama (Okayama University Dentistry and Pharmaceutical Sciences Graduate School of Medicine, Okayama-shi, Japan), and the details regarding generation and characterization of this antibody can be found in Otsuka et al. (2005).
Human Kidney Tissue.
Samples of human kidney cortex from nine individual donors (5 female and 4 male; 18–54 years of age) were received from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) Brain and Tissue Bank at the University of Maryland (Baltimore, MD). All tissues were removed within 9 h post mortem. The tissues from NICHD were used for real-time PCR and Western blotting. The sample of human renal cortex used for immunohistochemistry was obtained from Dr. Stephen H. Wright in the Department of Physiology at the University of Arizona, and all procedures were approved by the University of Arizona Human Subjects Research and Institutional Review Board.
Cloning of Transporters.
The open reading frames of human OAT1, OAT2, OAT3, and OCT2 were amplified from a human kidney cDNA library (Clontech, Mountain View, CA) using either Native pfu DNA polymerase or Platinum High Fidelity DNA polymerase and sequence-specific primers. The cloned PCR products were gel-purified and subcloned into either the pcDNA5/FRT/V5-His-TOPO (OAT2) or pEF/FRT/V5DEST (OAT1, OAT3, and OCT2) mammalian expression plasmids according to the manufacturer's instructions (Invitrogen). The sequence-specific primers included the native stop codons to prevent incorporation of the His and V5 tags (contained in the expression plasmids) into the transport proteins. Plasmid DNA was prepared using standard methods (QIAGEN), and sequences were confirmed by SeqWright (Houston, TX). The cloned sequences of OAT1 (GenBank accession number AF124373), OAT2 (OAT2-546aa, GenBank accession number NM_006672), OAT3 (GenBank accession number AB042505), and OCT2 (GenBank accession number NM_003058) were identical to those reported in the National Center for Biotechnology Information database.
Cell Culture and Stable Expression.
HEK Flp-In cells were grown in DMEM supplemented with 10% fetal calf serum, 1% penicillin-streptomycin, and 100 μg/ml Zeocin mixture in an atmosphere of 5% CO2/95% air at 37°C. HEK Flp-In cells were seeded into 100-mm dishes at a density of ∼0.02 million cells/cm2 and incubated overnight. Transfection procedures were done according to the manufacturer's recommendations (Invitrogen and F. Hoffmann-La Roche, Basel, Switzerland). In brief, FuGENE6 reagent (30 μl) was diluted in 400 μl of serum-free DMEM and incubated at room temperature for 5 min. Plasmid DNA (1 μg of expression plasmid and 9 μg of pOG44 plasmid) was added to the FuGENE6 mixture and incubated for an additional 15 min at room temperature. The DNA-FuGENE6 mixture was then added dropwise to the cells, and the culture dishes were incubated overnight at 37°C. Transfected cells were trypsinized and plated to ∼25% confluence in DMEM supplemented with 10% fetal calf serum and 1% penicillin-streptomycin. After a few hours, the medium was replaced with fresh medium containing 100 μg/ml hygromycin. Media were changed every 3 to 4 days until hygromycin-resistant colonies formed. The stable cell lines were then trypsinized and expanded.
RNA Isolation and Real-Time PCR.
Total RNA from cells and renal cortex was purified using the MagMAX-96 Isolation Kit in accordance with the manufacturer's protocol (Applied Biosystems). The concentration of total RNA was determined by UV spectrophotometry using an Eppendorf biophotometer. Total RNA (2.5 μg) was reverse transcribed using the iScriptTM cDNA Synthesis Kit according to the manufacturer's instructions (Bio-Rad Laboratories, Hercules, CA). The reverse transcription protocol included a step to remove genomic DNA. For absolute quantification, standard curves were generated for each transporter (OAT1, OAT2, OAT3, and OCT2) using the plasmids containing the respective transporter cDNAs. The plasmid containing the cDNA for GAPDH was obtained from OriGene (Rockville, MD). Over a cDNA concentration range of 2 × 10−2 to 2 × 10−7 nmol, the CT values ranged from 12 to 30, and this relationship was log linear. The R2 values for the standard curves were ≥0.98 (data not shown). The CT values for the unknown samples tested were within the boundaries of the standard curve. Transporter abundance was expressed relative to the amount of GAPDH (micromoles of transporter per mole of GAPDH). The comparative CT method was used to determine the relative levels of SGLT2 or ENT (ENT1–4) mRNA (with GAPDH as an endogenous reference) in the cultured cells and human kidney. For both absolute and relative quantification, cDNA (20 ng) from either cultured cells or human kidney was incubated (quadruplicate reactions) with TaqMan Universal PCR Master Mix and primer/probe mixture. Real-time PCR was run on the ABI Prism 7900 Sequence Detection System (Applied Biosystems) using the following thermal cycling parameters (40 cycles): 50°C (2 min), 95°C (10 min), 95°C (15 s), and 60°C (1 min). The specificity of the real-time PCR probes for OAT1, OAT2, OAT3, and OCT2 was tested using RNA isolated from the cell lines stably expressing the respective transporters. There was no amplification signal detected in control HEK cells, and there was no cross-reactivity observed (data not shown), suggesting that the probes were specific.
Crude Membrane Preparation and Western Blotting.
Crude membrane fractions from cultured cells and human kidney were prepared as described by Rius et al. (2010), with slight modification. All steps were conducted using ice-cold buffers. Cultured cells grown to confluence in a 10-cm dish were resuspended in 5 ml of lysis buffer (10 mM Tris-HCl and 250 mM glucose, pH 7.4) containing protease inhibitors [4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride, 104 μM; aprotinin, 80 μM; bestatin, 4 μM; trans-epoxysuccinic acid (E-64), 4 μM; leupeptin, 2 μM; and pepstatin, 1.5 μM; Sigma-Aldrich], followed by one freeze-thaw cycle. Samples of frozen renal cortex (0.5–1 mg) were resuspended in 5 ml of lysis buffer and were subsequently homogenized using a PowerGen 125 homogenizer for 1 min followed by 30 strokes with a Potter-Elvehjem homogenizer. The lysates were centrifuged at 1200g for 10 min at 4°C, and the resulting supernatants were centrifuged at 100,000g for 30 min at 4°C. The pellets were resuspended in 10 mM Tris-HCl (pH 7.4), and the protein concentration was determined using the bicinchoninic acid method (Thermo Fisher Scientific, Waltham, MA).
