


Abstract
UDP-glucuronosyltransferase (UGT)1A4 and UGT2B10 are the human UGT isoforms most frequently involved in N-glucuronidation of drugs. UGT2B10 exhibits higher affinity than UGT1A4 for numerous substrates, making it potentially the more important enzyme for metabolism of these compounds in vivo. Clinically relevant UGT2B10 polymorphisms, including a null activity splice site mutation common in African populations, can lead to large exposure differences for UGT2B10 substrates that may limit their developability as marketed drugs. UGT phenotyping approaches using recombinantly expressed UGTs are limited by low enzyme activity and lack of validation of scaling to in vivo. In this study, we describe the use of an efficient experimental protocol for identification of UGT2B10-selective substrates (i.e., those with high fraction metabolized by UGT2B10), which exploits the activity difference between pooled human liver microsomes (HLM) and HLM from a phenotypically UGT2B10 poor metabolizer donor. Following characterization of the approach with eight known UGT2B10 substrates, we used ligand-based virtual screening and literature precedents to select 24 potential UGT2B10 substrates of 140 UGT-metabolized drugs for testing. Of these, dothiepin, cidoxepin, cyclobenzaprine, azatadine, cyproheptadine, bifonazole, and asenapine were indicated to be selective UGT2B10 substrates that have not previously been described. UGT phenotyping experiments and tests comparing conjugative and oxidative clearance were then used to confirm these findings. These approaches provide rapid and sensitive ways to evaluate whether a potential drug candidate cleared via glucuronidation will be sensitive to UGT2B10 polymorphisms in vivo.
SIGNIFICANCE STATEMENT The role of highly polymorphic UDP-glucuronosyltransferase (UGT)2B10 is likely to be underestimated currently for many compounds cleared via N-glucuronidation due to high test concentrations often used in vitro and low activity of UGT2B10 preparations. The methodology described in this study can be combined with the assessment of UGT versus oxidative in vitro metabolism to rapidly identify compounds likely to be sensitive to UGT2B10 polymorphism (high fraction metabolized by UGT2B10), enabling either chemical modification or polymorphism risk assessment before candidate selection.
Introduction
Enzymes belonging to the UDP-glucuronosyltransferase (UGT) superfamily catalyze the glucuronidation of molecules containing nucleophilic functional groups (e.g., phenols, carboxylic acids, hydroxyls, amines, azoles, and thiols), facilitating their elimination from the body. The role of these enzymes in the metabolism of candidate drug molecules has been of increasing interest in recent years, especially with the efficient optimization of cytochrome P450 (P450)–mediated clearance and more frequent introduction of nitrogen-containing heterocycles into drug candidates (Gomtsyan, 2012). The object of the present study is the hepatic UGT2B10 isoform, which is specialized in N-glucuronidation reactions (Kaivosaari et al., 2011). Scientific interest in this isoform has risen with increasing appreciation of its relevance to drug metabolism and availability of enzymology tools to study UGT2B10 involvement in metabolism (Fowler et al., 2015; Kazmi et al., 2015). Some well-known UGT2B10 substrates include RO5263397 (Fowler et al., 2015), nicotine and its derivate cotinine (Chen et al., 2007; Pattanawongsa et al., 2016), chlorcyclizine and cyclizine (Lu et al., 2018), amitriptyline (Zhou et al., 2010; Kato et al., 2013), medetomidine (Kaivosaari et al., 2008), and desloratadine (Kazmi et al., 2015). The case of RO5263397 illustrates the importance of UGT2B10 substrate identification prior to drug development. A 136-fold above-average systemic exposure was observed in the plasma of a healthy volunteer subject after low-dose oral administration of RO5263397 (Fowler et al., 2015). A UGT2B10 splice site polymorphism was discovered, which conferred a UGT2B10 null activity phenotype in this individual. This splice site polymorphism (rs2942857) occurred at allelic frequencies of 45%, 8%, and <1% in individuals of African, Asian, and Caucasian origin, respectively, in data from the 1000 genomes program (Fowler et al., 2015). Additional UGT2B10 polymorphisms have also been described that result in reduced enzyme activity phenotypes (Chen et al., 2007, 2010; Berg et al., 2010). Clinical development of a UGT2B10-selective substrate may therefore be challenging, dependent upon the contribution of glucuronidation to overall clearance, the extent of first-pass metabolism, and the therapeutic index of the drug.
UGT2B10 is not the only enzyme frequently involved in N-glucuronidation reactions. UGT1A4 exhibits overlapping selectivity for many UGT2B10 substrates, and UGT1A3 may also catalyze the N-glucuronidation of some drugs (Kato et al., 2013). Compared with UGT2B10, UGT1A4 is a better understood and more comprehensively studied enzyme and has been assigned as the main contributor to many N-glucuronidation reactions. UGT2B10 and UGT1A4 have been shown not to correlate strongly [P > 0.05 for protein abundance (Achour et al., 2017); P = 0.10 for activity (Badée et al., 2019a)]. Many N-glucuronidated drug molecules are substrates of both UGT2B10 and UGT1A4 (Chen et al., 2007), but their kinetics may differ substantially. RO5263397, amitriptyline, imipramine, and diphenhydramine showed biphasic kinetic profiles in human liver microsomes with UGT2B10 taking the role of higher-affinity isoform (Kato et al., 2013; Fowler et al., 2015). In such cases, the high-affinity isoform contributes more to clearance in vivo due to the relatively low circulating drug concentrations. As many UGT phenotyping studies in the literature have been performed at concentrations of 10 µM or higher, favoring UGT1A4 activity, and some studies have not included UGT2B10 in the panel of enzymes assessed, there is a possibility that the role of UGT2B10 has been underestimated or overlooked (Lu et al., 2017; Cho et al., 2019; Xia et al., 2019). This is especially likely for drugs developed more than 10 years ago when UGT2B10 was considered an “orphan enzyme” with no assigned functionality and not commercially available (Kaivosaari et al., 2007).
In a recent study, we determined the activity for 10 human hepatic UGT isoforms using a panel of individual donor human liver microsome (HLM) samples and identified two donors that exhibited a UGT2B10 poor metabolizer phenotype (Badée et al., 2019a). Such donors are unusual, but not rare (e.g., one of six African American donors and one of six Hispanic donors in our recent study). Microsomes from these donors exhibited strikingly lower UGT2B10 activities compared with other donors and the 150-donor pooled liver microsomes, suggesting their utility in the detection of UGT2B10 substrates. One of these donors [H0295, henceforth referred to as HLM(-) in this manuscript] was selected because it exhibited very low UGT2B10 activity, whereas other UGT activities were retained at high levels, minimally 40% those of pooled HLM (Supplemental Fig. 1).
In this study, we describe a methodology for rapid identification of UGT2B10-selective substrates (i.e., those with high fraction metabolized by UGT2B10) comparing the glucuronidation activities of pooled HLM and HLM(-) samples. The method was validated using known UGT2B10 substrates and used to screen for new UGT2B10-selective substrates within a pool of likely candidates coming from ligand-based virtual screening and structural similarity searches of drugs known to be metabolized via glucuronidation. Finally, the methodology was compared with recombinant UGT phenotyping approaches to demonstrate its applicability in UGT2B10-selective substrate identification.
