


Abstract
Veverimer is a polymer being developed as a potential treatment of metabolic acidosis in patients with chronic kidney disease. Veverimer selectively binds and removes hydrochloric acid from the gastrointestinal tract, resulting in an increase in serum bicarbonate. Veverimer is not systemically absorbed, so potential drug-drug interactions (DDIs) are limited to effects on the absorption of other oral drugs through binding to veverimer in the gastrointestinal tract or increases in gastric pH caused by veverimer binding to hydrochloric acid. In in vitro binding experiments using a panel of 16 test drugs, no positively charged, neutral, or zwitterionic drugs bound to veverimer. Three negatively charged drugs (furosemide, aspirin, ethacrynic acid) bound to veverimer; however, this binding was reduced or eliminated in the presence of normal physiologic concentrations (100–170 mM) of chloride. Veverimer increased gastric pH in vivo by 1.5–3 pH units. This pH elevation peaked within 1 hour and had returned to baseline after 1.5–3 hours. Omeprazole did not alter the effect of veverimer on gastric pH. The clinical relevance of in vitro binding and the transient increase in gastric pH was evaluated in human DDI studies using two drugs with the most binding to veverimer (furosemide, aspirin) and two additional drugs with pH-dependent solubility effecting absorption (dabigatran, warfarin). None of the four drugs showed clinically meaningful DDI with veverimer in human studies. Based on the physicochemical characteristics of veverimer and results from in vitro and human studies, veverimer is unlikely to have significant DDIs.
SIGNIFICANCE STATEMENT Patients with chronic kidney disease, who are usually on many drugs, are vulnerable to drug-drug interactions (DDIs). The potential for DDIs with veverimer was evaluated based on the known site of action and physicochemical structure of the polymer, which restricts the compound to the gastrointestinal tract. Based on the findings from in vitro and human studies, we conclude that veverimer is unlikely to have clinically significant DDIs.
Introduction
Metabolic acidosis is a serious complication of chronic kidney disease (CKD) and accelerates its progression (Wesson et al., 2020). Patients with CKD generate metabolic acids and have a reduced capacity to excrete acid through the kidneys (Alpern and Sakhaee, 1997; Scialla and Anderson, 2013; Goraya and Wesson, 2017). Correction of metabolic acidosis in CKD slows kidney disease progression (de Brito-Ashurst et al., 2009; Mahajan et al., 2010; Phisitkul et al., 2010; Goraya et al., 2013; Goraya et al., 2014; Garneata et al., 2016; Dubey et al., 2018), improves muscle mass and function (de Brito-Ashurst et al., 2009; Abramowitz et al., 2013; Dubey et al., 2018), and improves bone health (Domrongkitchaiporn et al., 2002).
Veverimer is being developed as a once-daily treatment of metabolic acidosis in patients with CKD. Veverimer is an orally administered, nonabsorbed, insoluble, free-amine polymer that combines high capacity and selectivity to bind and remove hydrochloric acid (HCl) from the gastrointestinal (GI) tract, resulting in an increase in serum bicarbonate (Bushinsky et al., 2018; Wesson et al., 2019a,b). Acid binding and removal using a nonabsorbed polymer is a novel approach to treating metabolic acidosis. Within the GI tract, the polymer restores the ability to excrete acid from the body. This mechanism of action is fundamental to the effectiveness of veverimer in treating metabolic acidosis and is distinct from the mechanisms of other drugs, such as proton pump inhibitors (PPIs) and histamine H2-receptor antagonists, that affect gastric pH but do not have an effect on systemic acid-base balance. The effect of veverimer on acid binding, as assessed by a change in gastric pH, was evaluated in this study both in the presence and absence of a PPI. The effect of veverimer on a background of PPI use was a relevant question because these drugs affect gastric pH and are commonly used.
Veverimer was designed to bind HCl with high capacity and specificity. After ingestion, veverimer is protonated, and the positively charged polymer selectively binds the smallest anion in the GI tract, chloride, with little or no binding of other anions (Klaerner et al., 2020). The high HCl-binding capacity is a function of the amine content of the polymer, whereas the high specificity for chloride binding is the result of extensive crosslinking within the polymer beads that excludes anions larger than chloride (e.g., phosphate, citrate, bile acids, and short-chain and long-chain fatty acids) and minimizes interaction between the polymer and other concomitantly administered oral drugs (Klaerner et al., 2020).
Polypharmacy is common in CKD, and drug-drug interactions (DDIs) are a continuing clinical concern (Rama et al., 2012; Sommer et al., 2020). We used a directed approach based on the known physical and chemical characteristics of veverimer to analyze its potential for DDIs (Fig. 1). Veverimer is too large to be systemically absorbed (Klaerner et al., 2020), and therefore, its potential for DDIs is limited to effects on the absorption of other orally administered drugs either through 1) binding to veverimer or 2) transient increases in gastric pH caused by veverimer binding to HCl. In this study we tested the hypothesis that the potential for veverimer to interact with other orally administered drugs is low, which is consistent with the physical and chemical properties of this novel polymer.

Strategy to assess potential for veverimer drug-drug interactions. We used a directed approach based on the known physical and chemical characteristics of veverimer to analyze its potential for DDIs. Veverimer is too large to be systemically absorbed; therefore, its potential for DDIs is limited to effects on the absorption of other orally administered drugs either through 1) binding to veverimer or 2) transient increases in gastric pH caused by veverimer binding to HCl. To identify candidate drugs for testing with veverimer in human DDI studies, we conducted in vitro studies to identify the characteristics most likely to lead to binding to veverimer, and we evaluated the effect of veverimer on gastric pH in healthy volunteers. Results from these studies informed the selection of drugs for human DDI studies.
Materials and Methods
The protocols for all five clinical trials reported herein were approved by the relevant US institutional review boards [Chesapeake Research Review, Inc. (Columbia, MD) for the furosemide, aspirin, and warfarin DDI studies and the gastric pH study and Advarra (Columbia, MD) for the dabigatran DDI study]. All participants provided written, informed consent prior to trial initiation. The trials were conducted at Celerion in accordance with the principles of Good Clinical Practice and the Declaration of Helsinki. The furosemide, warfarin, and dabigatran DDI studies and the gastric pH study were conducted in Tempe, AZ, and the aspirin DDI study was conducted in Lincoln, NE.
In Vitro Assessment of Direct Drug Binding to Veverimer: Effect of Size Assessed Using Anionic Probes
Since the free-amine veverimer polymer becomes positively charged upon binding to hydrogen ions, negatively charged probe molecules of increasing molecular mass (36.5–234.2 Da; Supplemental Table S1) were used to assess the role of size exclusion in binding of molecules to veverimer. Veverimer (450 mg) was incubated in 20 mM aqueous solutions (100 mL) of individual probe molecules for 0.5, 2, 4, 6, and 24 hours. Phosphoric acid and HCl incubation periods were limited to 6 hours and 4 hours, respectively. At each time point, a 400-µL aliquot was filtered, diluted 10-fold, and analyzed by ion chromatography to calculate the number of millimoles of acid bound per gram of veverimer.
In Vitro Assessment of Direct Drug Binding to Veverimer: Test Drug Panel
An in vitro test system was designed to assess the potential for binding interactions between veverimer and a set of test drugs that included 14 oral medications used in patients with CKD as well as two water-soluble vitamins (Table 1). The test drug panel included prototypical drugs from 14 distinct drug classes that ranged in size from 129 to 482 Da; were positively charged (N = 5), neutral/zwitterionic (N = 4), or negatively charged (N = 7); and comprised all four Biopharmaceutics Classification System classes, covering a range of solubilities and permeabilities.