The crude membrane fractions were diluted in an equal volume of 2× Laemmli sample buffer (Bio-Rad Laboratories) containing 2.5% β-mercaptoethanol (final concentration); the final protein concentration did not exceed 5 μg/μl. After the samples were heated to 100°C for 5 min, the proteins were separated on 10% Tris-glycine gels (Invitrogen) and then transferred to polyvinylidene fluoride membranes. After a 1-h incubation in blocking buffer (LI-COR Biosciences, Lincoln, NE) at room temperature, the membranes were incubated overnight at 4°C in blocking buffer containing 0.5 μg/ml rabbit anti-human OAT2 antibody and 2.3 μg/ml mouse anti-human β-actin antibody (Sigma-Aldrich). The membranes were then rinsed extensively in washing buffer (PBS containing 0.05% Tween 20) followed by incubation for 1 h at room temperature in blocking buffer containing IRDye 680LT goat anti-mouse antibody and IRDye 800CW goat anti-rabbit antibody (1:10,000 dilutions, LI-COR Biosciences). The membranes were then washed with PBS containing 0.05% Tween 20 supplemented with 0.01% SDS, and immunoreactivity was detected with an Odyssey Infrared Imaging System (LI-COR Biosciences). Semiquantitative densitometry analysis of immunoreactivity was determined from the images using Image calc (C. H. A. van de Lest, Netherlands Asthma Foundation, Amersfoort, Netherlands) as described in Pelis et al. (2001). In brief, an immunoreactive band on the Western blot was selected, and Image calc scanned the selected part from top to bottom, thus determining the average 8-bit gray scale value located on each horizontal line. The average 8-bit gray scale values on each horizontal line were then summed to obtain cumulative 8-bit gray scale values for each particular band. Several areas on the Western blot that did not contain immunoreactivity were used as a background subtraction.
Immunohistochemistry.
Human renal cortex was fixed overnight at 4°C in a periodate-lysine-paraformaldehyde solution containing 75 mM l-lysine, 10 mM sodium periodate, 37 mM Na2HPO4 (pH 7.4), and 2% paraformaldehyde. After equilibration in 70% ethanol, the tissues were embedded in paraffin, and 5-μm sections were cut and placed on microscope slides. The tissue sections were deparafinized by a routine procedure and an antigen retrieval procedure was applied. In brief, the specimens were microwaved in 10 mM citric acid monohydrate (pH 6.1) on a high-power setting for 5 min followed by 6 min in the microwave on a low-power setting. After specimens were cooled to room temperature, they were washed in PBS, followed by incubation in 10% normal goat serum (Invitrogen) for 1 h at room temperature. The specimens were then incubated overnight at 4°C with rabbit anti-OAT1, rabbit anti-OAT2, or rabbit anti-OAT3 antibodies diluted to 5 μg/ml in 10% normal goat serum. The rabbit anti-human MATE1 antibody was diluted 1:200 in 10% normal goat serum. After extensive washing, the specimens were incubated for 1 h at room temperature with an AlexaFluor 488 goat anti-rabbit antibody (Invitrogen) diluted 1:400 in 10% normal goat serum. The specimens were then washed with PBS and incubated (room temperature for 10 min) in PBS containing 5 μg/ml propidium iodide to stain the nuclei. The specimens were washed, and immunoreactivity was examined using a confocal microscope (Nikon PCM 2000 scan head fitted to a Nikon E800 microscope) at the Arizona Cancer Center.
Transport Experiments.
Cells were grown to confluence in 24-well poly-d-lysine-coated plates (BD Biosciences, San Jose, CA). All transport experiments were conducted at 37°C using a transport solution containing Hanks' balanced salt solution supplemented with 5 mM HEPES and 1 mM CaCl2 (pH 7.4), along with the appropriate radiolabeled compound (antiviral drug or probe substrate). The probe substrates used for OAT1, OAT2, OAT3, and OCT2 were PAH, cGMP, ES, and MPP, respectively. The concentration of radiolabeled compound and the time point used for transport studies are indicated in the figure legends. After the transport period the transport solution was aspirated, and the cells were rinsed with three changes of ice-cold PBS. The cells were solubilized with lysis buffer (0.4 ml of 0.5 N NaOH containing 0.1% SDS). The resulting cell lysate was neutralized with 0.2 ml of 1 N HCl, and 0.5 ml of cell lysate was used for liquid scintillation counting. Protein concentration was determined using the bicinchoninic acid method. For kinetic analysis, transport experiments were conducted at initial rates using low concentrations (well below the Km values) of radiolabeled compound in the presence of increasing concentrations of unlabeled compound. The concentration range of unlabeled compounds was typically 0 to 1000 μM. However, higher concentrations of acyclovir had to be used when OAT1- and OAT3-mediated acyclovir transport kinetics were assessed because of the high Km values associated with their transport (Table 2). The cellular accumulation of compound (at each concentration tested) in the parental (control) HEK Flp-In cells was subtracted from uptake into the cells expressing the individual transporters; this was done to account for potential endogenous transport activity in the control cells. Kinetic parameters, Km (Michaelis constant) and Jmax (maximal rate of transport), were determined as described previously (Cheng et al., 2011).
Data Analysis.
Individual transport observations were performed in triplicate for each experiment, and observations were confirmed, at least three times, in separate experiments using cells of a different passage. All data are presented as means ± S.E.M. Statistical analysis was performed using a two-tailed Student's t test, and differences were considered significant when P < 0.05 (GraphPad Prism version 5.00; GraphPad Software Inc., San Diego, CA).