Materials and Methods
Materials
Alamethicin, amitriptyline hydrochloride, trifluoperazine, sertraline hydrochloride, NADPH, and uridine 5′-diphosphoglucuronic acid (UDPGA, trisodium salt) were purchased from Sigma-Aldrich (St. Louis, MO); cyclizine hydrochloride, asenapine, dothiepin, and (S)-(-)-nicotine were purchased from Toronto Research Chemicals (North York, ON, Canada); rupatadine fumarate, (±)-tioconazole, levamisole hydrochloride, lamotrigine, azatadine dimaleate, (±)-alprenolol, imidafenacin, and varenicline were purchased from Med Chem Express (Sollentuna, Sweden); (S)-(-)-cotinine, cidoxepin, tamoxifen, lidocaine, cyprohetadine, cyclobenzaprine, oxymetazoline, brucine, trazodone, carbamazepine, norclozapine, (±)-sulconazole, (±)-bifonazole, desloratadine, RO5263397, loxapine, clozapine, and (±)-propranolol were supplied by the internal Roche compound repository (Basel, Switzerland); (±)-chlorcyclizine was purchased from Cayman (Ann Arbor, MI); and (±)-miconazole was purchased from MP Biomedicals (Illkirch-Graffenstaden, France). Pooled HLMs (lot 38290, 150 donor pool 75 male, 75 female) and the recombinant UGTs 1A1, 1A3, 1A4, 1A6, 1A9, 2B4, 2B7, 2B10, 2B15, and 2B17 were purchased from Corning. HLM from individual donor (commercial code: H0295, lot 0510181, female, 54 year, African American) were purchased from XenoTech (Kansas City, KS). Tris buffer was supplied by Invitrogen (Carlsbad, CA).
Screening Assay
Thirty compounds (amitriptyline, cyclizine, chlorcyclizine, trifluoperazine, brucine, tradozone, cyclobenzaprine, dothiepin, cidoxepin, asenapine, cyproheptadine, clozapine, loxapine, bifonazole, alprenolol, miconazole, tamoxifen, lamotrigine, oxymetazoline, tioconazole, propranolol, sulconazole, azatadine, rupatadine, carbamazepine, imidafenacin, lidocaine, varenicline, sertraline, and norclozapine) were dissolved in DMSO as stock solutions. Pooled HLM and HLM(-) were prepared in Tris-HCl buffer (100 mM, pH 7.5) containing 10 mM MgCl2 at microsomal protein concentrations of 0.55 mg/ml (final reaction HLM concentration: 0.5 mg/ml) and then were pretreated with alamethicin (10 µg/mg protein) for 30 minutes on ice. Incubations were performed using a Tecan Fluent automated liquid handler (Badée et al., 2019b) with final compound concentrations of 1 µM (solvent content of 0.5% v/v DMSO) and warmed to 37°C over 5 minutes in a 96-deep well plate. Compounds were incubated individually in these and subsequent experiments to ensure inhibitory effects of UGT2B10 substrates such as amitriptyline (Pattanawongsa et al., 2016) did not interfere in the metabolism assessments. Reactions were initiated by the addition of 40 µl 10× concentrated UDPGA stock solution prewarmed to 37°C. Final UDPGA concentration was 5 mM, and total reaction volume was 400 µl. After 0.5, 3.5, 6.5, 10, 15, 30, 45, and 90 minutes of incubation, 40 µl each incubation mixture was transferred into 384-well-deep well plates preloaded with 80 µl quench solutions (cold acetonitrile containing 500 ng/ml of D6-midazolam as internal standard). Samples were chilled on ice for 30 minutes before centrifugation (20,000g, 10 minutes). The supernatant of each sample was analyzed by liquid chromatography (LC)–tandem mass spectrometry (MS/MS). All reaction rate calculations were performed using multiple timepoint data, and following linear initial rate conditions.
Characterization of N-Glucuronidation
Probe Substrates.
RO5263997, dexmedetomidine, azatadine, cyproheptadine, bifonazole, asenapine, and cidoxepine (final test concentration: 1 µM); trifluoperazine and amitriptyline (final test concentration: 5 µM); cyclizine, chlorcylizine, dothiepin, desloratadine, and cyclobenzaprine (final test concentration: 10 µM); and nicotine and cotinine (final test concentration: 100 µM) were used. Test concentrations for each of these substrates were at or below Michaelis–Menten constant values reported for HLM in the literature. UGTs 1A1, 1A3, 1A4, 1A6, 1A9, 2B4, 2B7, 2B10, 2B15, and 2B17 (final concentration: 0.5 mg/ml), HLM(-), and pooled HLM (final concentration: 1.0 mg/ml) were prepared in Tris-HCl buffer (100 mM, pH 7.5) containing 10 mM MgCl2, respectively. HLMs were pretreated with alamethicin (10 µg/mg protein) for 30 minutes on ice before incubation. Incubations were made up by spiking test compounds (solvent content of 0.5% v/v DMSO) into prepared enzyme solutions and warmed to 37°C over 5 minutes in a 96-well-deep well plate. Reactions were initiated by the addition of 40 µl 10× concentrated UDPGA stock solution prewarmed to 37°C. Final UDPGA concentration was 5 mM, and total reaction volume was 400 µl. After 0.5, 3.5, 6.5, 10, 15, 30, 45, and 90 minutes of incubation, 40 µl each incubation mixture was transferred into 384-well-deep well plates preloaded with 80 µl quench solution (cold acetonitrile containing 500 ng/ml D6-midazolam as internal standard). Samples were chilled on ice for 30 minutes before centrifugation (20,000g, 10 minutes). The supernatant of each sample was analyzed by LC-MS/MS. Incubations were performed in triplicate, and all reaction rate calculations were performed using multiple timepoint data, and following linear initial rate conditions.
Metabolic Stability Using NADPH as Cofactor
In This Assay.
Azatadine, cyproheptadine, bifonazole, asenapine, cidoxepine, dexmedetomidine, RO5263397, chlorcylizine, and cyclizine (final test concentration: 1 µM); amitriptyline (final test concentration: 5 µM); and dothiepin and cyclobenzaprine (final test concentration: 10 µM) were tested. The pooled HLM at 1.1 mg/ml (final reaction HLM concentration: 1.0 mg/ml) were prepared in 100 mM potassium phosphate buffer (pH 7.4). Incubations were made up by spiking test compounds with solvent content of 0.5% v/v DMSO into prepared HLM solutions and warmed to 37°C over 5 minutes in 96-deep well plates. Reactions were initiated by the addition of 40 µl 10× concentrated NADPH stock solution prewarmed to 37°C. Final NADPH concentration was 1 mM, and total reaction volume was 400 µl. After 0.5, 3.5, 6.5, 10, 15, 25, 35, and 45 minutes of incubation, 40 µl each incubation mixture was transferred into 384-well-deep well plates preloaded with 80 µl quench solutions (cold acetonitrile containing 500 ng/ml of D6-midazolam as internal standard). Samples were chilled on ice for 30 minutes before centrifugation (20,000g, 10 minutes). The supernatant of each sample was analyzed by LC-MS/MS. Incubations were performed in triplicate, and all reaction rate calculations were performed using multiple timepoint data, and following linear initial rate conditions.