Drugs tested for in vitro binding to veverimer
In vitro binding assays were conducted using seven matrices mimicking the pH and ionic conditions of the GI tract: simulated gastric fluid (SGF) with or without additional 60 mM HCl and without pepsin (pH 1–1.2); 50, 100, or 200 mM acetate buffer (pH 4.5); and simulated intestinal fluid (SIF) with or without an additional 50 mM phosphate (PO4) and without pancreatin (pH 6.8). The matrices with higher buffering capacity (SGF + 60 mM HCl, 100 mM and 200 mM acetate buffer, SIF + 50 mM PO4) were used to maintain the pH of the incubation mixture around the target pH values of 1.2, 4.5, and 6.8, respectively, in the presence of 9.0 mg/mL veverimer. The likelihood of identifying a drug interaction would be greatest at a high concentration of veverimer (4.5 g or 9.0 g) and a low concentration of the test drug. The maximum clinical dose of veverimer is anticipated to be 9 g once daily (QD). A volume of 1 liter was used for dispersion of an orally administered drug in the upper GI tract (Read et al., 1980; Metcalf et al., 1987; Thelen et al., 2011).
Binding to veverimer was assessed in six replicates by measuring free test drug concentration after a 3-hour incubation on a benchtop shaker at 37°C in the presence or absence of veverimer (4.5 mg/mL or 9.0 mg/mL). After incubation, samples were allowed to settle for 5 minutes and were filtered with a 0.45-µm polyvinylidene fluoride (PVDF) filter plate unit (allopurinol, aspirin, gliclazide, metoprolol tartrate, lisinopril, riboflavin, thiamine hydrochloride, and trimethoprim) or a 0.45-µm PVDF syringe filter (amlodipine besylate, ethacrynic acid, furosemide, metformin hydrochloride, rosuvastatin, spironolactone, valsartan, and warfarin). Syringe filters were used for test drugs that adsorbed to PVDF filter plates. Filtrates were transferred to glass vials for analysis by high-performance liquid chromatography with UV absorbance detection.
The 90% confidence intervals of test drug recovery with veverimer (population 1) and without veverimer (population 2) were calculated as follows.
1. Analyses were performed on natural log-transformed variables.
2. The 90% confidence intervals were calculated as:  , wherein D̅ is the difference between the means of the two populations, sD̅ is the S.D. of the difference between the means, and ttwo−sided 90%,DF is the t-value for a two-sided 90% confidence interval with DF, degrees of freedom. Assuming equal variances between the two populations, the S.D. of the difference between the means is:
, wherein D̅ is the difference between the means of the two populations, sD̅ is the S.D. of the difference between the means, and ttwo−sided 90%,DF is the t-value for a two-sided 90% confidence interval with DF, degrees of freedom. Assuming equal variances between the two populations, the S.D. of the difference between the means is:

wherein s12 is the variance of n1 observations from population 1, s22 is the variance of n2 observations from population 2, and DF = n1 + n2 – 2.
The lower bound is calculated with the minus sign, and the upper bound is calculated with the plus sign. These bounds are 90% intervals on the differences of the natural log-transformed variables. Exponentiating differences in logs results in ratios. Thus, the 90% confidence interval can be expressed in % Ratios as 100 exp(lower bound), 100 exp(upper bound).
Based on information available for other nonabsorbed drugs [Velphoro (sucroferric oxyhydroxide), 2013; Auryxia (ferric citrate), 2014; Veltassa (patiromer), 2015], a threshold was set for test drug interaction with veverimer at ≥30% binding (i.e., test drugs, for which both the upper and lower bounds of the 90% confidence intervals (CI) on the ratio of probe recovery in the presence of veverimer to that without veverimer were <30%, were deemed to have no significant binding to veverimer under the conditions tested).
Results from these in vitro studies informed the selection of drugs for human DDI studies.
Evaluation of Potential Veverimer Drug Binding in the Presence of Chloride
Because veverimer is an HCl-binding polymer, the potential impact of chloride levels on in vitro binding of veverimer to test drugs was assessed. Five representative test drugs from the set of 14 oral medications, one each with a positive (trimethoprim) or neutral (allopurinol) charge and three with a negative charge (aspirin, ethacrynic acid, and furosemide), were assessed for veverimer binding in six test matrices supplemented with chloride. Chloride concentrations in the upper GI tract vary between 70 and 170 mM, with an average concentration of approximately 100 mM (Fordtran and Locklear, 1966; Binder, 2012). For binding experiments conducted in the presence of physiologic chloride levels, additional chloride was added to the test matrices to achieve a final chloride concentration of approximately 170 mM in SGF and 100 mM in acetate buffer and in SIF.
Effect of Veverimer on Gastric pH in Healthy Volunteers with and without Omeprazole
An open-label, in-patient, randomized, crossover, 2-stage study in healthy volunteers was conducted to assess the effect of veverimer on gastric pH (Supplemental Fig. S1). The first stage evaluated the magnitude and duration of the effect under both fasted and fed conditions. The second stage evaluated the effect under fasted and fed conditions in the presence of the proton pump inhibitor omeprazole. The same subjects were to be enrolled in stage 1 and stage 2; subjects who withdrew from the study after stage 1 were replaced to ensure that the same number of subjects (N = 40) participated in each stage. The sample size was estimated based on the integrated acidity variability when no drug was administered to show no difference and ensure that the effect observed with veverimer was due to the drug administration and not the natural variability of the pH levels in healthy subjects. The number of subjects enrolled was based on in-house data and conservative assumptions regarding intrasubject and intersubject coefficients of variation to obtain a power of at least 80%, which was defined as the probability of having a 90% CI for a treatment ratio within the range of 80.00–125.00%.
Both stage 1 and stage 2 had a randomized (1:1:1:1), 4-period, 4-way crossover design. One of four study drug treatments was administered on day 1 of each of four treatment periods in each stage. Periods 1 and 2 and periods 3 and 4 were each conducted over 2 consecutive days; period 2 and period 3 were separated by a 1-day rest period. As prespecified in the protocol, after completion of period 4 in stage 1 and review of the preliminary intragastric pH data, the decision was made to conduct stage 2.
Stage 2 began with a 6-day run-in period (days −6 to −1), during which subjects received omeprazole QD. Baseline 22-hour intragastric pH monitoring was performed beginning on day −1. After completion of the run-in period, subjects received omeprazole QD through day 1 of period 4. Key entry criteria for the study are provided in Supplemental Section 3.1.1.1.
The study included nine pH-monitoring periods: four in stage 1 and five in stage 2. On day 1 of each treatment period in stage 1 as well as on day −1 of the run-in period and on day 1 of each treatment period in stage 2, gastric pH was measured and recorded continuously for 22 hours using a microelectrode probe. See Supplemental Section 3.1.1.2 for further details.
In stage 1, subjects were domiciled at the clinical research unit (CRU) from day −1 of period 1 through completion of study procedures on day 2 of period 4. In stage 2, subjects were domiciled at the CRU on day −7 through completion of study procedures on day −6 of the run-in period and again from day −2 of the run-in period through completion of study procedures on day 2 of period 4. Adverse event (AE) collection continued for 14 days after completion of pH monitoring.
In stage 1 of the study, subjects received the following treatments:
Treatment A: water fasted. Ninety (90) mL of water administered at hour 0 on day 1 under fasted conditions.
Treatment B: water fed. Ninety (90) mL of water administered at hour 0 on day 1 within 15 minutes after the start of a standard breakfast.
Treatment C: veverimer fasted. Nine (9) g veverimer administered as an oral suspension with 90 mL of water at hour 0 on day 1 under fasted conditions.
Treatment D: veverimer fed. Nine (9) g veverimer administered as an oral suspension with 90 mL of water at hour 0 on day 1 within 15 minutes after the start of a standard breakfast.
In stage 2, omeprazole (40 mg delayed-release capsule) was administered QD with 240 mL of water approximately 1 hour prior to a standard meal at the end of the afternoon or early evening on day −6 through day −1 of the run-in period and from day 1 of period 1 through day 1 of period 4 (within ±1 hour of omeprazole dosing time on day −6 of the run-in period).
In stage 2 of the study, subjects received the following treatments:
Treatment E: water fasted. Ninety (90) mL of water administered at hour 0 on day 1 under fasted conditions.
Treatment F: water fed. Ninety (90) mL of water administered at hour 0 on day 1 within 15 minutes after the start of a standard breakfast.