Results
Given that evidence for the presence of OAT2 in the human kidney is somewhat limited, in particular at the protein level, we initially sought to characterize mRNA and protein expression of the transport protein in the human renal cortex. To confirm that the kidney tissue samples used for these studies were in fact from the cortical region, the level of SGLT2 mRNA expression was examined by real-time PCR analysis. SGLT2 expression is restricted to the early proximal tubule (primarily the S1 segment), which resides in the outer cortex (Kanai et al., 1994). The average percentage difference in SGLT2 mRNA expression in eight of the nine donor tissues was only 17 ± 13%, and, thus, these eight samples were used for examining transporter mRNA and protein expression. A low level of SGLT2 mRNA in one of the samples (female subject) precluded its use in further analyses. Not surprisingly, the level of OAT1, OAT2, OAT3, and OCT2 mRNA was nearly undetectable in the donor tissue that had a low level of SGLT2. Despite similar levels of SGLT2, the absolute abundance of OAT2 mRNA varied as much as 17-fold across the eight donors examined (Fig. 2A). This high variability in OAT2 mRNA expression was largely due to the low level of expression in subject 6. OAT2 transcript levels only varied 2- to 3-fold across the other seven donors. Although the data set was small (4 females and 4 males), no significant effect of gender on OAT2 mRNA expression was observed (3268 ± 962 μmol of OAT2/mol GAPDH for males versus 2111 ± 967 μmol of OAT2/mol GAPDH for females).
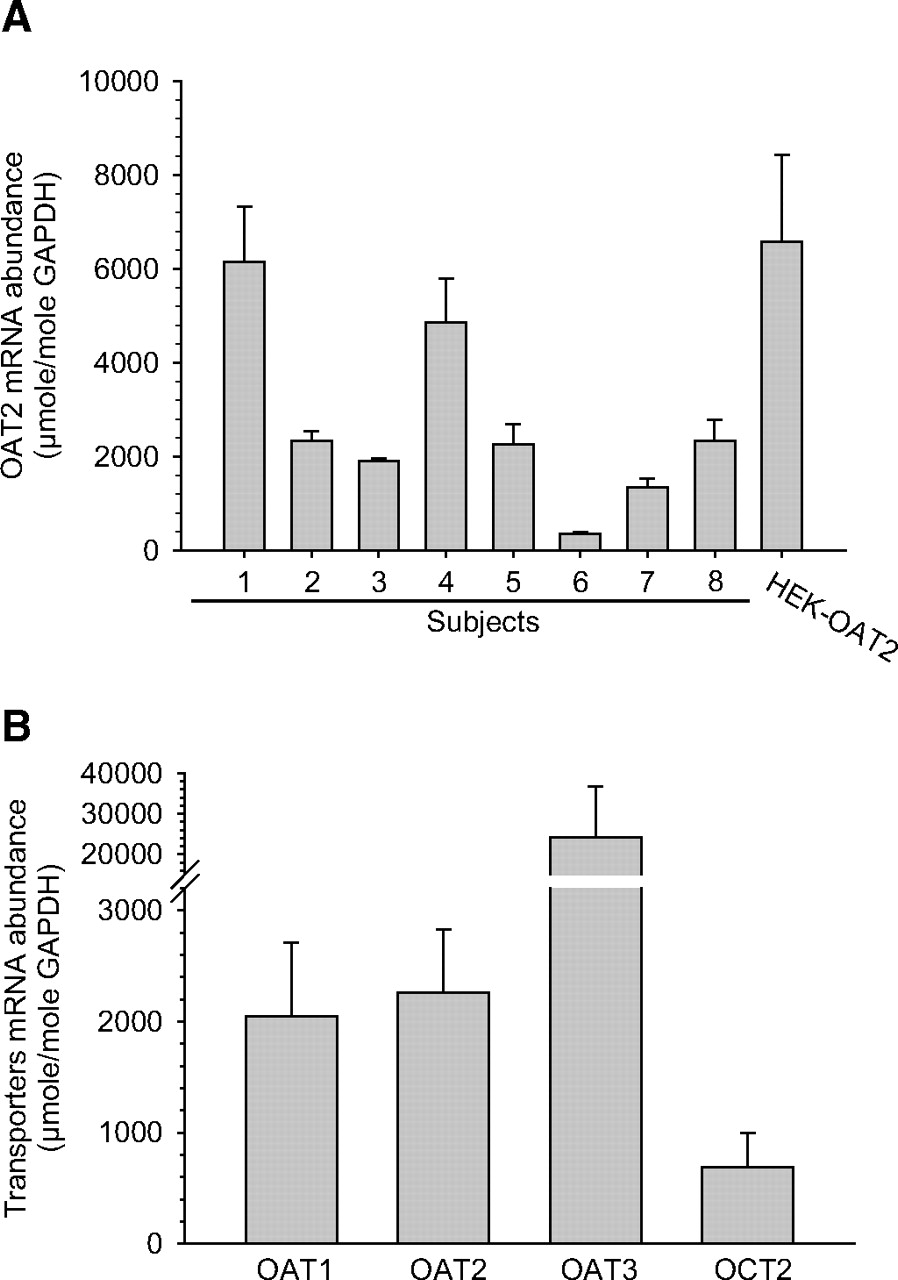
Characterization of OAT2 mRNA expression by real-time PCR. A, absolute mRNA abundance of OAT2 relative to GAPDH (micromoles of OAT2 per mole of GAPDH) in samples of renal cortex from eight individual human subjects and in HEK cells stably expressing OAT2. The transcript was not detected in control HEK cells (data not shown). Data are means ± S.E.M. of four separate reactions. B, absolute mRNA abundance of OAT1, OAT2, OAT3, and OCT2 relative to GAPDH in samples of renal cortex from eight human subjects. Data are means ± S.E.M. of the eight subjects.
The amount of OAT2 mRNA was also compared with transcript levels of OAT1, OAT3, and OCT2, because these transporters are known to be clinically relevant to drug uptake into renal proximal tubule cells. OAT1 and OAT2 mRNA levels were approximately equal to each other, and both were ∼3-fold greater than the levels of OCT2 mRNA (Fig. 2B). The levels of OAT3 mRNA were the highest among the transport proteins examined, ∼10-fold higher than those for OAT1 and OAT2 (Fig. 2B).