LC-MS/MS Analysis
The LC-MS/MS system comprised two of ultra-high performance liquid chromatography pumps (Shimadzu LC-30AD), an autosampler (Shimadzu Nexera ×2), and an API 6500 (AB-Sciex) mass spectrometer. The chromatography conditions, analyte retention times, and MS/MS analysis parameters are reported in Supplemental Methods and Supplemental Table 1.
In Silico Selection of Potential UGT2B10 Substrates
All 578 compounds listed (at the time of searching) in the University of Washington Drug-Drug Interaction (DDI) database (Hachad et al., 2010) as substrates for one or more UGT enzymes (other than UGT2B10) were collected. These included substances belonging to different classes (e.g., drugs, herbal medicaments, food, additives, etc.). We then used the open DRUGBANK database to extract available structure data format (SDF) structures for these compounds. The information from the two databases was merged using the commercial name available in the two databases compared against all different synonymous combinations available in the DRUGBANK database. For 219 molecules, the SDF format structures were available and converted to SMILES strings. Molecules containing no nitrogen atoms were removed, leaving 140 molecule structures that were uploaded in SDF format to FLAP (Fingerprints for Ligands and Proteins; Molecular Discovery) (Baroni et al., 2007; Sciabola et al., 2019) to generate the molecular interaction fields (MIFs). We used four probes to compute GRID MIFs with a resolution of 0.75 Å. The four probes were H, DRY, N1, and O, which describe the molecular volume, hydrophobicity, hydrogen bond donor, and hydrogen bond acceptor characteristics, respectively. The molecular structure was uploaded with a maximum number of 50 conformers and with root mean square conformer overlap of 0.30. This parameter represents a critical value to minimize the count of molecular conformations during FLAP database generation. Desloratadine, amitriptyline, cyclizine, RO5263397, nicotine, and dexmedetomidine were uploaded to the FLAP database in the same way as reported for the test set, and were used as target structure templates. Ligand-based virtual screening was performed using the default parameters and using the most accurate setting conditions provided in the FLAP software. The HDRYN1 parameter, which considers the different three-dimensional contribution of hydrophobic, volume, and hydrogen bond acceptor, was used to assess similarity between database drug structures and known substrate templates, and a similarity score was generated. Then the molecules were sorted in ascending order of HDRYN1 score, and 20 compounds with the highest scores using the six UGT2B10 templates were selected. A frequency number was assigned, which counts how many times a certain molecule was in the top 20 ranking using the six different templates. Finally, compounds with a frequency number score two or more (two times in the top 20 ranking) were selected for further study.
Tested compounds selected by two different in silico modes and respective UGT2B10 metabolism selectivity using HLM(-)
In addition, compounds not described as UGT substrates in the University of Washington DDI database but still present in the DRUGBANK database might still have been of interest. Therefore, we employed a simple substructure searching methodology using the chemical scaffolds of amitriptyline and desloratadine. A SMARTS query representing the chemical scaffold of the two templates was generated and compared with the molecules in the DRUGBANK database. The compounds selected with this substructure method are shown in Table 2. The data mining described previously was performed using Knime (version 3.5.3, KNIME AG, Zurich, Switzerland).
Data Analysis
Substrate Depletion (Pooled HLM Incubations).
The natural logarithm of the peak area ratios (compound peak area/internal standard peak area) were plotted against time, and the slope of the linear regression was determined using Microsoft Excel 2010 (Redmond, WA). The elimination rate constant (k) was calculated according to: The intrinsic clearance [Clint (microliters per minute per milligram protein)] was calculated (with protein concentration in mg/ml) as:
The intrinsic clearance [Clint (microliters per minute per milligram protein)] was calculated (with protein concentration in mg/ml) as: The combination of automated liquid-handling systems for experimental conduct and sample preparation, the employment of optimized LC-MS/MS methods, and the use of multiple timepoint data from each incubation enabled depletion of >10% initial drug concentration to be detected with confidence. Lower limits for Clint were determined on this basis, taking into account incubation time and microsomal protein concentration in the incubations.
The combination of automated liquid-handling systems for experimental conduct and sample preparation, the employment of optimized LC-MS/MS methods, and the use of multiple timepoint data from each incubation enabled depletion of >10% initial drug concentration to be detected with confidence. Lower limits for Clint were determined on this basis, taking into account incubation time and microsomal protein concentration in the incubations.
Relative Rates of Metabolite Appearance.
In the absence of glucuronide standards for calibration, metabolite appearance was assessed using the peak area ratio (glucuronide metabolite peak area/internal standard peak area) in the HLMs and recombinants. The relative rates of metabolite formation were assessed as increase in peak area ratio per minute incubation time per unit protein concentration. These data were normalized with the rate of formation by pooled HLM set as 100%. The Clint values determined by substrate depletion and the percentage activity values determined using glucuronide generation data were reported as the mean ± S.D. from triplicate determinations. All graphs reported in this work were drawn using GraphPad Prism v7.04 (GraphPad Software, San Diego, CA).
Estimation of Fraction Metabolized.
Fraction metabolized via glucuronidation, fm(gluc), was calculated following the method reported by Kilford et al. (2008). For estimation of fraction of glucuronidation contributed by UGT2B10, it was assumed that UGT1A4 and UGT2B10 were the only enzymes significantly contributing to metabolism for the drugs in question. Although UGT2B10 contribution can be estimated as the ratio of metabolite formation rate in the HLM(-) and pooled HLM preparations, correction for the reduced activity of UGT1A4 in HLM(-) was also required. Therefore, the fraction of glucuronidation assigned to UGT2B10 was calculated as follows:
For estimation of fraction of glucuronidation contributed by UGT2B10, it was assumed that UGT1A4 and UGT2B10 were the only enzymes significantly contributing to metabolism for the drugs in question. Although UGT2B10 contribution can be estimated as the ratio of metabolite formation rate in the HLM(-) and pooled HLM preparations, correction for the reduced activity of UGT1A4 in HLM(-) was also required. Therefore, the fraction of glucuronidation assigned to UGT2B10 was calculated as follows: The overall fraction metabolized via UGT2B10 was then calculated as follows:
The overall fraction metabolized via UGT2B10 was then calculated as follows:
Results
Qualification of the HLM(-) Versus Pooled HLM Assay for Identification of UGT2B10 Substrates.