Treatment G: veverimer fasted. Nine (9) g veverimer administered as an oral suspension with 90 mL of water at hour 0 day 1 under fasted conditions.
Treatment H: veverimer fed. Nine (9) g veverimer administered an oral suspension with 90 mL of water at hour 0 on day 1 within 15 minutes after the start of a standard breakfast.
Safety was monitored throughout the study with clinical laboratory evaluations, reporting of AEs, physical examination, vital signs, and 12-lead ECGs.
Information on subject disposition is provided in Supplemental Table S2. Demographics and key baseline information for the study population are presented in Supplemental Table S3. Most subjects were White (80.0% in stage 1; 77.5% in stage 2), and approximately half were of Hispanic or Latino ethnicity (57.5% in each stage). Most subjects were female (67.5% in stage 1; 65.0% in stage 2), and the mean age was 33.4 years in stage 1 and 35.5 years in stage 2.
Results from this study informed the selection of additional drugs for human DDI studies.
Human DDI Studies.
To characterize the potential for clinically relevant DDIs due to binding of other orally administered drugs to veverimer, human DDI studies were conducted with the two drugs that showed the highest binding with veverimer in vitro: aspirin and furosemide. Given the transient effect of veverimer on gastric pH, human DDI studies were also conducted to evaluate the potential for veverimer to affect the bioavailability of drugs with pH-dependent solubility; the victim drugs evaluated in these studies were the weak acids furosemide and warfarin and the weak base dabigatran (Supplemental Table S4).
Human DDI studies were open-label, in-patient, randomized, crossover studies that examined the effect of veverimer on the pharmacokinetics (PK) profile of each of the four victim drugs. The treatments administered comprised victim drug alone, victim drug coadministered with veverimer, and victim drug separated from veverimer by 1–3 hours, and subjects were randomly assigned to treatment sequence (Supplemental Figs. S2 and S3). Key entry criteria for the studies are provided in Supplemental Section 3.1.2.1.

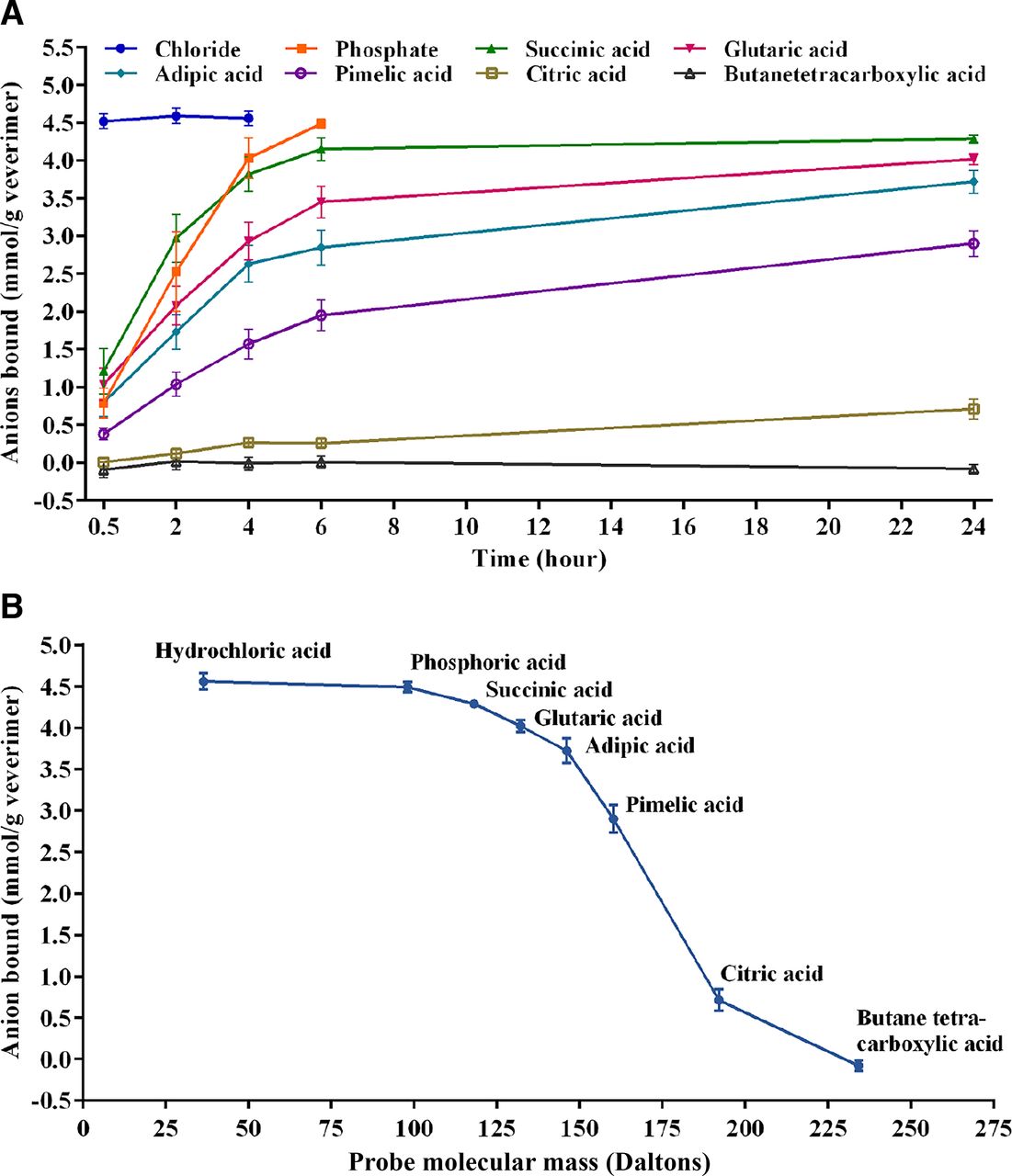
Binding of negatively charged probes to veverimer. (A) Binding kinetics for negatively charged probes. (B) Binding of the probe molecules to veverimer as a function of size. N = 9, mean ± S.D.
To maximize the possibility of observing binding to veverimer, the highest anticipated daily dose of veverimer (9 g) and the lowest feasible clinical dose of the victim drugs were administered (20 mg furosemide, 81 mg aspirin, 2 mg warfarin, 150 mg dabigatran). Study treatments were administered during treatment periods in which subjects were confined in a CRU where they were fed a standardized diet with an out-patient washout period between treatment periods and a 14-day follow-up period after the final study treatment. In each treatment period, serial blood samples for PK analysis were collected after administration of the victim drug; the sampling time periods and washout periods between treatments were based on the half-lives of the victim drugs (furosemide: 12-hour sampling time and ≥4-day washout period; aspirin: 24-hour sampling time and ≥3-day washout period; warfarin: 168-hour sampling time and ≥21-day washout period; dabigatran: 72-hour sampling time and ≥7-day washout period). Sample sizes were determined separately for each study (furosemide: 52 subjects; aspirin: 51 subjects; warfarin: 15 subjects; dabigatran: 84 subjects). All analytes [furosemide, acetylsalicylic acid, salicylic acid, R-warfarin, S-warfarin, free dabigatran, total (free + conjugated) dabigatran] were measured in plasma using validated liquid chromatography with tandem mass-spectrometric detection methods.
The primary PK endpoint of each study was the area under the concentration-time curve (AUC) from time 0 to the last observed nonzero concentration (AUC0-t), AUC from time 0 extrapolated to infinity (AUC0-inf), and Cmax for the victim drug after a single oral dose administered with and without veverimer.