Interindividual variability in the OAT2 protein level in the human liver has been shown to be 10-fold, and, thus, we examined OAT2 protein expression in the samples of renal cortex by Western blot. The anti-OAT2 antibody used reacted on Western blots with OAT2 from HEK-OAT2 cells, whereas no immunoreactivity was detected in crude membrane preparations from HEK cells expressing OAT1 or OAT3 (Fig. 3A). OAT2 protein from crude membrane preparations of human renal cortex and HEK-OAT2 cells migrated to ∼70 kDa when separated on SDS-polyacrylamide gel electrophoresis gels, and was detected in each of the human renal cortex samples examined (Fig. 3B). Semiquantitative densitometric analysis indicated that OAT2 protein varied 3-fold (Fig. 3C).

Characterization of OAT2 protein by Western blot. A, specificity of the anti-OAT2 antibody was determined by probing a Western blot containing protein from crude membrane fractions prepared from control HEK cells or HEK cells expressing OAT1, OAT2, or OAT3 with the antibody; 25 μg of crude membrane fraction were used for separation on SDS-polyacrylamide gel electrophoresis gels. B, immunoreactivity of OAT2 was detected using an anti-OAT2 antibody on Western blots containing crude membrane fractions (100 μg) prepared from the renal cortex of eight human subjects. OAT2 from the human kidney samples had a migration pattern on the Western blot similar to that of OAT2 stably expressed in HEK cells. The Western blot was also probed with an anti-β-actin (∼42 kDa) antibody, which served as a loading control. The molecular mass makers (in kilodaltons) are shown. C, densitometric analysis of OAT2 immunoreactivity from the Western blot shown in Fig. 3B. Note: β-actin immunoreactivity was not used in the densitometric analysis. Immunoreactivity is expressed as cumulative gray scale. h, human.
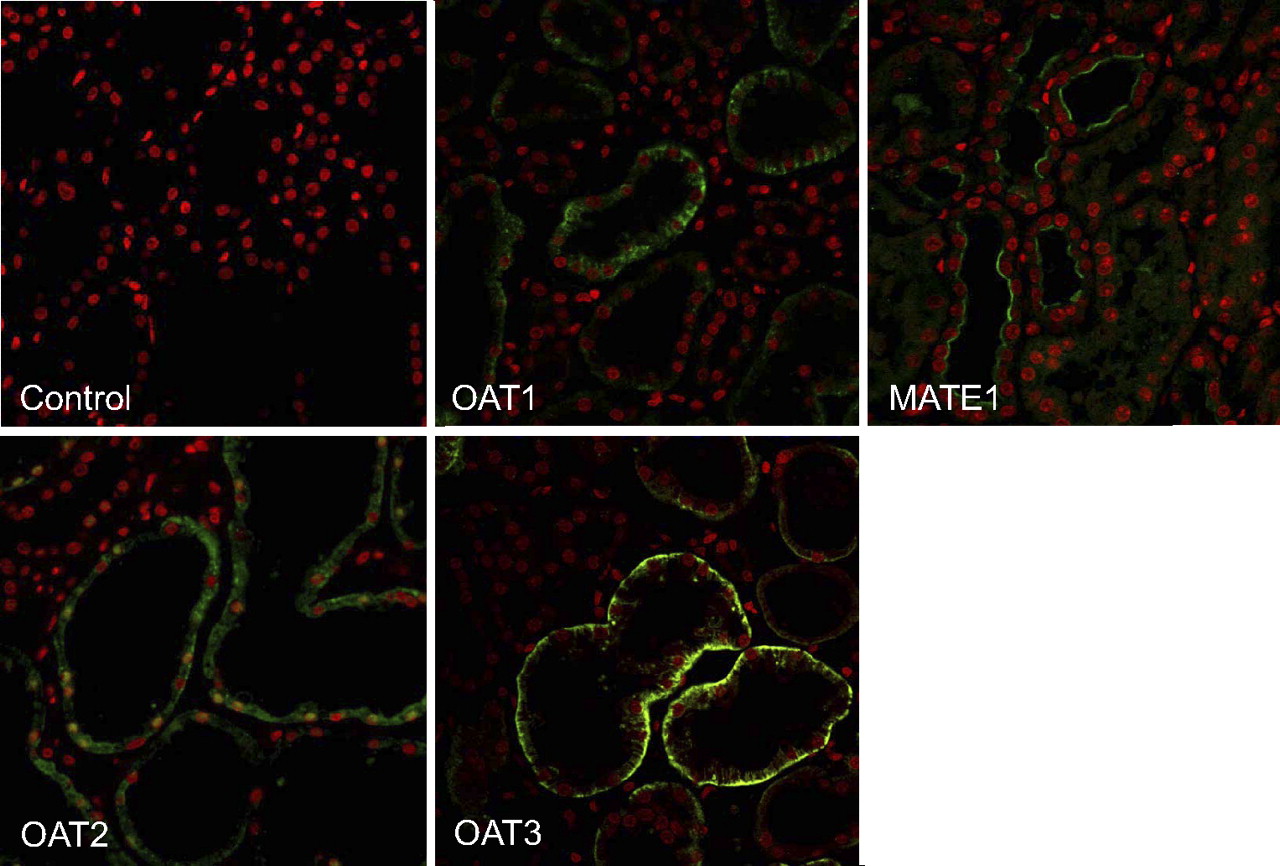
The expression of OAT2 protein in the human renal cortex was also examined by immunohistochemistry along with expression of OAT1, OAT3, and MATE1 (Fig. 4). No immunoreactivity was detected when primary antibodies were omitted (control). Immunoreactivity against all three OAT transport proteins was detected in tubules of the renal cortex, and the pattern of staining was consistent with their basolateral localization. This result is in contrast to the immunoreactive profile of MATE1, which was restricted to the apical membrane of cortical tubules. Not all of the tubules were immunoreactive for each of the proteins examined.

Immunolocalization of OAT1, OAT2, OAT3, and MATE1 in human renal cortex. Sections of human renal cortex were incubated with rabbit anti-human OAT1, OAT2, OAT3, or MATE1 antibodies followed by incubation with an Alexa Fluor 488 goat anti-rabbit antibody (green). The control sample was incubated with Alexa Fluor 488 goat anti-rabbit antibody only. Nuclei were stained with propidium iodide (red). Images were taken at 400× magnification.