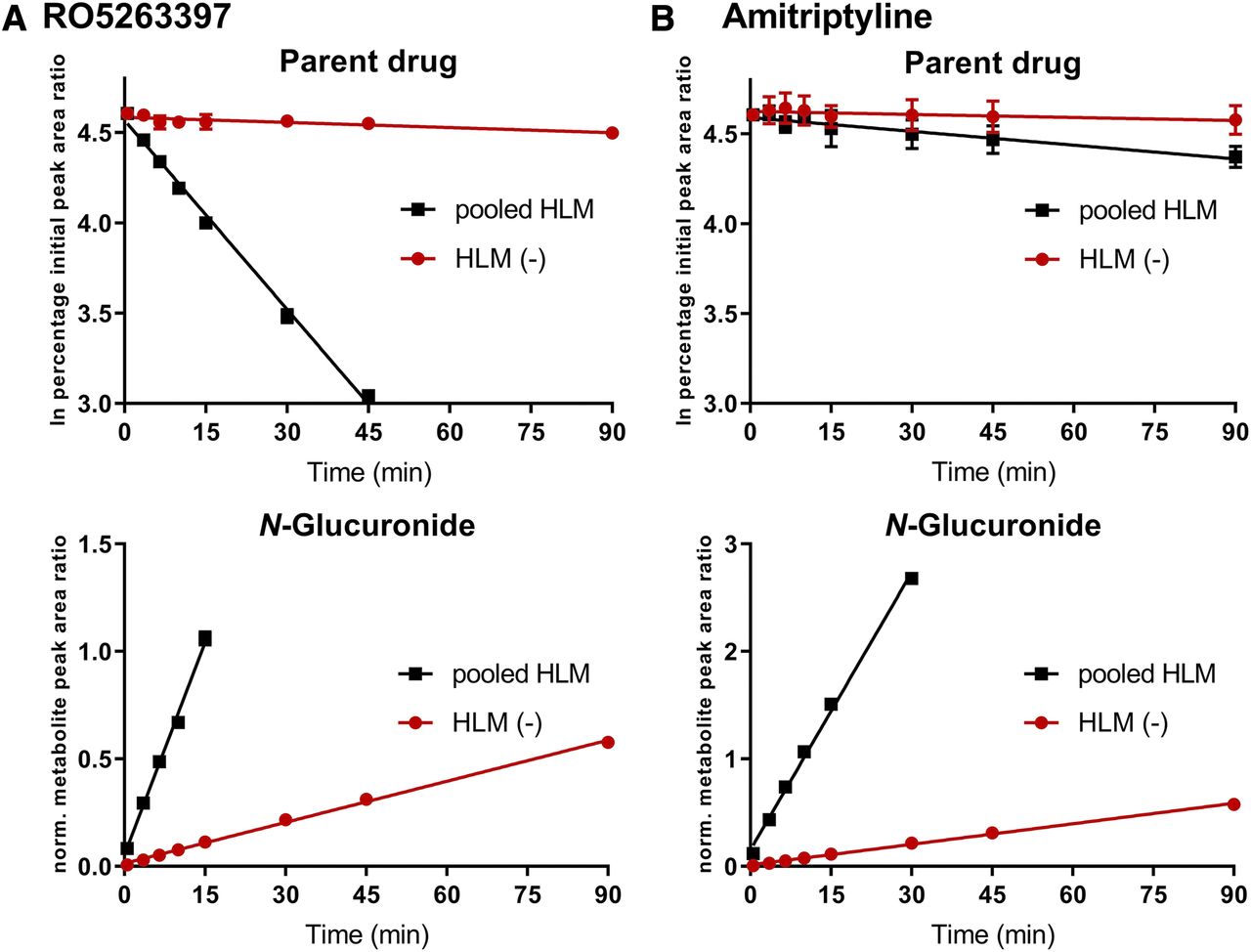
Chemical structures of known and investigated UGT2B10 substrates can be seen in Fig. 1. RO5263397 and amitriptyline were tested using HLM(-) and pooled HLM to assess the utility of the proposed methodology for UGT2B10 substrate detection using compounds of moderate and low UGT turnover, respectively (Fig. 2). RO5263397 had a dramatically lower rate of depletion in the HLM(-) samples compared with those from pooled HLM (<1.1 µl/min per milligram vs. 35 µl/min per milligram). Similarly, the glucuronide metabolite formation rate for RO5263397 by HLM(-) was only 3% that of pooled HLM. Calculation of relative amitriptyline depletion rates could not be made accurately due to low turnover [Clint < 1.1 µl/min per milligram for HLM(-) vs. 2.5 µl/min per milligram for pooled HLM, Table 1]. In contrast, the much greater sensitivity of amitriptyline glucuronide formation measurements showed that HLM(-) was only 14% as active as pooled HLM.

Chemical structures of UGT2B10 substrates and other molecules tested in this study.

Differential activity of HLM(-) with low UGT2B10 activity and pooled HLM for known UGT2B10-selective substrates RO5263397 and amitriptyline. Depletion of parent drug and metabolite formation rate by pooled HLM (black squares) and individual HLM(-) donor (red circles) for RO5263397 (A) and amitriptyline (B). All data points are mean ± S.D. of triplicate determinations. Error bars are not visible where values are smaller than plot symbols. Values not shown were outside of chart ranges manually selected to show low-activity microsomal data.
Intrinsic clearance by pooled HLM and relative rates of glucuronidation by HLM(-), UGT2B10, and UGT1A4 for known UGT2B10, newly identified UGT2B10 substrates, and control compounds
Relative activity factors for UGT1A4 (trifluoperazine) = 0.31, for UGT2B10 (RO5263397 and dexmedetomidine) = 4.3, and for UGT2B10 (cyclizine, chlorcyclizine, nicotine, cotinine) = 11–15.9.
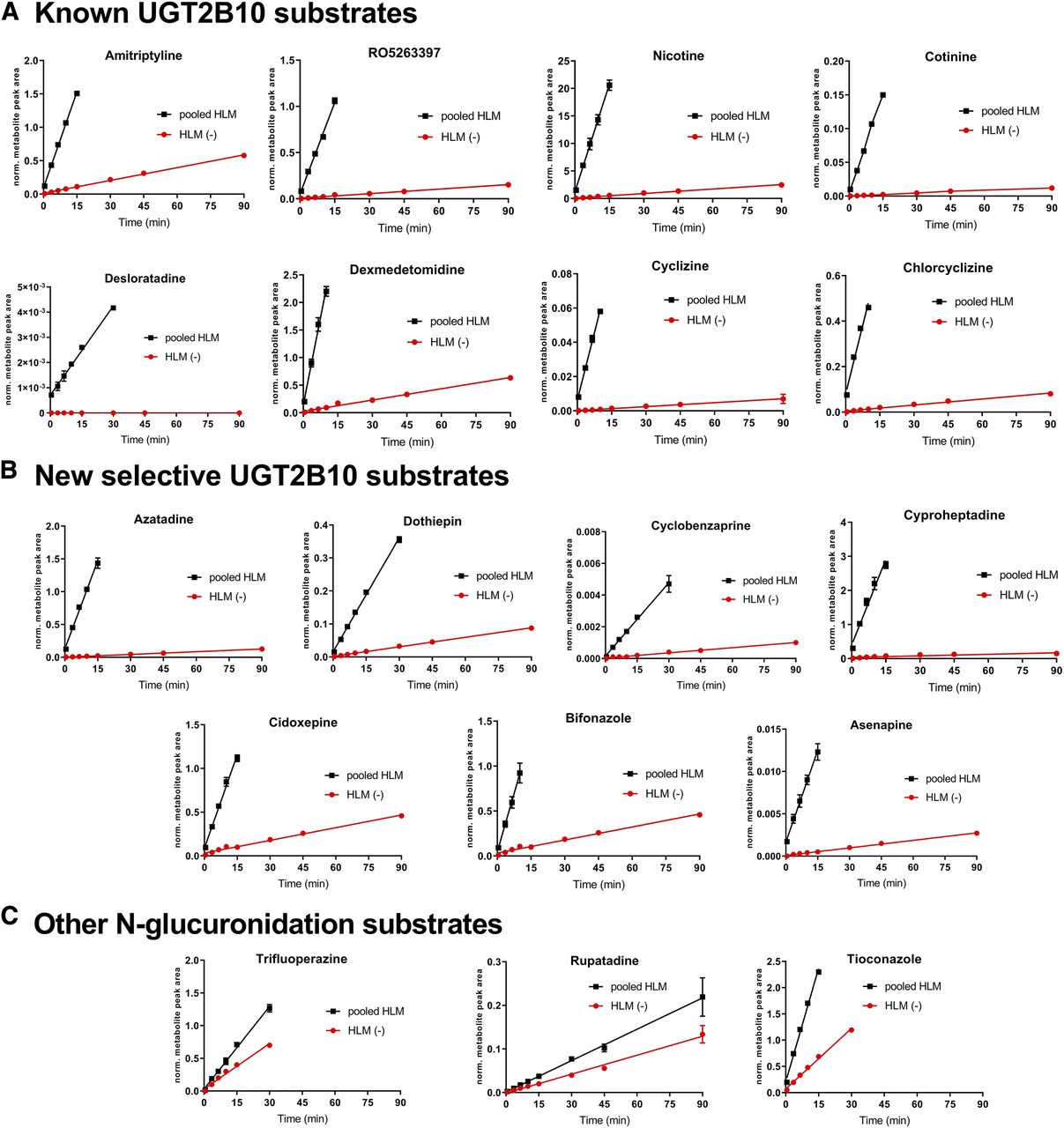
Six other known UGT2B10 substrates (nicotine, cotinine, dexmedetomidine, desloratadine, cyclizine, and chlorcyclizine, Fig. 1) were tested in a similar manner. These drugs exhibited moderate (10–30 µl/min per milligram; dexmedetomidine, cyclizine, and chlorcyclizine) or low (<10 µl/min per milligram; nicotine, cotinine, and desloratadine) Clint in pooled human liver microsomes (Table 1). Relative rates of glucuronide formation for all six UGT2B10 substrates were in the range of 1% to 3% in HLM(-) compared with pooled HLM (Fig. 3A). Amitriptyline exhibited the highest percentage activity (14%) in HLM(-) compared with the pooled HLM, indicating that the other compounds were more selective UGT2B10 substrates. This suggested a practical threshold of 15% pooled HLM activity to use when categorizing compounds as potential selective UGT2B10 substrates using this assay procedure.

Relative rates of glucuronide formation in incubations with pooled HLM and individual HLM(-) donor with low UGT2B10 activity. Mean ± S.D. metabolite peak area ratio values plotted against incubation time for (A) eight known UGT2B10-selective substrates, (B) seven new identified selective substrates, and (C) other drugs metabolized by N-glucuronidation. Incubation time course experiments were performed in triplicate. Red circles show data for HLM(-) individual donor lacking UGT2B10 activity. Black squares show data for pooled HLM.
Computational Search for Likely UGT2B10 Substrate Drugs.
