


Abstract
The mitochondrial amidoxime reducing component is a recently discovered molybdenum enzyme in mammals which, in concert with the electron transport proteins cytochrome b5 and NADH cytochrome b5 reductase, catalyzes the reduction of N-oxygenated structures. This three component enzyme system plays a major role in N-reductive drug metabolism. Belonging to the group of N-hydroxylated structures, hydroxamic acids are also potential substrates of the mARC-system. Hydroxamic acids show a variety of pharmacological activities and are therefore often found in drug candidates. They can also exhibit toxic properties as is the case for many aryl hydroxamic acids formed during the metabolism of arylamides. Biotransformation assays using recombinant human proteins, subcellular porcine tissue fractions as well as human cell culture were performed. Here the mARC-dependent reduction of the model compound benzhydroxamic acid is reported in addition to the reduction of three drugs. In comparison with other known substrates of the molybdenum depending enzyme system (e.g., amidoxime prodrugs) the conversion rates measured here are slower, thereby reflecting the mediocre metabolic stability and oral bioavailability of distinct hydroxamic acids. Moreover, the toxic N-hydroxylated metabolite of the analgesic phenacetin, N-hydroxyphenacetin, is not reduced by the mARC-system under the chosen conditions. This confirms the high toxicity of this component, as it needs to be detoxified by other pathways. This work highlights the need to monitor the N-reductive metabolism of new drug candidates by the mARC-system when evaluating the metabolic stability of hydroxamic acid-containing structures or the potential risks of toxic metabolites.
Introduction
Hydroxamic acids are a class of substances with a variety of biologic activities, including antibiotic (Barb et al., 2007; Halouska et al., 2014) and anti-inflammatory activities (Brogden et al., 1975), along with inhibitory properties toward metalloproteinases (Dalvie et al., 2008; Verma, 2012) and histone-deacetylases (HDACs) (Dokmanovic et al., 2007; Zhang and Zhong, 2014). The latter is of particular interest, as inhibitors of HDACs show antineoplastic activities and are currently applied in cancer treatment with new candidates under development (Wagner et al., 2010; Zhang and Zhong, 2014). Examples are vorinostat (suberoylanilide hydroxamic acid) which is marketed as Zolinza for the treatment of T-cell lymphoma and Panabinostat (Farydak) which has just been approved (Raedler, 2016).
Applying compounds with hydroxamic acids in therapy requires a reasonable level of metabolic stability. One expectable metabolic pathway, besides conjugation reactions, is hydrolysis to the corresponding carboxylic acid (Liu et al., 2014). As shown in Fig. 1, another likely metabolic conversion is the reduction of hydroxamic acids to the corresponding amide (Lowenthal, 1954; Hirsch and Kaplan, 1961; Kitamura and Tatsumi, 1985; Kiesel et al., 2013). For example, metabolism studies of CP544439, a hydroxamic acid-containing matrix metalloproteinase inhibitor, revealed the formation of the amide, the glucuronide and the carboxylic acid as the main metabolites of the orally administered drug (Dalvie et al., 2008). Besides their pharmacological advantages, aryl hydroxamic acids have been shown to possess toxic and mutagenic properties (Miller et al., 1961; Vaught et al., 1981). For example, the analgesic drug “Phenacetin” is N-hydroxylated during metabolism to yield N-hydroxyphenacetin (Hinson and Mitchell, 1976; Wirth et al., 1980). Phenacetin was withdrawn from the market because it was found to induce severe renal papillary necrosis and tumors of the renal pelvis and bladder in humans (Liu et al., 1972; Bengtsson et al., 1978). N-hydroxyphenacetin has been held responsible for these severe effects as it conducts similar pathways to many aromatic amines leading to the formation of DNA adducts (Vaught et al., 1981). In vivo and in vitro, N-hydroxyphenacetin is metabolized to Phenacetin (Fischbach and Lenk, 1985). The enzymatic basics of the reductive metabolism remain undetermined. The involvement of the cytochrome P450 isoform 2S1in this reductive metabolism could be excluded (Wang and Guengerich, 2013).