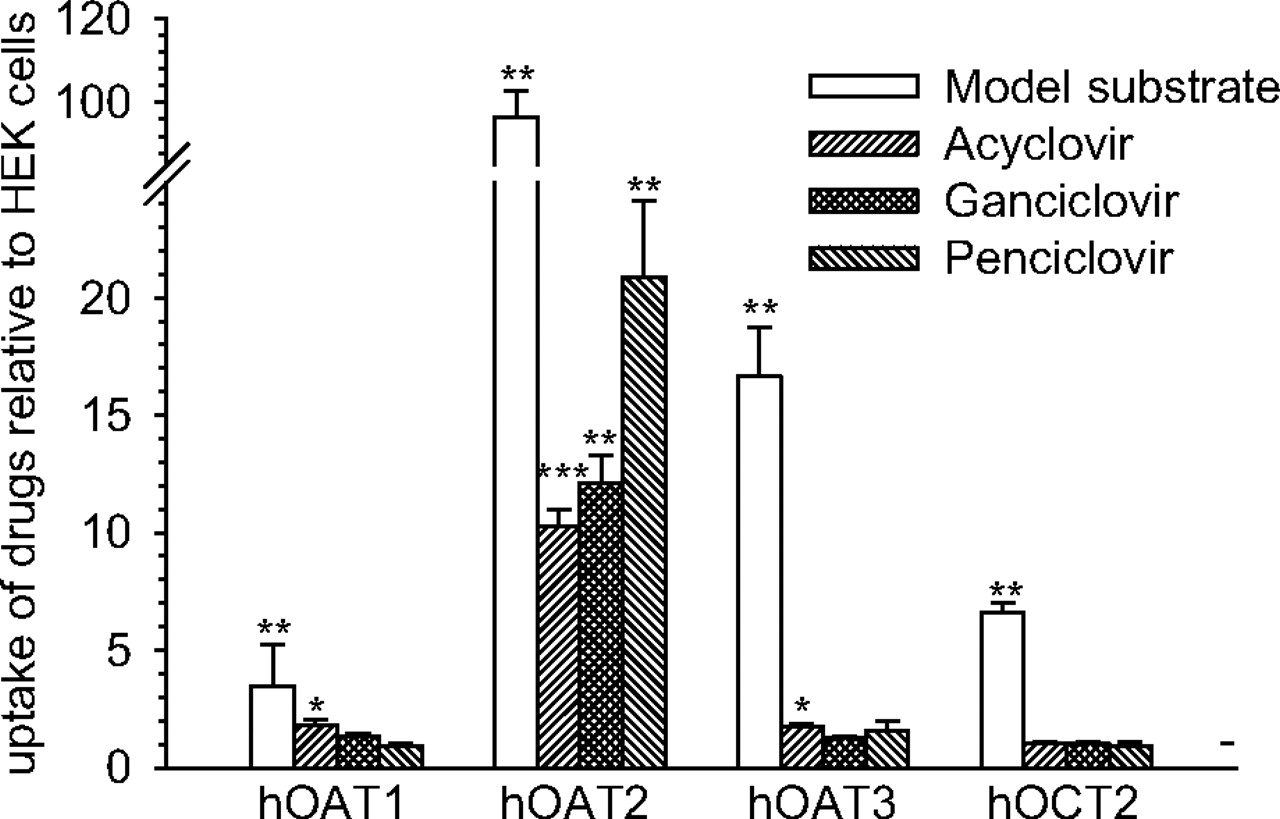
The ability of OAT2 to transport acyclovir, ganciclovir, and penciclovir was tested in HEK cells stably expressing the transport protein. OAT1, OAT3, and OCT2 were also tested for their ability to transport the antivirals. All of the transport proteins were functional in the cell lines tested. The uptake of PAH, cGMP, ES, and MPP was stimulated by expression of OAT1 (3.5-fold), OAT2 (97-fold), OAT3 (17-fold), and OCT2 (6.5-fold), respectively, compared with that in control HEK cells (Fig. 5). The uptake of acyclovir, ganciclovir, and penciclovir was 10-, 12-, and 21-fold higher into HEK-OAT2 cells compared with control HEK cells (Fig. 5). The uptake of acyclovir into HEK-OAT1 and HEK-OAT3 cells was also significantly greater (∼1.7-fold) than uptake into control HEK cells. In contrast, OAT1, OAT3, and OCT2 did not mediate ganciclovir or penciclovir transport, and there was no evidence of OCT2-mediated acyclovir transport (Fig. 5).