All molecules listed in the University of Washington DDI Database as substrates for “UGT” or specifically UGTs 1A1, 1A3, 1A4, 1A6, 1A9, 1A10, 2B4, 2B7, 2B15, and 2B17 were selected. These compounds were further filtered using the open source DRUGBANK database (8710 total molecules) to select commercial drugs with available structures from the previous list. In addition, the compounds without a nitrogen atom were filtered out. The final list was composed of 140 drugs (Supplemental Table 2). Ligand-based virtual screening was performed using six template structures (amitriptyline, RO5263397, nicotine, dexmedetomidine, cyclizine, and desloratadine), leading to a list of 26 likely UGT2B10 substrates, 19 of which were available for testing (Table 2). In addition, a substructure searching method was used to identify compounds in the DRUGBANK database that were not in the University of Washington DDI database. Five compounds were selected: doxepin, dothiepin, and cyclobenzaprine (amitriptyline template) and azatadine and rupatadine (desloratadine template). Cidoxepin, the pure cis regioisomer of doxepin, was tested in place of doxepin. In conclusion, 24 potentially selective UGT2B10 substrates (19 from ligand-based virtual screening and five from substructure searching) were chosen for screening as UGT2B10-selective substrates (Supplemental Table 3). The selection approach is shown schematically in Supplemental Fig. 3.
Screening for UGT2B10-Selective Substrates.
Twenty-four compounds were selected using in silico approaches, as described above, and supplemented with three selective UGT2B10 substrates (amitriptyline, chlorcyclizine, and cyclizine) and three UGT1A4 substrates (trifluoperazine, trandozone, and brucine) identified in the literature (Kato et al., 2013; Lu et al., 2017, 2018) (Table 2). Figure 4 shows the relative metabolite formation rates determined in singlicate and normalized with values for pooled HLM set as 100%. Trifluoperazine, brucine, and trazodone (indicated with an asterisk) are known UGT1A4 substrates. Seven compounds (cyclobenzaprine, dothiepin, cidoxepin, cyproheptadine, azatadine, bifonazole, and asenapine) exhibited a much lower glucuronidation rate using HLM(-) with percentage activities below 15% of pooled HLM (dashed red bars in Fig. 4). These compounds were flagged as potential UGT2B10-selective substrates for further study. The solid black bars show compounds with greater than 15% activity, indicating that they were not selective UGT2B10 substrates. Compounds for which we could not detect metabolism using either HLM(-) or pooled HLM are not shown in Fig. 4 but are listed in Table 2. These compounds were as follows: carbamazepine, imidafenacin, lidocaine, norclozapine, varenicline, and sertraline.

Screening assay using pooled HLM and HLM(-) to detect potential UGT2B10-selective substrates. Percentage pooled HLM N-glucuronidation rates shown for HLM(-) individual donor lacking UGT2B10. Molecules indicated with an asterisk are nonselective UGT2B10 substrates. Data are from singlicate incubations with initial reaction rates calculated using multiple timepoint measurements. Compounds reported with hashed red and solid black bars represent relative activity less and higher than 15% of pooled HLM activity (dotted line), respectively.
Further Characterization and Confirmation of UGT2B10 Substrate Selectivity.
The seven new potential selective UGT2B10 substrates (cyclobenzaprine, cyproheptadine, dothiepin, cidoxepin, azatadine, asenapine, and bifonazole) were assessed further. The rates of metabolite appearance in the HLM(-) incubations compared with the pooled HLM were less than 15% for all compounds in this follow-up experiment, confirming the screening experiment results (Fig. 3B; Table 1). Cidoxepin, cyproheptadine, azatadine, and asenapine exhibited very low percentage activities (1%–5%), indicating high UGT2B10 selectivity, similar to RO5263397 (3%) and other highly selective UGT2B10 substrates (dexmedetomidine, cyclizine, chlorcyclizine, nicotine, and cotinine). Cyclobenzapine, dothiepine, and bifonazole had more moderate percentage activities [8%–12%, similar to amitriptyline (14%)]. Trifluoperazine, rupatadine, and tioconazole were also tested as known non-UGT2B10 substrates (Table 1). They showed activities >15% in HLM(-) compared with the pooled HLM, in accordance with the screening assay (Fig. 3C). The compound structures of new UGT2B10 substrates and compounds found not to be selective UGT2B10 substrates assessed are shown in Fig. 1.