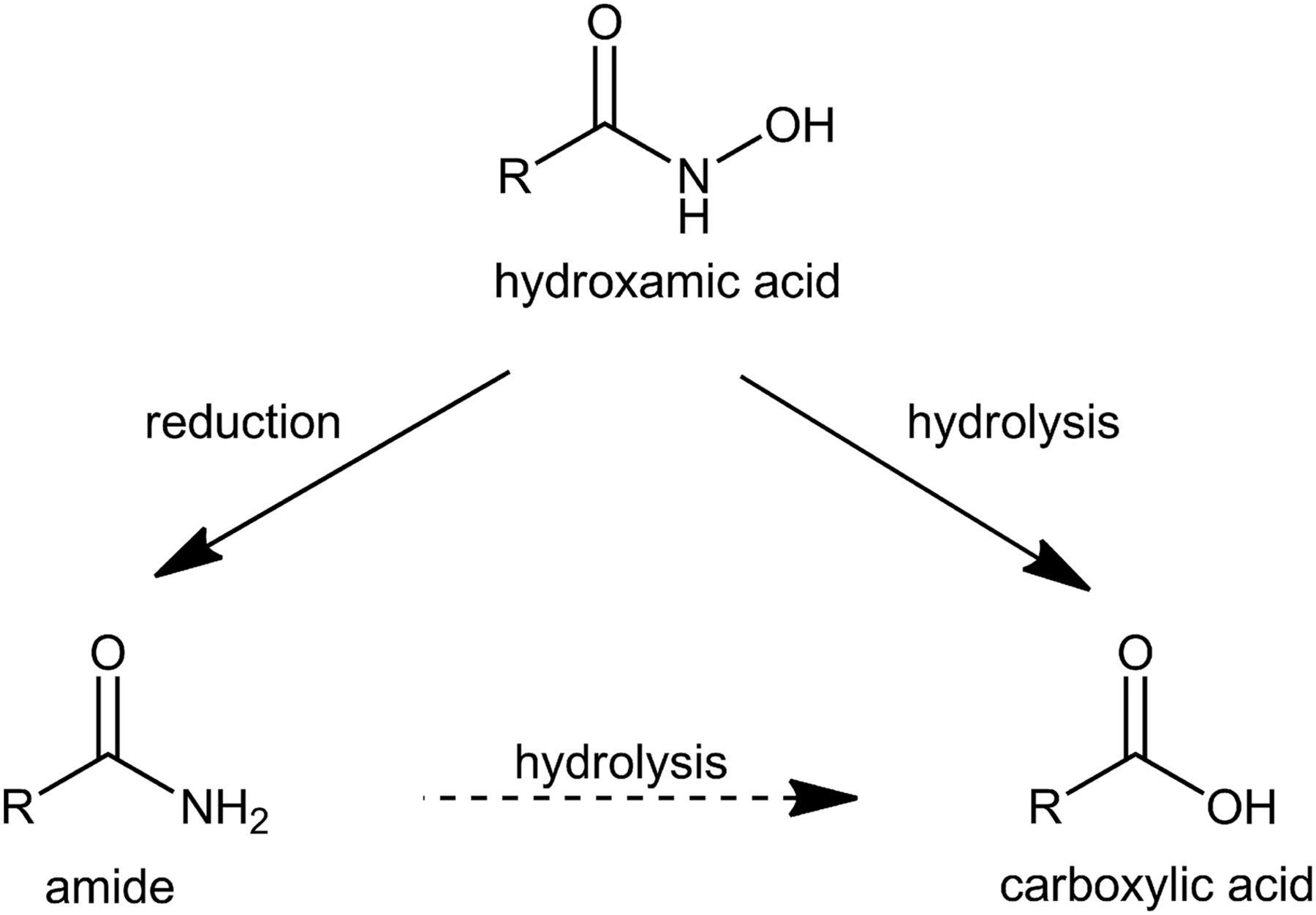
Metabolism of hydroxamic acids. Hydroxamic acids can either undergo reduction to the corresponding amide or hydrolysis to the corresponding carboxylic acid. A hydrolysis of the amide to the carboxylic acid might be possible.