For analyses of PK parameters, treatment A (victim drug alone) was the reference treatment, and the test treatments were treatments B, C, and D (victim drug administered with veverimer). Comparisons of interest were treatment B/treatment A, treatment C/treatment A, and treatment D/treatment A. A mixed-model ANOVA was used to analyze the natural logarithm (ln)-transformed AUC0-t, AUC0-inf, and Cmax of the victim drug. The ANOVA model included sequence, treatment, and period as fixed effects and subject nested within sequence as a random effect. Estimates of least-squares means (LSMs) for victim drug (treatment A, reference), victim drug + veverimer (treatment B, treatment C, or treatment D test), difference in LSM and 90% CIs of the difference between the test and the reference LSM were first constructed in the logarithmic scale. Upon back-transformation, the geometric least-squares mean (GLSM), geometric least-squares-mean ratio (GLSMR), and 90% CI of GLSMR were calculated for AUC0-t, AUC0-inf, and Cmax. The GLSMRs and 90% CIs of GLSMRs were expressed as percentages relative to treatment A, consistent with the two one-sided tests. The sample size in each study was calculated using a power of at least 80% and an α error of 5%. The power was defined as the probability of having a 90% CI for at least one of the treatment ratios within the interval of 80.00%–125.00%. A true ratio between 95% and 105% was assumed, and the intrasubject CV for each analyte was provided by the clinical research organization responsible for conduct of the studies.
Two pharmacodynamics (PD) parameters were assessed in the warfarin DDI study: the area under the international normalized ratio (INR) versus time curve from time 0 to the 168-hour time point after warfarin dosing (INR AUC0-168) and the maximum observed INR measurement (INRmax).
All DDI studies included assessment of the safety and tolerability of veverimer when coadministered with the victim drug. Safety was evaluated from 12-lead ECGs, measurements of vital signs and clinical laboratory parameters, assessment of AEs, and physical examinations.
Information on the disposition of subjects in the human DDI studies is summarized in Supplemental Table S6. A total of 52 subjects were randomized, enrolled, and received at least one dose of study drug in the furosemide DDI study. The furosemide PK analysis set comprised 51 subjects; one subject vomited during administration of the first dose of veverimer in treatment period 1 and was withdrawn from the study. Per protocol, because the subject did not have ≥2 consecutive time points with measurable furosemide concentrations, he was excluded from the furosemide PK analysis set.
In the aspirin DDI study, a total of 51 subjects were randomized, enrolled, and received at least one dose of study drug and were included in the aspirin PK analysis set. Forty-eight subjects completed the study; three subjects withdrew early for diverse reasons (i.e., mild headache, difficulty with study blood draws, personal reasons).
In the warfarin DDI study, a total of 15 subjects were randomized, enrolled, received at least one dose of study drug, and completed the study. The warfarin PK and PD analysis sets comprised 15 subjects who complied sufficiently with the protocol and displayed evaluable PK and PD profiles, respectively. A subset analysis was prespecified that excluded obviously aberrant PK profiles based on the presence of outlier values in one or more of the key PK parameters, which are defined as those falling below the first quartile or above the third quartile by more than 1.5 times the interquartile range, in the event that outlier values were noted for the PK parameters AUC0-t, AUC0-inf, or Cmax, for R- or S-warfarin. The initial PK and statistical assessments included a PK profile for one subject [treatment A (warfarin alone)] that diverged dramatically from the remainder of the data. It was readily apparent on visual inspection of the concentration-time profiles that there was a unique issue in this individual on this dosing occasion (i.e., the subject did not appear to have ingested the drug), and that this profile should be excluded from the analysis. A cause for the effective absence of warfarin blood levels in this subject on this occasion could not be identified. The decision to perform subset analyses with the aberrant treatment A profile removed from the warfarin PK analysis set was supported by the statistical assessment described above.
In the warfarin DDI study, Plasma R- and S-warfarin levels were below the limit of quantitation (i.e., <1.00 ng/mL) in all samples collected prior to dosing in period 1. Quantifiable R- and S-warfarin concentrations were observed in all predose plasma samples in periods 2 and 3, which was consistent with carryover from the prior dose. This suggests that the 21-day washout period was insufficient. However, individual predose concentrations were low, ranging from approximately 1.5%–6.8% of the subsequent Cmax for R-warfarin, with nine instances exceeding 5%. For S-warfarin, predose concentrations ranged from approximately 2.0%–8.6% of Cmax, with four instances exceeding 5%. Therefore, it is highly improbable that the measurable predose concentrations had a discernable impact on the PK data.
In the dabigatran DDI study, a total of 84 subjects were randomized, enrolled, and received at least one dose of study drug. Of the 84 subjects in the dabigatran PK analysis set, 81 subjects (96.4%) completed the study, receiving all doses of each study drug; three subjects had incomplete PK data; one subject was discontinued for a protocol violation and had no PK data for treatment D; and three subjects withdrew early causing one to miss treatment D and the other to miss treatments C and D. The available data for all subjects were included in the PK analyses.
Demographic and baseline characteristics of the subjects enrolled in the DDI studies are summarized in Supplemental Table S7. The average age of the subjects in the studies ranged from 33 to 39 years and the study populations ranged from 47% to 80% male. The majority (80%–90%) of the patients studied were White, 2%–79% were Hispanic or Latino, and 6%–13% were Black or African American.
Results
Binding of Veverimer to Anionic Probes and Representative Test Drugs In Vitro.
The binding kinetics for the anionic probe molecules to veverimer are shown in Fig. 2A, and binding of the probe molecules to veverimer as a function of size is illustrated in Fig. 2B. The rate of binding as well as the total amount of probe bound to the polymer were inversely proportional to the size of the probe, suggesting that smaller negatively charged molecules preferentially bound to the polymer over larger negatively charged molecules. Anionic probe molecules >200 Da did not bind to veverimer.
In in vitro binding experiments using a panel of 16 test drugs, none of the five test drugs that are positively charged across the physiologic pH range of the GI tract (i.e., amlodipine, metformin, metoprolol, thiamine, and trimethoprim) bound to veverimer under any condition (Fig. 3). Similarly, none of the four test drugs that are neutral or zwitterionic (i.e., allopurinol, riboflavin, spironolactone, and lisinopril) bound to veverimer under any condition (Fig. 3). The remaining seven test drugs (aspirin, ethacrynic acid, furosemide, valsartan, rosuvastatin, warfarin, and gliclazide) are weak acids; of these, only furosemide, aspirin, and ethacrynic acid bound to veverimer, and these only did so in the acetate buffer (pH 4.5) matrix. Under the pH conditions of the SGF matrix (pH 1–1.2), the weak acids are neutral (Table 1), and none bound to veverimer (Fig. 3). Although the weak acids are negatively charged in the SIF matrix (pH 6.8), none bound to veverimer, likely because 50–100 mM of the small anion, phosphate, was available to compete for veverimer binding sites (Fig. 3).

In vitro binding of test drugs to veverimer. The potential for binding interactions with veverimer was assessed in vitro using a set of test drugs that included 14 oral medications and two water-soluble vitamins and test matrices mimicking the pH of various GI compartments. (A) 4.5 mg/mL veverimer in SGF; (B) 9.0 mg/mL veverimer in SGF + 60 mM HCl; (C) 4.5 mg/mL veverimer in 50 mM acetate buffer; (D) 9.0 mg/mL veverimer in 100 mM acetate buffer; (E) 4.5 mg/mL veverimer in SIF; (F) 9.0 mg/mL veverimer in SIF + 50 mM PO4. The dashed line represents the threshold for test drug interaction with veverimer. PO4, phosphate. N = 6, mean ± 90% confidence interval.
The three test drugs that bound to veverimer in the acetate buffer matrix (aspirin, ethacrynic acid, and furosemide) all have similar pKa (3.5, 3.5 and 3.62, respectively) and are negatively charged in the acetate buffer matrix. Another test drug, valsartan, has a similar pKa (3.9) but did not bind to veverimer (Fig. 3). Valsartan has a molecular mass of 435.5 Da, whereas the other three test drugs are relatively small, with molecular masses ranging from 180.2 to 330.7 Da, suggesting that the size cutoff for a negatively charged compound to bind to veverimer was between 330 and 435 Da. Furosemide (MM = 330.7) is similar in size to gliclazide (MM = 323.4) and warfarin (MM = 308.3), but of these three weak acids, only furosemide bound to veverimer (Fig. 3). Importantly, only furosemide (pKa = 3.62) but neither gliclazide (pKa = 5.80) nor warfarin (pKa = 4.94) are negatively charged in the acetate buffer matrix.