UGT Phenotyping Using Recombinant UGTs.
The relative rates of N-glucuronidation by recombinantly expressed UGTs 1A4 and 2B10 were determined in parallel with the HLM pool and HLM(-) characterization assessments (Supplemental Fig. 2). For all of the previously known UGT2B10 substrates, recombinant UGT2B10 activity was low (<23%) compared with HLM (Table 1). The rate of amitriptyline glucuronidation was higher using recombinant UGT1A4 than UGT2B10 (28% and 17% vs. pooled HLM, respectively); and for chlorcyclizine the rates were almost the same. Dexmedetomidine showed twice the activity with recombinant UGT2B10 compared with UGT1A4. RO5263397, cotinine, and cyclizine showed ∼4- to 6-fold higher UGT2B10 activity than UGT1A4. Nicotine showed a more than 13-fold higher recombinant UGT2B10 activity. Desloratadine metabolite formation could not be detected using either UGT1A4 or UGT2B10 due to the very low turnover rate. UGT2B10-selective substrates therefore exhibited glucuronide formation rates equal to or higher than those of UGT1A4 preparations in these experiments. Relative activity factors for the selective UGT2B10 substrates ranged from 4.3 (RO5263397 and dexmedetomidine) to 16 (cyclizine), indicating a high degree of substrate dependency in relative activity factor value. Further calculations and extrapolations were not made, due to this uncertainty.
Table 1 shows the relative rates of UGT1A4 and UGT2B10 glucuronidation activity of the selective UGT2B10 substrates identified using the HLM(-) assay: cyclobenzaprine, dothiepin, cidoxepin, cyproheptadine, azatadine, bifonazole, and asenapine. The drugs cidoxepin, azatadine, and cyproheptadine exhibited higher activity in recombinant UGT2B10 than UGT1A4 (Supplemental Fig. 2; Table 1). In comparison, cyclobenzaprine, dothiepin, and, in particular, bifonazole and asenapine showed a higher metabolite formation rate using recombinant UGT1A4 than UGT2B10.
To ensure that other important UGT activities had not been overlooked for these compounds, incubations were subsequently performed using an additional eight hepatic UGTs (UGTs 1A1, 1A3, 1A6, 1A9, 2B4, 2B7, 2B15, and 2B17). This experiment confirmed that UGT1A4 and UGT2B10 were essentially the only enzymes active in N-glucuronidation of the new UGT2B10 substrate molecules (Fig. 5). As negative controls, three non-UGT2B10–selective substrates, trifluoperazine, rupatadine, and tioconazole, were also tested. These compounds exhibited higher activity in UGT1A4 than UGT2B10; in particular, trifluoperazine and rupatadine were metabolized by the recombinant UGT1A4 faster than pooled HLM (Table 1). The activity in the recombinant UGT1A4 was about 15-fold more than UGT2B10 for tioconazole and more than 300-fold higher for trifluoperazine (Supplemental Fig. 2). In addition, the nonselective substrate rupatadine showed the highest activity in the UGT1A4 preparation (>16-fold higher than pooled HLM activity) and was metabolized by UGTs 1A3, 1A1, and 1A9 with activities of 100%, 19%, and 13% that of pooled HLM, respectively (Fig. 5). Isoform-selective positive control substrates (Badée et al., 2019b) were incubated in parallel for each enzyme and confirmed that the respective enzyme preparations were active (data not shown).

Heatmap showing glucuronidation activity of 10 individual hepatic UGT enzyme preparations toward identified UGT2B10 substrates. Mean values from triplicate glucuronidation rate determinations for 10 UGT isoforms reported as a percentage of mean pooled HLM activity. The incremental activity in the recombinants is reported with a gradient color from light blue to red. The activities reported with a cross showed an activity lower than the experimental limit of detection. UGT1A4 and UGT2B10 were the only active enzymes in the metabolism of six compounds (cyclobenzaprine, cyproheptadine, azatadine, asenapine, doxepin, dothiepin). For bifonazole, UGT1A3 was also active. For rupatadine (indicated with an asterisk), UGTs 1A1, 1A3, and 1A9 were also active, and UGT2B10 essentially inactive.
Oxidative Metabolic Clearance.
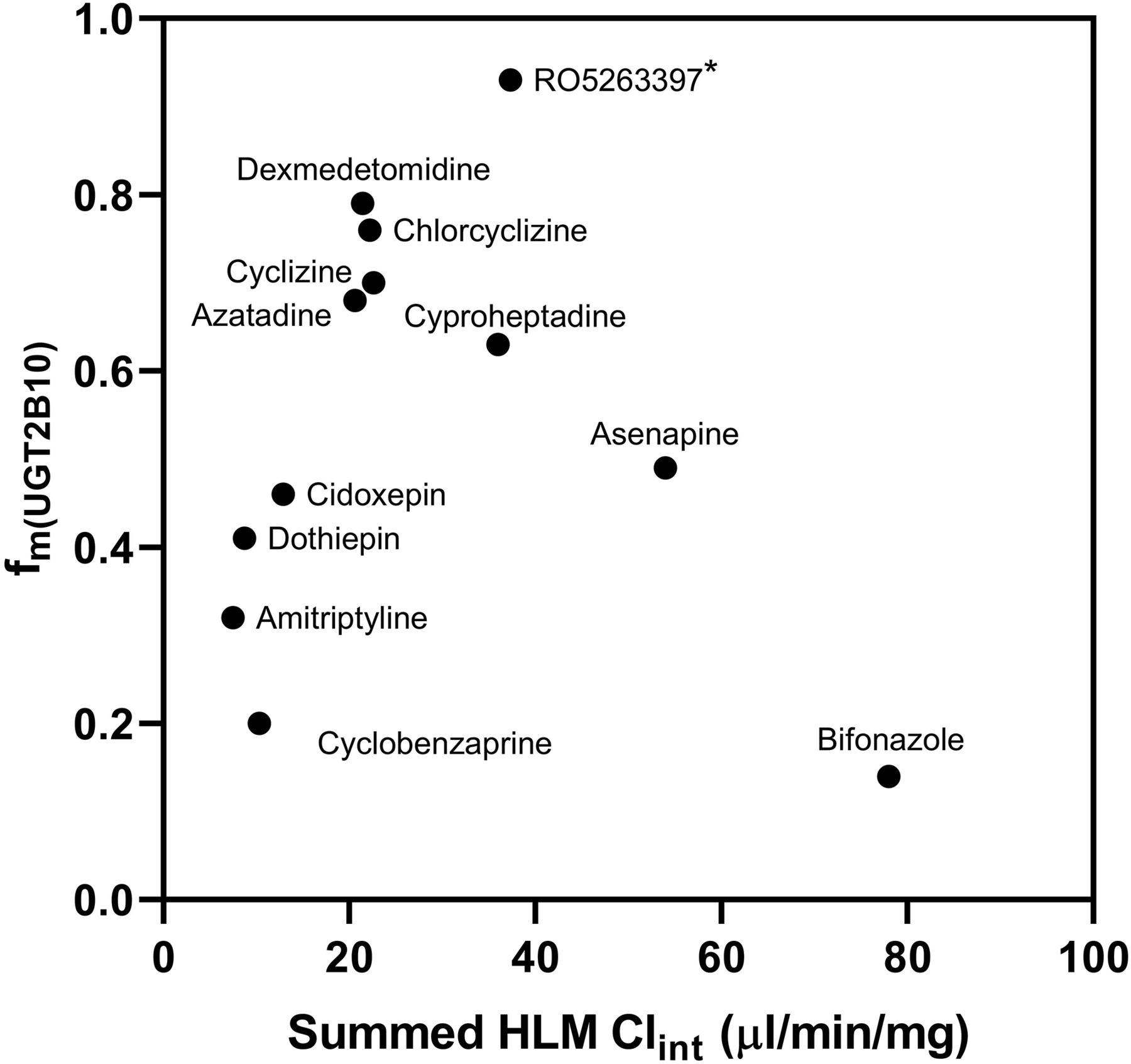
The Clint of cyclobenzaprine, dothiepin, cidoxepin, asenapine, azatadine, cyproheptadine, and bifonazole as well as known UGT2B10 substrates was assessed using pooled HLM supplemented with NADPH as cofactor (no UDPGA) to indicate the relative importance of oxidation and glucuronidation to metabolic clearance (Fig. 6). The same HLM concentration (1 mg/ml) was used for both glucuronidation and oxidation Clint assessments. Desloratadine, nicotine, and cotinine were not tested because we had not been able to determine the Clint under glucuronidation conditions. Oxidative Clint values ranged from <2.3 (RO5263397) to 67 ± 7 µl/min per milligram (bifonazole). The quantitative results are reported in Table 3, in which the total clearance was calculated as the sum of the individual Clint by UGTs and CYPs to estimate the UGT percentage contribution in the metabolism of the drugs (Kilford et al., 2008). The fm(UGT2B10) values were very high (>0.93) for RO5263397; high (>0.6) for dex-medetomidine chlorcyclizine, cyclizine, azatadine, and cyproheptadine; and moderate (0.4–0.6) for asenapine, dothiepin, and cidoxepin. The contribution of UGT2B10 in the overall metabolism of amitriptyline, bifonazole, and cyclobenzaprine was low (fm(UGT2B10) < 0.32).