It is well accepted that the mitochondrial amidoxime-reducing component “mARC”, together with the electron transport proteins cytochrome b5 and NADH-cytochrome b5 reductase, forms an N-reductive three component enzyme system located in the outer mitochondrial membrane which plays a major role in N-reductive drug metabolism. It has been shown after in vitro reconstitution and in cell culture that the mARC-system is responsible for the reduction of various N-hydroxylated compounds like amidoximes, N-hydroxyguanidines or sulfhydroxamic acids, hydroxylamines and N-oxides (Plitzko et al., 2013; Ott et al., 2015). Our laboratory has recently shown that mARC is a mitochondrial, molybdenum-containing enzyme (Havemeyer et al., 2006). All of the currently analyzed and completely annotated mammalian genomes code for two mARC proteins (mARC1 and mARC2) which share a high degree of sequence identity to each other (Wahl et al., 2010). Though the endogenous function of mARC is still not fully understood, mARC proteins are assumed to be involved in detoxification of mutagenic and toxic aromatic hydroxylamines like N-hydroxylated DNA-base analogs (Krompholz et al., 2012; Plitzko et al., 2015). The involvement in energy and NO metabolism has been discussed (Kotthaus et al., 2011; Neve et al., 2012; Jakobs et al., 2014; Sparacino-Watkins et al., 2014). In this study we examined whether the mARC-system is also capable of reducing hydroxamic acids and is thus involved in the metabolic conversion of this substance class. As shown in Fig. 2, we assayed the N-reduction of the model substrate benzhydroxamic acid and of four relevant pharmaceutical compounds (vorinostat, bufexamac and CP544439 as drugs and N-hydroxyphenacetin as a toxic drug metabolite) by performing biotransformation assays in the reconstituted recombinant human N-reductive system and in subcellular porcine liver fractions. Additionally, by performing metabolism studies and RNAi-mediated down-regulations of mARC in HEK-293 cells, the physiologic relevance of the N-reduction of hydroxamic acids in human cell metabolism was evaluated.