In the presence of physiologically relevant concentrations of chloride (100 mM), the three test drugs (aspirin, ethacrynic acid, furosemide) that bound to veverimer in acetate buffer were unable to bind, likely because of preferential binding of the small chloride ion (Fig. 4). Drugs previously shown not to bind veverimer (allopurinol, trimethoprim) also did not bind to the polymer in the presence of 100 mM chloride.
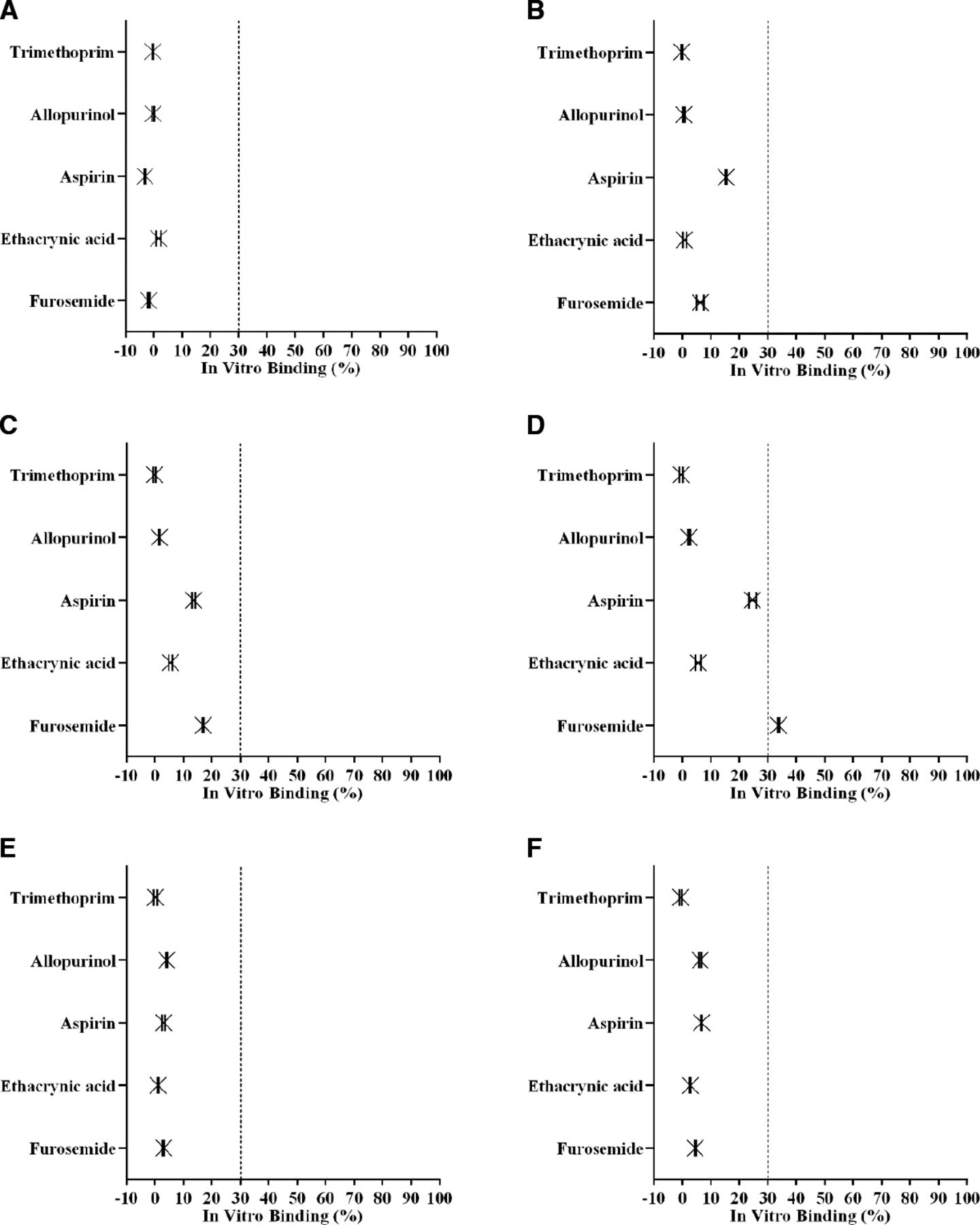
In vitro binding of test drugs to veverimer in the presence of physiologically relevant concentrations of chloride. Five representative test drugs from the set of 14 oral medications, one each with a positive (trimethoprim) or neutral (allopurinol) charge and three with a negative charge (aspirin, ethacrynic acid, and furosemide) were assessed for veverimer binding in six test matrices supplemented with 100 mM chloride. (A) 4.5 mg/mL veverimer in SGF + 100 mM chloride; (B) 9.0 mg/mL veverimer in SGF + 100 mM chloride; (C) 4.5 mg/mL veverimer in 50 mM acetate buffer with 100 mM chloride; (D) 9.0 mg/mL veverimer in 100 mM acetate buffer with 100 mM chloride; (E) 4.5 mg/mL veverimer in SIF with 100 mM chloride; (F) 9.0 mg/mL veverimer in SIF + 50 mM PO4 with 100 mM chloride. The dashed line represents the threshold for test drug interaction with veverimer. PO4, phosphate. N = 6, mean ± 90% confidence interval.
Effects of Veverimer on Gastric pH with and without a Proton Pump Inhibitor.
The magnitude and duration of effect of veverimer on gastric pH were measured continuously in vivo in healthy volunteers using a microelectrode pH probe.
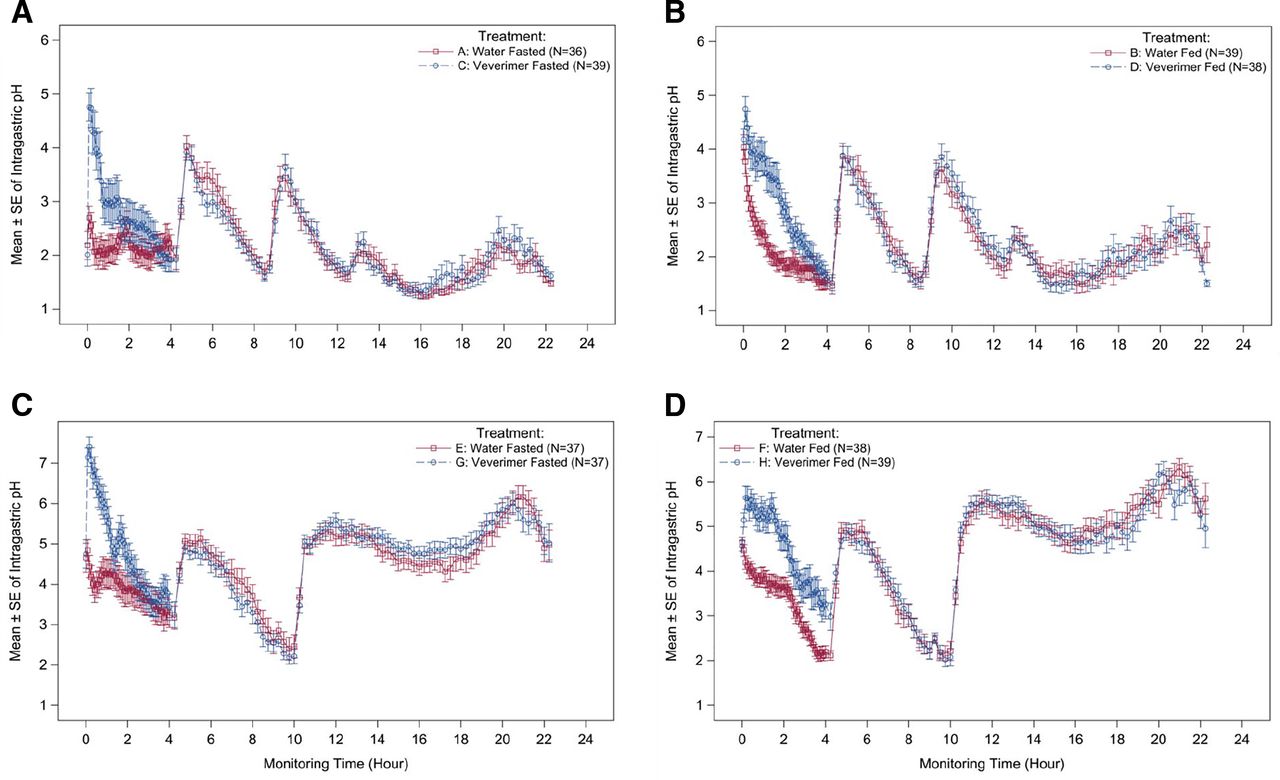
Veverimer increased gastric pH by approximately 3 pH units and approximately 1.5 pH units in fasted and fed subjects, respectively (Fig. 5, A and B). The increase in gastric pH peaked within 1 hour after veverimer dosing and returned to baseline after approximately 1.5 hours and approximately 3 hours under fasting and fed conditions, respectively. The effect on gastric pH after administration of veverimer was similar in the presence and absence of a proton pump inhibitor (omeprazole) (Fig. 5, C and D). Supplemental Figures S4 and S5 show the independent effects of food or omeprazole (in the absence of veverimer) on gastric pH.