Comparison of Clint related to oxidative metabolism vs. glucuronidation and total glucuronidation vs. UGT2B10-mediated glucuronidation. Relative importance of UGT2B10 metabolism vs. total Clint (microliters per minute per milligram). fm(UGT2B10) is plotted against sum of HLM Clint determined in the presence of metabolism cofactors for oxidation and glucuronidation reactions, NADPH and UDPGA. A high fm(UGT2B10) indicates a high risk of increased drug exposure in UGT2B10 poor metabolizer individuals. When combined with high Clint, the risk is further increased for orally administered compounds due to likely reduced first-pass hepatic extraction of the compound before systemic circulation. Asterisk denotes known underestimation of fm(UGT2B10) due to low P450 contribution to clearnce.
Intrinsic clearance (microliters per minute per milligram) measured for known and new UGT2B10 substrates using UDPGA and NADPH as a cofactor and fm(gluc) and fm(UGT2B10)
Discussion
Limitations of Current Approaches in Identifying Selective UGT2B10 Substrates.
Current UGT phenotyping approaches are mainly qualitative and use individual UGT preparations to identify enzymes that may contribute to metabolism. Known limitations include the lack of availability for some enzymes, lack of data about the UGT expression level in the preparations (precluding calculation of intersystem extrapolation factors), poor activity of some recombinantly expressed enzymes (e.g., UGT1A10 and UGT2B10) (Kurkela et al., 2003; Court, 2005; Patten, 2006; Zhou et al., 2010; Oda et al., 2015), and highly substrate-dependent relative activity factors (values ranged from 4 to 16 for UGT2B10 substrates in this study). Where several enzymes can contribute to drug metabolism, there is generally a low risk of polymorphic drug exposure. However, where a candidate drug is only metabolized by one or two isoforms, further work is needed to quantify the polymorphism risk. Although N-glucuronidation by UGT1A4 and UGT2B10 may represent a simplified version of this problem, quantitative assessment of contributions to metabolism remains challenging. Where literature data exist, reports often indicate compounds for which UGT2B10 has the potential to contribute to metabolism but without establishing the importance of the enzyme to clearance in vivo, for example by considering other clearance pathways. Methodological considerations, such as low/unquantified activity of UGT2B10 preparations and use of incubation concentrations above Michaelis–Menten constant and many times higher than those experienced in vivo contribute to the difficulty in extrapolation. Both of these elements tend toward assignment of UGT1A4 as the more important enzyme.
Opportunity for an Alternative Testing Methodology.
Recently, we profiled the UGT activities of a panel of pediatric and adult individual donor HLM samples with substrates for 10 hepatic UGTs (Badée et al., 2019a). In the course of this work, two phenotypic UGT2B10 poor metabolizer donor samples were identified. One of these [donor code H0295, “HLM(-)”] has been used in the present study. The percentage activity for RO5263397 and amitriptyline in HLM(-) were 3% and 14% compared with that of the pooled HLM, respectively (Fig. 2). In contrast, UGT1A4 probe substrate trifluoperazine had a percentage activity of 59% in HLM(-) compared with pooled HLM. On the basis of experiments performed using 5 µM amitriptyline, we established a threshold using the HLM(-) donor of 15% pooled HLM activity below which a compound could be classified as highly likely to be a selective UGT2B10 substrate and therefore potentially high fm(UGT2B10). We first demonstrated the utility of this methodology using RO5263397 (Fig. 2A) and then with a further six known UGT2B10 probe substrates (nicotine, cotinine, dexmedetomidine, desloratadine cyclizine, and chlorcyclizine) (Fig. 3A). Each of these eight compounds exhibited the same activity pattern: low or very low rates of glucuronide generation by HLM(-) compared with pooled HLM, all relative rates being <15%. Indeed, with the exception of amitriptyline, all relative rates were in the 1% to 3% range, more than 30-fold lower than pooled HLM (Table 1). For further UGT phenotyping studies, we could therefore recommend the use of RO5263397, dex-medetomidine, cyclizine, and chlorcyclizine on the basis of their UGT2B10 selectivity under glucuronidation conditions.
Screening for UGT2B10 Substrates.
We set out to discover new selective substrates of UGT2B10: compounds for which the majority of glucuronidation is catalyzed by UGT2B10 at clinically relevant concentrations. For such compounds, it might be expected that UGT2B10 polymorphisms will affect the exposure if conjugative metabolism is the main route of clearance from the body (Berg et al., 2010; Chen et al., 2010; Fowler et al., 2015). Ligand-based virtual screening and substructure searching approaches were performed using different UGT substrate structures as templates, resulting in a test set of 24 compounds (Table 2). As FLAP calculates the MIFs using the GRID force field, the molecules are selected by comparing the common three-dimensional chemical features of test and template molecules rather than substructure similarity. A substructure searching method using desloratadine and amitriptyline as templates was also applied to identify additional compounds from the DRUGBANK database. These compounds were of unknown UGT metabolism (not contained in University of Washington DDI database). The substructure searching method had a very high hit rate due to close chemical similarity of identified drugs with known UGT2B10-selective substrates. In contrast, the ligand-based virtual screening approach enabled identification of molecules not obviously UGT2B10 substrates, albeit at a substantially lower hit rate (Table 2).