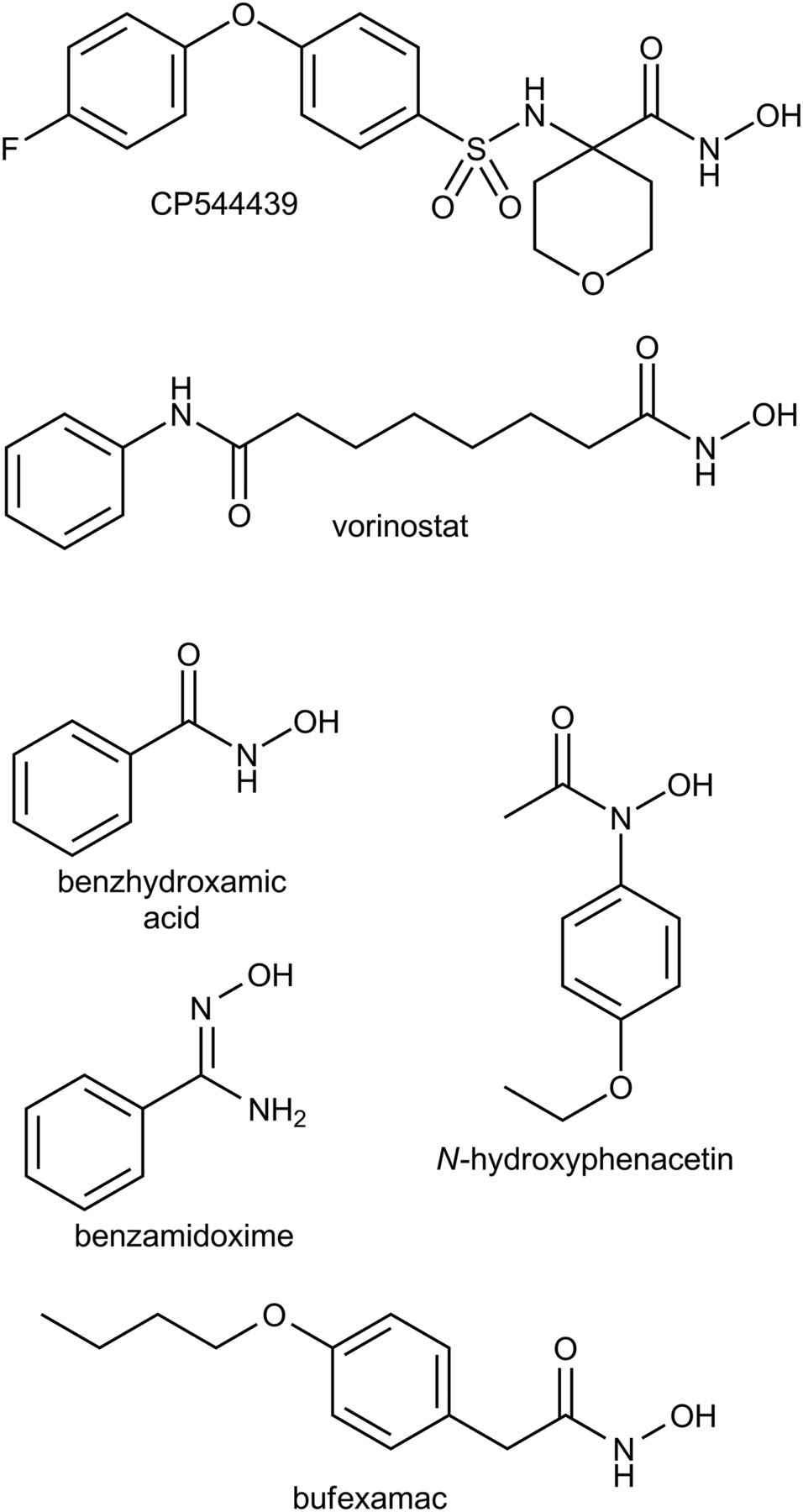
Structures of all studied compounds.
Materials and Methods
Reagents and Cell Lines
Unless otherwise stated all chemicals were purchased from Carl Roth GmbH & co. KG (Karlsruhe, Germany), Sigma Aldrich (St. Louis, MO) or Fluka (Buchs, Switzerland) and used without further purification. Methanol (HPLC grade) was from JT Baker (Deventer, Netherlands). Acetonitrile (HPLC grade) was from Honeywell (Seelze, Germany). Benzoic acid and chlorosulfonic acid were from Merck KGaA (Darmstadt, Germany). Ethyl 4-aminotetrahydro-2H-pyran-4-carboxylate hydrochloride and 4-fluorophenoxybenzene were from abcr (Karlsruhe, Germany). 4-nitrophenetole, 4-butoxyphenylacetic acid and sodium 1-octanesulfonate were from TCI (Zwijndrecht, Belgium). HEK-293 human embryonic kidney cells were purchased from Cell Lines Service (Eppelheim, Germany). Opti-MEM, minimum essential medium, sodium pyruvate solution, sodium bicarbonate, minimum Eagles’s medium nonessential amino acids, FBS, trypsin, L-glutamine, PBS, Lipofectamine RNAiMAX, Stealth Select RNAi siRNA targeting human MOSC1 (MOSC1HSS127704), and Stealth Select RNAi siRNA negative control were obtained from Invitrogen (Carlsbad, CA). ONTARGETplus SMARTpool siRNA targeting human MOSC2 was purchased from Thermo Scientific (Waltham, MA). Complete protease inhibitor cocktail was acquired from Roche Applied Science (Mannheim, Germany). Benzamidoxime was synthesized from benzonitrile and hydroxylamine (Krüger, 1885). For the synthesis of CP544439 (4-((4-(4-fluorophenoxy)phenyl)-sulfonylamino)tetrahydropyran-4-carbohydroxamic acid) and its metabolites see Supplemental Data.