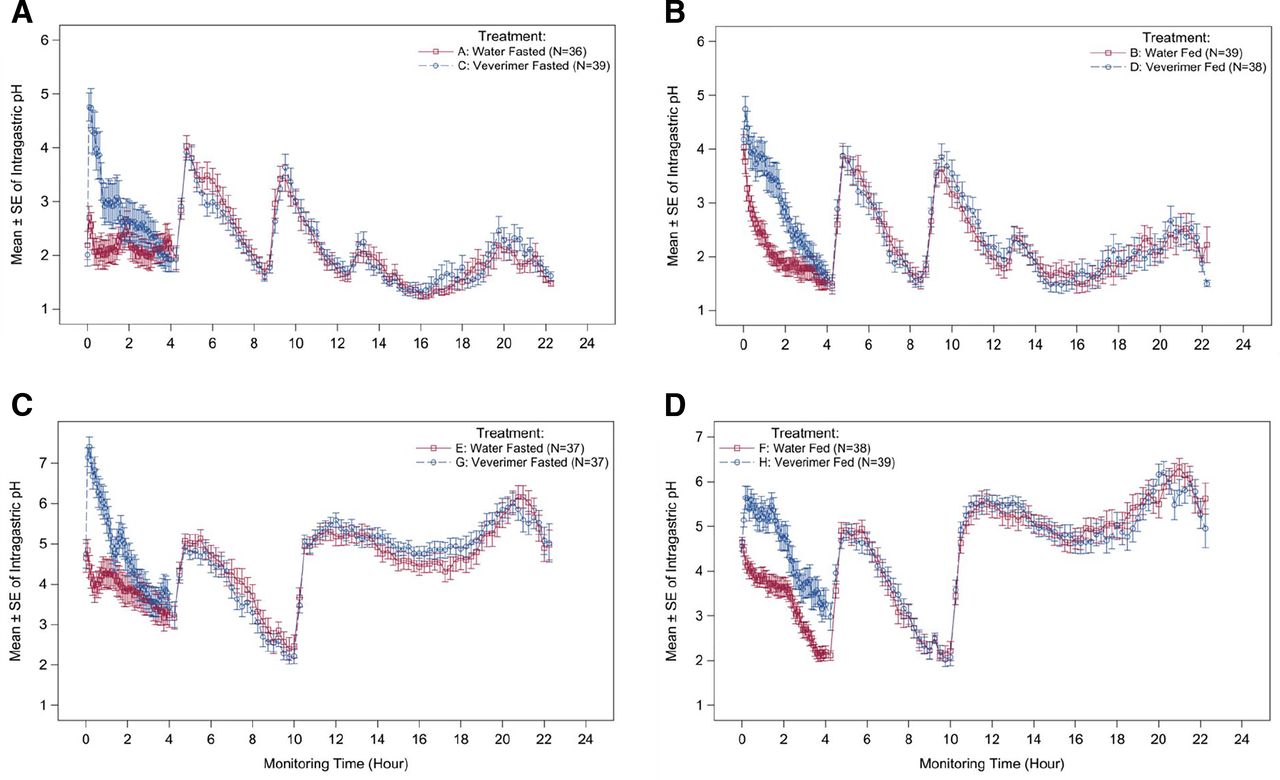
Effect of veverimer on gastric pH in the absence and presence of a proton pump inhibitor. The magnitude and duration of effect of veverimer on gastric pH was measured continuously in vivo in healthy volunteers using a microelectrode pH probe. Ingestion of veverimer or water occurred at ?hour 0, just after initiation of pH monitoring. In experiments assessing the fed condition, breakfast was eaten within 15 minutes prior to hour 0. Other meal/snack times occurred at ?hour 4, hour 9 (or 10), and hour 13. Omeprazole was administered at hour 9. (A) Veverimer versus water control under fasted conditions (without PPI); (B) veverimer versus water control under fed conditions (without PPI); (C) veverimer versus water control under fasted conditions (with PPI); (D) veverimer versus water control under fed conditions (with PPI).
A total of 12 of 40 subjects (30%) in stage 1 and 13 of 40 subjects (32.5%) in stage 2 had one or more AEs during the study. No deaths or other serious AEs were reported, and no subject discontinued the study because of an AE. The most common AEs observed in the veverimer treatment periods (i.e., reported by more than one subject in either study stage in the veverimer-fasted and veverimer-fed treatment periods combined) were headache, nausea, and oropharyngeal pain. All AEs were mild (22.5% of subjects in stage 1 and 30% of subjects in stage 2) or moderate (7.5% of subjects in stage 1 and 2.5% of subjects in stage 2); there were no severe AEs reported for any treatment group. There were no treatment effects noted in this study on clinical laboratory or vital signs parameters, physical examination findings, or ECG intervals.
Human Drug-Drug Interaction Studies.
Human DDI studies were conducted with drugs that demonstrated the greatest potential for direct interaction with veverimer in vitro (furosemide, aspirin) and those with susceptibility to gastric pH changes (furosemide, warfarin, dabigatran).
No DDIs were observed between veverimer and any of the drugs tested, either with concomitant administration (Fig. 6) or with 1–3-hour dosing separation intervals (Table 2; Supplemental Figs. S6–S19; Supplemental Table S8). For furosemide, acetylsalicylic acid, salicylic acid, R-warfarin, and S-warfarin, GLSM ratios and 90% CIs of Cmax, AUC0-t, and AUC0-inf for the test treatments (veverimer coadministered with or separated by 3 hours from the victim drug) all fell within the range of 80.00%–125.00%. When veverimer and dabigatran were coadministered, the 90% CIs of GLSM ratios for free and total dabigatran Cmax were within the range of 80.00%–125.00%, whereas the lower bound of the 90% CIs for AUC0-t and AUC0-inf fell slightly below 80.00% for free (77.3% and 77.6%, respectively) and total (77.1% and 77.7%, respectively) dabigatran, with GLSM ratio point estimates of 82.4% to 82.8% for both analytes. The GLSM ratios and 90% CIs for free and total dabigatran Cmax, AUC0-t, and AUC0-inf were within 80.00%–125.00% when veverimer and dabigatran doses were separated by 1 or 2 hours.