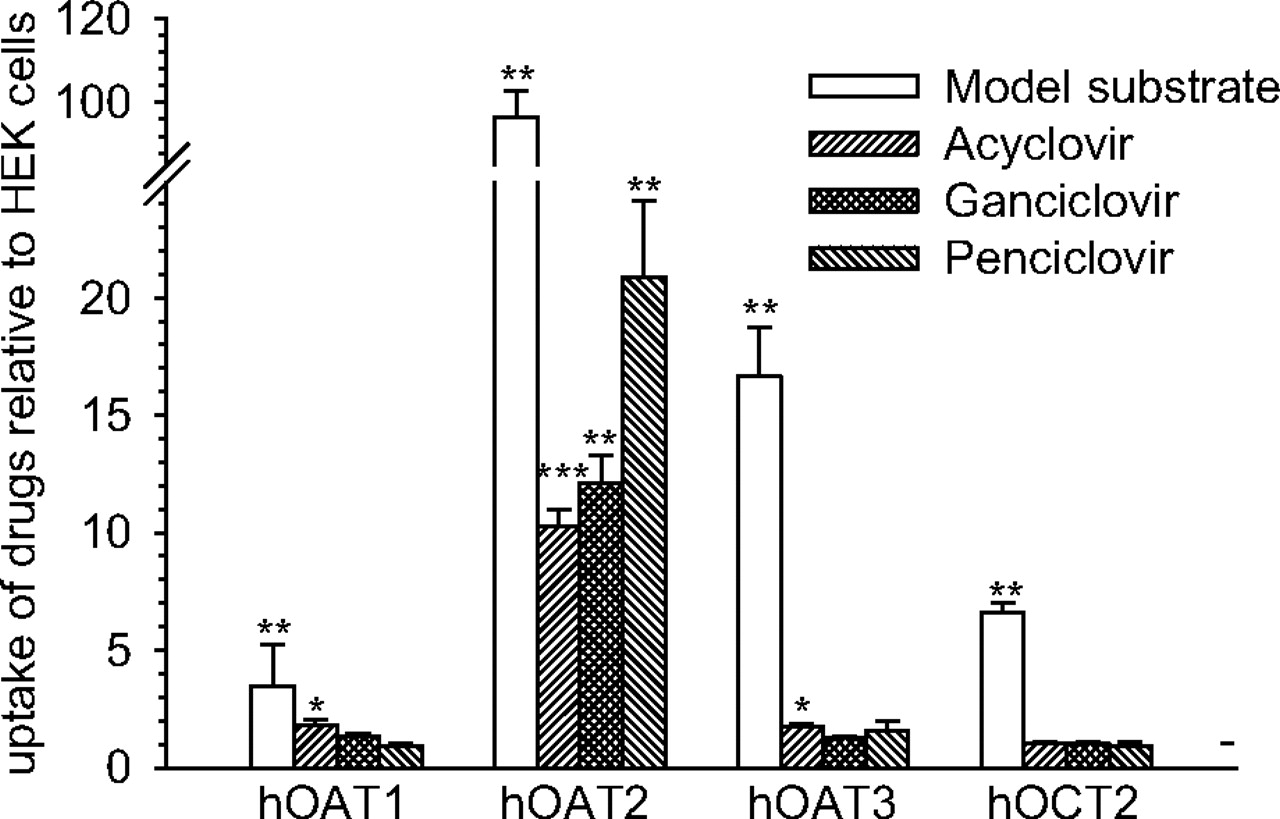
Transporter-mediated uptake of acyclovir, ganciclovir, and penciclovir. The uptake of tritium-labeled acyclovir (0.06 μM), ganciclovir (0.19 μM), and penciclovir (0.07 μM) was conducted in control HEK cells or HEK cells expressing OAT1, OAT2, OAT3, or OCT2 for 10 min at 37°C (n = 4 different passages; mean ± S.E.M.). Probe substrate uptake by OAT1 (PAH, 0.12 μM), OAT2 (cGMP, 0.05 μM), OAT3 (ES, 0.01 μM), and OCT2 (MPP, 0.009 μM) was also conducted to ensure that the transport proteins were active in the respective cell lines. Uptake is expressed relative to uptake into control HEK cells (dashed horizontal line). Significant differences from control HEK cells: *, P < 0.05; **, P < 0.01; ***, P < 0.001, using a two-tailed Student's t test. h, human.
Given the robust transport activity observed, the kinetics of acyclovir, ganciclovir, and penciclovir transport by OAT2 were examined (Fig. 6). In initial experiments, uptake of the antivirals into control HEK cells was reduced in the presence of the ENT inhibitor nitrobenzylthioinosine (100 μM), suggesting that ENTs are endogenously expressed in HEK cells and that they are capable of transporting the antivirals (data not shown). However, ENT1, ENT2, ENT3, and ENT4 mRNA levels were not different between control HEK cells and HEK-OAT2 cells, suggesting that ENT activity did not contribute to the stimulation in antiviral uptake caused by the expression of OAT2. Regardless, to obtain accurate kinetics of antiviral transport by OAT2, the uptake of acyclovir, ganciclovir, and penciclovir into control HEK cells was subtracted from uptake into HEK-OAT2 cells at each concentration of antiviral tested. Before kinetic studies were performed, time courses of drug uptake were conducted (Fig. 6, insets), and an initial rate time point was chosen for kinetic analyses (see legend to Fig. 6 for details). OAT2-mediated transport of [3H]acyclovir, [3H]ganciclovir, and [3H]penciclovir was reduced by increasing concentrations of the respective unlabeled compounds by a process adequately described by the Michaelis-Menten equation for competitive interaction of labeled and unlabeled substrate (Groves et al., 1998). The Km values associated with OAT2-mediated acyclovir, ganciclovir, and penciclovir transport were 94.4 ± 7.6, 264 ± 24, and 277 ± 10 μM, respectively (Table 1). The efficiency (Jmax/Km) of OAT2-mediated acyclovir and ganciclovir transport was similar (6–7 μl · min−1 · mg protein−1), but 3- to 4-fold lower than the efficiency of penciclovir transport (24 μl · min−1 · mg protein−1) (Table 1). For comparison, the efficiency values for probe substrate transport by OAT1 (PAH), OAT3 (ES), and OCT2 (MPP) were 11, 26, and 52 μl · min−1 · mg protein−1, respectively (Table 1). In a separate set of experiments (Table 2), we examined the kinetics of OAT1- and OAT3-mediated acyclovir transport. The kinetics of OAT1-mediated PAH transport and OAT3-mediated ES transport were run in parallel on cells of the same passage. The Km values (shown in Table 2) associated with PAH transport by OAT1 (28 μM) and ES transport by OAT3 (17 μM) were in line with the Km values reported in Table 1. However, the Jmax values were slightly higher, suggesting that OAT expression was comparatively high in this passage. Despite this observation, the efficiency values for acyclovir transport by OAT1 (0.5 μl · min−1 · mg protein−1) and OAT3 (1.0 μl · min−1 · mg protein−1) were low and ≥30-fold less than the transport efficiencies observed for probe substrate transport by OAT1 and OAT3.

Kinetics of OAT2-mediated antiviral drug transport. The intracellular uptake of tritium-labeled acyclovir (A), ganciclovir (B), and penciclovir (C) into HEK-OAT2 cells was conducted in the presence of increasing concentrations of the respective unlabeled drugs (n = 4 different passages; mean ± S.E.M.). The antiviral uptake shown in the kinetic experiments was specific to OAT2 because uptake into control HEK cells (conducted in parallel) was subtracted from uptake into HEK-OAT2 cells at each concentration tested. The time courses of tritium-labeled antiviral uptake by HEK-OAT2 cells is shown in the insets. All kinetic experiments were conducted at initial rates (2 min). The kinetic parameters Jmax and Km were determined as indicated under Materials and Methods (values shown in Table 1).