Our screening experiment confirmed the expected UGT2B10 selectivity for amitriptyline, cyclizine, and chlorcylizine. Trifluoperazine, brucine, and trazodone were not selective UGT2B10 substrates (Fig. 4). The drugs noted as likely selective UGT2B10 substrates were assessed in more detail (Table 1). We confirmed that cidoxepin (the pure cis regioisomer of doxepin), dothiepin, and cyclobenzaprine were likely selective UGT2B10 substrates. Cyproheptadine has previously been reported to be metabolized by UGT1A3 and 1A4 (Green and Tephly, 1998), at a time when recombinantly expressed UGT2B10 was not commercially available. In our experiments, cyproheptadine appeared as a selective UGT2B10 substrate. Bifonazole has been shown to be metabolized by UGT1A4 (as well as by UGTs 1A3 and 2B7) in a UGT phenotyping experiment that employed 12 different UGTs but did not include UGT2B10 (Bourcier et al., 2010); however, from our data, bifonazole has the characteristics of a selective UGT2B10 substrate. The antipsychotic drug asenapine and the first-generation antihistamine and anticholinergic azatadine were also identified as compounds with UGT2B10 selectivity.
To further qualify the new findings, the identified UGT2B10-selective substrates were tested in a UGT phenotyping experiment using 10 different hepatic UGT enzymes. UGT2B10 and UGT1A4 were the most active enzymes in metabolism (Fig. 5). In the context of this study, which includes characterization of relative UGT1A4 and UGT2B10 activities for seven well-characterized UGT2B10 substrates, it can be seen that drugs for which recombinant UGT2B10 activity exceeded that of UGT1A4 and were not metabolized by other UGT isoforms will be highly selective UGT2B10 substrates. Such a relative activity approach can work if the individual UGT activities are well characterized and preparations are of sufficient activity. However, it is qualitative in nature, and the 4-fold range of relative activity factor values that could be calculated highlights potential difficulties for more quantitative extrapolation. In contrast, the HLM methodology enables a much more rapid assessment using fewer reagents, at lower cost and with greater measurement sensitivity. Indeed, for the highly stable UGT2B10 substrate desloratadine, it was only possible for us to make the assessment using HLM due to the low turnover by recombinant UGTs. For the known selective UGT2B10 substrates such as RO5263397, nicotine, cotinine, dexmedetomidine, and cyclizine, the recombinant activity profiles confirm the data from the HLM(-) assay. Moreover, there is also agreement for the recently identified UGT2B10 substrates azatadine, cyproheptadine, and cidoxepin. Although dothiepin and cyclobenzaprine showed higher turnover by UGT1A4, their activity profiles were very similar to the close structural analog amitriptyline. Some discrepancy with expected and measured UGT1A4 activities was seen for asenapine and bifonazole. However, asenapine has been reported to have a 14% reduced clearance by people of African descent, likely reflecting UGT2B10 involvement (Citrome, 2014). Xu et al. (2016) used asenapine together with four UGT1A4 substrate probes for UGT1A4 IC50 profiling in HLM. There was discrepancy in IC50 values using asenapine compared with the other substrates from 2- to more than 10-fold, suggesting glucuronidation of asenapine by another enzyme, such as UGT2B10.
Potential for UGT2B10-Driven Polymorphic Exposure.
The potential for polymorphic exposure in patients treated with compounds whose glucuronidation is UGT2B10-selective is dependent upon the relative importance of additional clearance pathways. Principal among these are CYP-mediated oxidative metabolism and direct excretion of unchanged drug. Estimates of fm(UGT2B10) have been made for the selective substrates and plotted in Fig. 6 against summed microsomal Clint (indicative of first-pass hepatic extraction). RO5263397 exhibited the highest polymorphism risk, combining very high fm(UGT2B10) with moderate-high Clint. In contrast, the UGT2B10-related polymorphic exposure risk for amitriptyline was low, as other metabolism pathways contribute more than glucuronidation to metabolic clearance (low fm(UGT2B10)). Further examination of clinical study data for azatadine, cyproheptadine, cyclizine, chlorcyclizine, and dexmedetomidine, which have relatively high fm(UGT2B10) values, may be useful to assess whether there have been examples of higher drug exposure in some study subjects due to UGT2B10 poor metabolizer status.
Conclusions
With this work we have shown the utility of an innovative experimental protocol exploiting phenotypically poor metabolizer individual donor human liver microsomes to identify UGT2B10-selective substrates. This methodology can be used as an effective alternative to UGT phenotyping using a large panel of recombinantly expressed UGT enzymes. The approach is applicable to other drug-metabolizing enzymes for which individual donors can be identified with pronounced activity differences and where we currently lack selective inhibitors. The assay offers more sensitivity (due to higher turnover by pooled HLM than rec UGT2B10) and an easier to interpret readout due to the threshold approach. In combination with assessment of HLM-mediated oxidative metabolism, the assay can be used as a rapid UGT2B10 risk assessment tool for estimating fm(UGT2B10). Figure 6 shows a straightforward graphical way to represent such data for compound selection discussions and provides several relevant drug molecule examples that may be used for comparison with new candidate drugs. In our study, we demonstrated for the first time that drugs such as dothiepin, cidoxepin, cyclobenzaprine, cyproheptadine, asenapine, azatadine, and bifonazole are mainly glucuronidated by UGT2B10 in vitro and could be appropriate molecules to use in UGT2B10 phenotyping experiments and correlation analyses using panels of individual donor human liver microsomes. These substrates could be used in addition to the established RO5263397 and dexmedetomidine and would be superior in selectivity to amitriptyline. However, for experiments to evaluate the activity of UGT2B10 in hepatocytes, RO5263397 is the most suitable molecule, combining extremely high selectivity (fm(UGT) > 0.94 and fm(UGT2B10) > 0.93) with moderate-high turnover and known clinical UGT2B10 relevance.
Acknowledgments
We gratefully acknowledge helpful feedback on this manuscript from Neil Parrott and Dr. Kenichi Umehara (Hoffmann-La Roche, Basel, Switzerland) and Prof. Abby Collier (University of British Columbia, Vancouver, Canada). We also thank Drs. Christoph Funk, Franz Schuler, and Adrian Roth (Hoffmann-La Roche, Basel, Switzerland) for sponsoring these investigations.
Authorship Contributions
Participated in research design: Milani, Qiu, Molitor, Cruciani, Fowler.
Conducted experiments: Milani, Qiu, Molitor, Badée.
Performed data analysis: Milani, Qiu, Fowler.
Wrote or contributed to the writing of the manuscript: Milani, Badée, Cruciani, Fowler.
Footnotes
- Received September 23, 2019.
- Accepted December 2, 2019.
↵1 Current affiliation: Modelling and Simulation, PK Sciences, Novartis Institutes for BioMedical Research, Basel, Switzerland.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- Clint
- intrinsic clearance
- P450
- cytochrome P450
- fm
- fraction metabolized
- HLM
- human liver microsome
- LC
- liquid chromatography
- MIF
- molecular interaction field
- MS/MS
- tandem mass spectrometry
- SDF
- structure data format
- UDPGA
- uridine 5′-diphosphoglucuronic acid
- UGT
- UDP-glucuronosyltransferase
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}