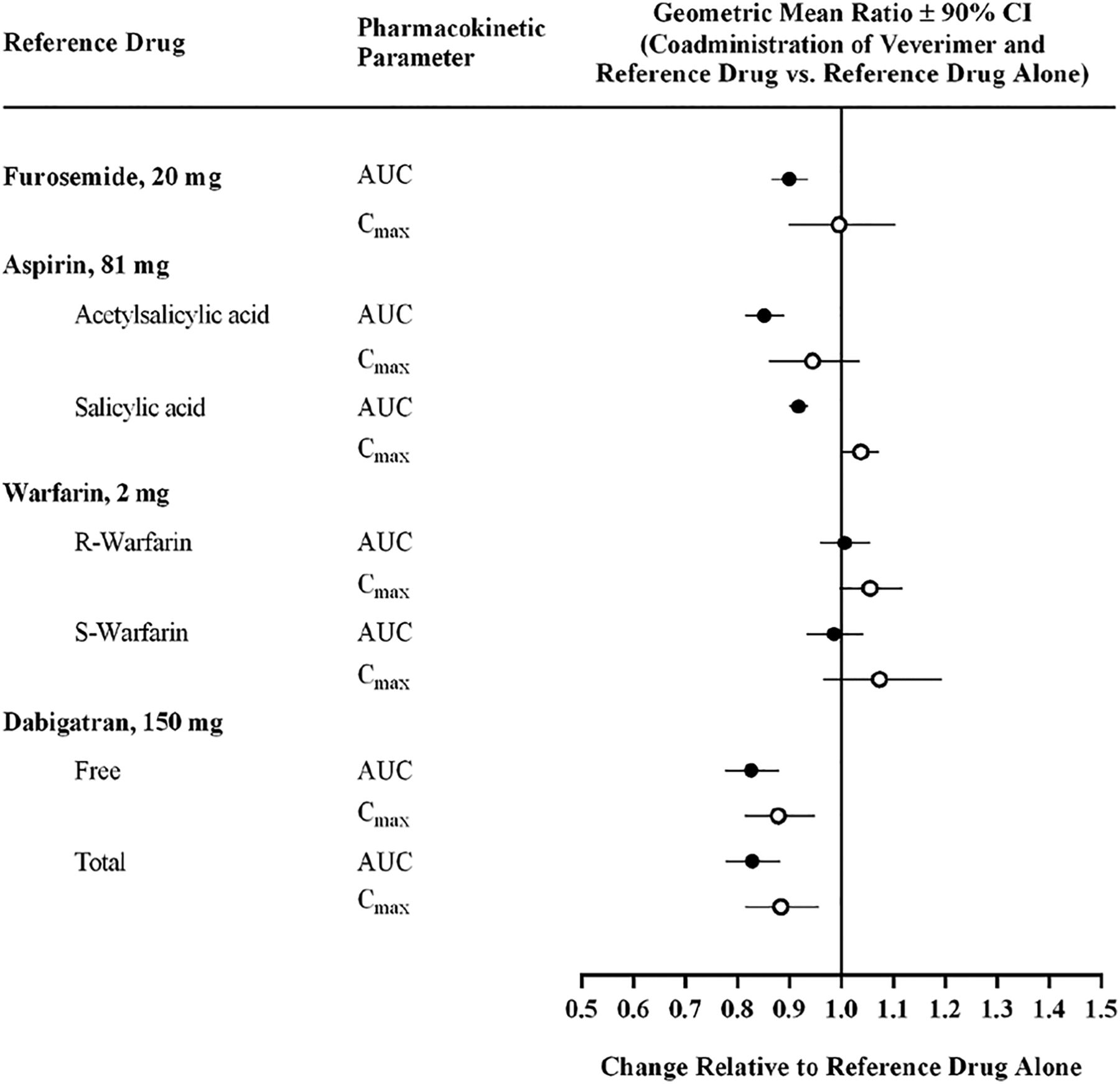
Effects of veverimer on the pharmacokinetic exposures of other orally administered medications. The effect of veverimer on the bioavailability of test drugs chosen to represent medications most likely to interact with veverimer via direct binding (furosemide, aspirin) or via indirect effects on gastric pH (furosemide, warfarin, dabigatran) was assessed in healthy volunteers.
Pharmacokinetic analyses: ANOVA comparison of geometric least-squares-mean ratios (90% confidence intervals)
Median INR AUC0-168 and INRmax values after administration of warfarin alone and coadministered with veverimer and administered between two doses of veverimer with a 3-hour dosing separation were 168 hours to 170 hours and 1.00 to 1.10, respectively (Supplemental Figs. S20 and S21; Supplemental Table S9). The 90% CIs for INR AUC0-168 and INRmax fell within the range of 80.00%–125.00% for both test/reference assessments (Supplemental Table S10).
In the furosemide DDI study, there were no deaths, serious AEs, or severe treatment-emergent AEs. Three subjects were withdrawn because of treatment-emergent AEs (mild postural dizziness, mild eosinophil count increase and mild white blood cell count increase, mild vomiting). In the aspirin DDI study, there were no deaths, serious AEs, or severe treatment-emergent AEs. All treatment-emergent AEs were mild. One subject withdrew because of a mild headache. In the warfarin DDI study, there were no deaths or treatment-emergent AEs leading to discontinuation of study drug. One subject had a serious AE [jaw fracture (due to trauma)] 10 days after study treatment in period 1. This event was assessed as unrelated to study drug. All other treatment-emergent AEs that occurred were mild or moderate and nonserious. In the dabigatran DDI study, there were no deaths, serious AEs, or severe treatment-emergent AEs. There were no treatment-emergent AEs that led to discontinuation of study drug.
There were no notable changes from baseline to any time point for hematology, serum chemistry, or urinalysis parameters or for vital signs or ECG parameters in any of the DDI studies.
Discussion
Reported here are results from in vitro and in vivo studies that evaluated potential interactions of veverimer with other orally administered drugs. We considered the physicochemical and biopharmaceutical properties and method of use of veverimer to design a rational, sequential, and tailored approach that examined possible mechanisms of interaction. Because the polymer is not absorbed from the GI tract, it is unlikely that it would alter the pharmacokinetics of concomitantly administered drugs through inhibition and/or induction of drug-metabolizing enzymes or transporters. Potential interactions with veverimer are restricted to those that could affect absorption of other drugs from the GI tract, such as through direct binding or indirect effects on bioavailability resulting from transient increases in gastric pH.
The mechanism by which veverimer transiently reduces acidity in the GI tract after ingestion involves protonation of the polyamine polymer with subsequent binding of chloride and the removal of HCl from the GI tract in the feces (Bushinsky et al., 2018). The highly crosslinked structure of veverimer confers a marked size exclusion selectivity to the negatively charged moieties that bind to the protonated polymer, strongly favoring binding of the smallest anions and restricting binding of larger anions (Klaerner et al., 2020). These properties were affirmed in studies reported here evaluating the in vitro binding of a range of anionic probe molecules and a diverse panel of test drugs clinically relevant to the CKD population. The results of these studies illustrated the predisposition of veverimer to bind negatively charged molecules, with an inverse relationship between size of the anion and its propensity for binding to the polymer (Fig. 2); data on furosemide, gliclazide, and warfarin suggested that size is secondary to negative charge in this regard. Although binding of aspirin, ethacrynic acid, and furosemide to veverimer was significant at pH 4.5 in acetate buffer, it was reduced or eliminated in the presence of physiologically relevant concentrations of chloride (100–170 mM) (Fig. 4). This is consistent with results from previous in vitro studies in matrices mimicking the lower GI tract demonstrating that veverimer preferentially bound chloride in the presence of competing organic and inorganic anions [e.g., acetate, phosphate, citrate, taurocholate, oleic acid (Klaerner et al., 2020)]. In the current study, chloride effectively competed for veverimer binding sites with the three negatively charged test drugs at chloride concentrations normally present in the GI tract, which predicted a low likelihood of clinically meaningful DDIs mediated by direct binding of even the smallest coadministered anionic drugs.
Consistent with the binding properties of veverimer, administration of the drug did not impact absorption of fat-soluble vitamins from the GI tract in rat and dog chronic toxicity studies (Supplemental Section 2). Since the essential nutrient requirements of rats and dogs are qualitatively similar to humans, the absence of clinically relevant binding of veverimer to any essential dietary substance in the chronic toxicology studies is expected to extrapolate to humans.