Kinetic parameters associated with substrate transport by OAT1, OAT2, OAT3, and OCT2
The actual kinetic experiments associated with OAT2-mediated antiviral transport are shown in Fig. 6. The concentration of unlabeled substrate ranged from 0 to 1000 μM. The kinetic parameters associated with OAT1- and OAT3-mediated acyclovir transport and probe substrate transport by OAT1 (PAH), OAT3 (ES), and OCT2 (MPP) are shown for comparison. Transport efficiency was determined by dividing the Jmax value by the Km value. Kinetics of OAT2-mediated acyclovir, ganciclovir, and penciclovir transport were conducted on the same day using cells of the same passage. n = 3 to 4 different passages; data are means ± S.E.M.
Kinetic parameters associated with acyclovir and probe substrate transport by OAT1 and OAT3
Transport efficiency was determined by dividing the Jmax value by the Km value. Kinetics of OAT1- and OAT3-mediated acyclovir transport and probe substrate transport were conducted on the same day using cells of the same passage. n = 1 passage.
Discussion
Oat2 was originally cloned from a rat liver cDNA library, and it was shown to be almost exclusively expressed in rat liver by Northern blotting, suggesting that it may be most relevant to hepatic transport processes (Simonson et al., 1994; Sekine et al., 1998). However, mRNA levels of OAT2 in human kidney and liver were comparable (Sun et al., 2001; Cropp et al., 2008). In the present study, mRNA corresponding to OAT2 was detected in the renal cortex of each of the human donor kidneys examined. Levels of the OAT2 transcript exhibited relatively large interindividual variability (up to 17-fold), despite similar levels of the proximal tubule marker, SGLT2. The absolute abundance of OAT2 mRNA was also compared with mRNA levels of renal uptake transporters known to be clinically relevant, namely OAT1, OAT3, and OCT2, with the following trend observed: OAT3 > OAT1 ≈ OAT2 > OCT2. This result is in agreement with an earlier finding that OAT3 mRNA levels are highest in the human kidney, followed by (in descending order) OAT1, OCT2,and OAT2 (Motohashi et al., 2002). In addition to mRNA, OAT2 protein was also detected in samples of renal cortex from each donor by Western blot but exhibited less interindividual variability (3-fold) than at the transcript level, indicating that OAT2 mRNA and protein levels are not necessarily reflective of each other. The observed apparent molecular mass of ∼70 kDa is slightly bigger than expected on the basis of the length of the amino acid sequence (∼55 kDa), which could be due to the presence of glycosylation; OAT2 contains three consensus sites for N-glycosylation. Although this is the first study to detect OAT2 in the human kidney by Western blot, Enomoto et al. (2002) had previously demonstrated that OAT2 localizes to basolateral membranes of tubules in the human renal cortex. Using immunohistochemistry, we also localized OAT2 protein, as well as OAT1 and OAT3, to basolateral membranes of tubule cells in the human renal cortex. The pattern of immunoreactivity was quite different from that of MATE1, which localizes to apical membrane of renal tubules (Otsuka et al., 2005). Taken together, these data confirm that both OAT2 mRNA and protein are expressed in the renal cortex of humans, and their abundance, at least at the mRNA level, is comparable to that of transport proteins known to play an important role in drug uptake into proximal tubule cells.
The human ortholog of OAT2 has two splice variants, with variant 2 (OAT2-548aa) containing an additional serine and glutamine at amino acid positions 132 and 133, respectively. The levels of mRNA for the two variants are approximately equal in the kidney (Cropp et al., 2008). In cloning OAT2 from a human kidney cDNA library, we obtained variant 1 (OAT2-546aa). This variant was stably expressed in HEK cells and supported robust cGMP transport, as had been reported previously (Cropp et al., 2008; Fork et al., 2011). In addition to cGMP, OAT2-546aa was previously found to transport a number of other guanine-containing compounds, including guanine itself, 2′-deoxyguanosine, guanosine monophosphate, guanosine diphosphate, and guanosine triphosphate (Cropp et al., 2008). This finding led to the hypothesis that variant 1 may transport therapeutic drugs containing a guanine moiety.
Acyclovir, ganciclovir, and penciclovir are guanine-containing antiviral drugs that are mainly excreted in the urine unchanged, and their urinary excretion greatly exceeds the glomerular filtration rate, indicating active tubular secretion (de Miranda et al., 1982; Fletcher et al., 1986; Fowles et al., 1992). The antivirals are hydrophilic (ClogP values ∼−2.5), making transport proteins important for their efficient movement across biological membranes, such as basolateral membranes of proximal tubule cells. The transport of the antivirals into HEK cells was stimulated >10-fold by expression of OAT2. In contrast, there was little to no mediated transport of the antivirals by OAT1, OAT3, or OCT2. Although members of the OAT family are most known for their ability to interact with relatively small organic anions (<500 g/mol), acyclovir, ganciclovir, and penciclovir (225–250 g/mol) are neutral at physiological pH. Most recently, Fork et al. (2011) showed that OAT2-546aa transports trigonelline, which is a small (137 g/mol) zwitterion. Together, these data indicate that the substrate specificity of OAT2-546aa is not restricted to compounds containing an anionic moiety but instead is relatively broad, as is the case with other drug transporters in the solute carrier family.
Takeda et al. (2002) had previously examined the interaction of acyclovir and ganciclovir (penciclovir was not examined) with OAT1, OAT2, OAT3, and OCT2 and found that only OAT1 was capable of mediating their transport; we also noted OAT1-mediated transport of acyclovir. The reason that Takeda et al. (2002) failed to observe OAT2-mediated transport of the antivirals is not clear, but it is possible that they used variant 2 (OAT2-548aa) in their studies (the actual sequence was not referenced) and that variant 2 has a substrate specificity different from that of variant 1. For example, dehydroepiandrosterone, estrone-3-sulfate, and paclitaxel (Taxol) were shown to be transported by variant 2 expressed in Xenopus laevis oocytes but were not transported by variant 1 in either HEK or Chinese hamster ovary cells (R. Pelis and Y. Cheng, unpublished observations). Further work is required to determine whether the two splice variants differ in their substrate specificity. In addition, it is not clear why we failed to observe OAT1-mediated ganciclovir transport, which was reported by Takeda et al. (2002). It is possible that differences in the expression systems used are a factor; they used mouse S2 cells for stable expression of the transport proteins.