The pharmacodynamic effects of veverimer on gastric acidity were evaluated in human volunteers to elucidate the extent and time course of gastric pH changes mediated by the intended removal of HCl from the GI tract by the polymer. Continuous monitoring of stomach acidity demonstrated a modest and transient increase in gastric pH after oral administration of veverimer. The magnitude of the gastric pH increase observed after ingestion of a meal or when the PPI omeprazole was given without food was similar to that seen with veverimer given in the fasted state. All three factors individually appeared to increase mean gastric pH by 2–4 pH units, although the time course of the effect on gastric pH differed. Although omeprazole, which had been dosed to steady state prior to the test, increased gastric pH throughout the 22-hour monitoring period, the effects of food and veverimer were short-lived (i.e., disappearing within 1–4 hours). In this way, the effect of veverimer on gastric pH more closely resembled the transient effect of food than the long-lasting effect of a PPI. Thus, the mechanism of action of veverimer (i.e., increasing serum bicarbonate by binding and removing HCl from the GI tract), as assessed by its effect on gastric pH, was unaffected by proton pump inhibition. These findings are consistent with clinical trials in patients with CKD and metabolic acidosis, which showed that the effect of veverimer on serum bicarbonate was similar in patients who were and were not receiving proton pump inhibitors or H-2 receptor blockers (Wesson et al., 2019b).
Data from the in vitro studies and in the gastric pH-monitoring investigations suggested a low likelihood of significant interactions between veverimer and concomitantly administered drugs mediated by direct binding or modulation of acidity in the gastric compartment. Human DDI studies were performed to test these hypotheses. Furosemide and aspirin were chosen as probe drugs to characterize the DDI potential of veverimer because these drugs showed the highest binding to veverimer in vitro. The human DDI studies were designed to maximize the potential for binding to the polymer by administering the highest therapeutic dose of veverimer with the lowest dose of the victim drug that would allow adequate characterization of its pharmacokinetic parameters. In addition, the transient increase in gastric pH caused by veverimer could represent a potential DDI risk for drugs with pH-sensitive solubility. Therefore, DDI studies were conducted using three drugs with pH-dependent solubility that are used in the veverimer target population (warfarin, furosemide, dabigatran).
No DDIs were observed between veverimer and any of the four victim drugs tested, either with concomitant administration or with dose separations of 1 to 3 hours. Administration of veverimer with the victim drugs did not result in a clinically relevant change in exposure of aspirin or its metabolite, salicylic acid, furosemide, or warfarin (R- and S-enantiomers) and did not alter the pharmacodynamic effect of warfarin, as indicated by international normalized ratio values. The 90% confidence intervals for GLSM ratios for all analytes under both dosing conditions were completely contained within a reference range of 80.00% to 125.00%, which are the boundaries used in evaluating bioequivalence (Food and Drug Administration, 2014). With both free and total dabigatran, GLSM ratio point estimates were reduced by less than 18% when coadministered with veverimer, with the effect lessening slightly with a widening of the dose separation interval (e.g., 1 and 2 hours). This minimal reduction is substantially less than that observed when dabigatran was coadministered with the PPI pantoprazole, which diminished dabigatran Cmax and AUC by 40% and 28%, respectively, changes that are not clinically meaningful (Zhang et al., 2014). Notably, the lack of effect on aspirin, furosemide, warfarin, and dabigatran exposures indicates that veverimer does not interact in a clinically relevant manner with small, negatively charged drugs or drugs that have pH-dependent solubility, demonstrating the lack of two potential mechanisms by which the veverimer polymer might foster DDIs.
In summary, the potential for DDIs with veverimer was evaluated based on the known site of action and physicochemical structure of the polymer, which restricts the compound to the GI tract and direct binding interactions. In vitro studies defined three parameters of test drugs that controlled potential direct DDIs with veverimer, including charge, molecular mass, and competition with chloride anion. Human DDI studies in healthy volunteers demonstrated that concomitant dosing of veverimer with drugs showing in vitro binding had no effect on test drug bioavailability; similarly, drugs with pH-sensitive solubility were not affected by coadministration with veverimer. Patients with CKD, who are usually on many drugs, are particularly vulnerable to DDIs (Sommer et al., 2020). Based on the physicochemical characteristics of veverimer and the findings from the in vitro and human studies presented here, we conclude that veverimer is unlikely to have clinically significant DDIs.
Acknowledgments
We thank Dr. David Bushinsky and Dr. Jerry Buysse for review of the manuscript. We thank the investigators, site personnel, and subjects who participated in the clinical trials; members of the Tricida clinical operations, regulatory and chemistry; manufacturing and controls teams; and contract research organization personnel at Celerion for execution of the studies.
Authorship Contributions
Participated in research design: Parsell, Shao, Guttendorf, Mathur, Li, Tabakman, Stasiv, Lee, Biyani, Klaerner.
Conducted experiments: Shao, Mathur, Wu, Tsao, Tabakman, Stasiv, Biyani.
Contributed new reagents or analytic tools: Wu, Tsao, Tabakman, Biyani.
Performed data analysis: Parsell, Shao, Guttendorf, Mathur, Li, Tabakman, Lee, Biyani, Klaerner.
Wrote or contributed to the writing of the manuscript: Parsell, Shao, Guttendorf, Mathur, Li, Tabakman, Stasiv, Lee, Biyani, Klaerner.
Footnotes
- Received January 8, 2021.
- Accepted April 12, 2021.
This work was funded by Tricida, Inc.
D.P., J.S., Y.S.W., L.T., S.T., Y.S., A.L., K.B., and G.K. were employees of Tricida, Inc. at the time of the work. R.G., V.M., and E.L. were paid consultants to Tricida, Inc. in connection with the development of this manuscript. D.P., J.S., V.M., Y.S.W., L.T., S.T., Y.S., A.L., K.B., and G.K. own stock or stock options in Tricida, Inc. D.P., J.S., V.M., S.T., Y.S., K.B., and G.K. are listed on patents related to work for Tricida, Inc. V.M. and G.K. are members of Tricida, Inc. advisory boards. G.K. is a member of the Board of Directors for Tricida, Inc. V.M. reports additional consulting fees from Tricida, Inc., Equillium, Myovant, Rigel, Corvidia, Acuta, Frazier, Intarcia, PTC Bio, and Sanifit outside the submitted work.
The work was partially published in abstract form at Shao J, Parsell P, Guttendorf R, Wu YS, Tsao L, Tabakman S, Stasiv Y, Lee A, Biyani K, Klaerner G (2020) Evaluation of the potential for drug interactions with veverimer. Kidney Week 2020 Virtual Meeting; 2020 Oct 22. American Society of Nephrology; and in Shao J, Parsell P, Guttendorf R, Wu YS, Tsao L, Tabakman S, Stasiv Y, Lee A, Biyani K, Klaerner G (2020) Evaluation of the potential for drug interactions with veverimer. European Renal Association – European Dialysis and Transplant Association 2020 Virtual Meeting; 2020 Jun 6–9. European Renal Association – European Dialysis and Transplant Association.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AE
- adverse event
- AUC
- area under the concentration-time curve
- AUC0-inf
- area under the concentration-time curve from time 0 extrapolated to infinity
- AUC0-t
- area under the concentration-time curve from time 0 to the last observed nonzero concentration
- CKD
- chronic kidney disease
- CI
- confidence interval
- CRU
- clinical research unit
- DDI
- drug-drug interaction
- GI
- gastrointestinal
- GLSM
- geometric least-squares mean
- GLSMR
- geometric least-squares-mean ratio
- HCl
- hydrochloric acid
- INR
- international normalized ratio
- INR AUC0-168
- area under the INR versus time curve from hour 0 to hour 168 postdosing
- INRmax
- maximum observed INR measurement
- LSM
- least-squares mean
- MM
- molecular mass
- PD
- pharmacodynamics
- PK
- pharmacokinetics
- PPI
- proton pump inhibitor
- PVDF
- polyvinylidene fluoride
- QD
- once daily
- SGF
- simulated gastric fluid
- SIF
- simulated intestinal fluid
- Copyright © 2021 by The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}