With the acknowledgment that transport efficiency (Jmax/Km) is dependent on expression level, it has been suggested that transport efficiencies determined in vitro with heterologous expression systems are probably irrelevant when less than 1 μl · min−1 · mg protein−1 (Schömig et al., 1998). The transport efficiencies of probe substrate transport by OAT1, OAT3, and OCT2 were greater than 10 μl · min−1 · mg protein−1. The efficiency with which the antiviral drugs were transported by OAT2 was comparatively high (6–24 μl · min−1 · mg protein−1), with penciclovir being the most efficiently transported of the three drugs tested. Although acyclovir was found to be a substrate of OAT1 and OAT3, the efficiency with which it was transported by OAT1 and OAT3 was relatively low (≤1 μl · min−1 · mg protein−1).
The Km values associated with acyclovir, ganciclovir, and penciclovir transport by OAT2 were 94, 264, and 284 μM, respectively. After a single oral therapeutic dose of the prodrugs valacyclovir, valganciclovir, and famciclovir to humans, the maximum plasma concentrations of the active drugs were 29.5 μM (acyclovir), 11.7 μM (ganciclovir), and 11.2 μM (penciclovir) (Boike et al., 1994; Soul-Lawton et al., 1995; Jung and Dorr, 1999). These concentrations are well below the Km values for their interaction with OAT2, and, thus, the transporter would not be saturated by therapeutic levels of the antivirals.
OAT2 has been shown to operate as an exchanger, with the Krebs' cycle intermediates succinate and fumarate serving as trans-substrates (Kobayashi et al., 2005). The intracellular concentration of these dicarboxylates are maintained at a high level in renal tubule cells compared with that in the interstitium owing to oxidative metabolism as well as the activity of the Na+-dependent dicarboxylate cotransporter 3 (Burckhardt et al., 2005). The outwardly directed concentration gradients for succinate and fumarate would be expected to trans-stimulate OAT2, thus concentrating acyclovir, ganciclovir, and penciclovir inside proximal tubule cells. In addition, the electrogenic exchange of an intracellular dicarboxylate (net charge of −2) for an extracellular neutral antiviral drug would be further stimulated by the inside negative membrane potential. Overall, OAT2-mediated uptake of acyclovir, ganciclovir, and penciclovir across the basolateral membranes of proximal tubule cells is expected to be an energetically favorable process. We also noted in our experiments that the antiviral drugs tested are substrates of ENTs, which are also expressed in the human renal cortex. However, ENTs are equilibrative transporters, and their activity at the basolateral membrane alone cannot explain the ability of the proximal tubule to actively secrete the neutral antiviral drugs. That is, in the presence of basolateral ENT activity alone, the intracellular concentration of the neutral antiviral drugs would at best approximate interstitial levels.
In addition to its expression in renal tubules OAT2 is expressed in the sinusoidal membranes of hepatocytes (Simonson et al., 1994). However, there is little to no hepatic metabolism or biliary clearance of acyclovir, ganciclovir, or penciclovir; we do have unpublished data showing that at least penciclovir is actively taken up into human hepatocytes in suspension. Thus, we speculate that a transporter(s) present in the apical membrane of proximal tubules that is not present (or with low endogenous expression) in the canalicular membrane of hepatocytes determines the final elimination pathway of these antivirals, i.e., renal as opposed to hepatic. In addition, a transporter(s) present in the sinusoidal membranes of hepatocytes must be capable of effluxing back into blood drug that had accumulated in hepatocytes.
In conclusion, the present study has provided a comprehensive profile of OAT2 mRNA and protein expression in the human renal cortex. OAT2 mRNA and protein were detected in each of the donor kidneys examined, and the transport protein was localized to basolateral membranes of tubules in the cortex. Furthermore, OAT2 was found to transport the antiviral drugs acyclovir, ganciclovir, and penciclovir with relatively high efficiency. In contrast, transport of the antivirals by OAT1, OAT3, and OCT2 was weak to absent. This is the first study to show that antiviral drugs eliminated almost exclusively in the urine are preferentially transported by OAT2, as opposed to the renal uptake transporters suspected of being clinically relevant, i.e., OAT1, OAT3, and OCT2. On the basis of these in vitro data, OAT2 probably contributes to active renal tubular secretion of acyclovir, ganciclovir, and penciclovir in vivo.
Authorship Contributions
Participated in research design: Cheng, Aleksunes, and Pelis.
Conducted experiments: Cheng, Vapurcuyan, and Shahidullah.
Contributed new reagents or analytic tools: Cheng and Shahidullah.
Wrote or contributed to the writing of the manuscript: Cheng, Vapurcuyan, Shahidullah, Alexsunes, and Pelis.
Acknowledgments
Human tissue was obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland (Baltimore, MD) (Contract HHSN275200900011C, ref. no. N01-HD-9-0011).
Footnotes
Y.C. was supported by a Novartis Institutes for Biomedical Research Postdoctoral fellowship grant.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
ABBREVIATIONS:
- OAT
- organic anion transporter
- OCT
- organic cation transporter
- PCR
- polymerase chain reaction
- MPP
- 1-methyl-4-phenylpyridinium
- ES
- estrone-3-sulfate
- PAH
- para-aminohippurate
- PBS
- phosphate-buffered saline
- DMEM
- Dulbecco's modified Eagle's medium
- HEK
- human embryonic kidney
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- ENT
- equilibrative nucleoside transporter
- SGLT2
- sodium-glucose transporter 2
- MATE1
- multidrug and toxin extrusion transporter 1
- NICHD
- Eunice Kennedy Shriver National Institute of Child Health and Human Development.
- Received August 2, 2011.
- Accepted December 21, 2011.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}