Synthesis of Vorinostat (Suberoanilohydroxamic Acid) and Metabolites
Chromatographic separations were performed on silica gel (15–40 mesh; Merck) using flash methodology. Reaction progress was monitored by analytical TLC on pre-coated silica gel (Kieselgel 60 F254) plates, and spots were detected by UV light (λ 254 nm). mp of the final target HDAC inhibitors were determined by the open capillary method on a Stuart-Scientific SMP3 electrothermal apparatus and are uncorrected. 1H NMR spectra were recorded in the indicated deuterated solvents on a Bruker Avance DRX 400 MHz spectrometer and 13C NMR on a Varian 100 MHz. Signals due to OH and NH protons were located by deuterium exchange with D2O. EI- and CI-mass spectra were measured with a TSQ700 mass spectrometer (Thermoelectron, Waltham, MA). ESI- and PCI-mass spectra were recorded with a LCQ-Advantage mass spectrometer. In all cases, spectroscopic data are in agreement with known compounds and assigned structures. HPLC purity determinations were performed on a JASCO HPLC system under isocratic conditions, using a Phenomenex Synergi Hydro RP-C18 column (250 × 4.6 mm, 4 µm particle size). Elution was performed using 0.05% of TFA in water/ACN 60/40 (v/v), at room temperature. The purity of all tested compounds was ≥98%, as measured by HPLC. Injection volumes were 2 µl, flow rate was 0.5 ml/min, detection was performed with UV (λ = 254 nm). All chromatographic and spectroscopic data were in accordance with literature data.
Synthesis of Vorinostat.
Vorinostat was synthesized according to Mai et al. (2001). Retention time for HPLC was 7.8 minutes.
Synthesis of DS92.
DS92 was synthesized by a modification of the procedure described in the literature (Suzuki et al., 2005). Suberic acid (5.00 g, 28.7 mmol) was slowly added to neat freshly distilled aniline (2.70 ml, 30.1 mmol) and the mixture was stirred at 185°C and left at the same temperature for 2 hour. The mixture was cooled to room temperature and a NaOH 2 N aqueous solution was added to set the pH of the mixture at ∼8. The suspension was filtered and the filtrate was acidified to pH∼2, obtaining a white precipitate that was collected and suspended in warm water (50°C). The insoluble part was filtered and washed with hot water, yielding DS92 as white pure precipitate (3.93 g, 55%). Rf = 0.35 (EtOAc/Cy, 8:2); Retention time for HPLC was 14.5 minute; mp = 124–126°C; 1HNMR (400 MHz, d6DMSO): δ 1.23–1.35 (m, 4H), 1.42–1.52 (m, 2H), 1.54–1.63 (m, 2H), 2.20 (t, J = 7.4 Hz, 2H), 2.29 (t, J = 7.4 Hz, 2H), 7.01 (tt, J = 7.6; J = 1.1 Hz, 1H), 7.28 (t, J = 7.6 Hz, 2H), 7.58 (dd, J = 8.7; J = 1.1 Hz, 2H), 9.85 (s, 1H), 11.97 (br s, 1H); LRMS (ESI) m/z 248.1 [M-H]-.
Synthesis of DS116.
DS116 was obtained by a fast two-step synthesis starting from the carboxylic acid derivative DS92, which is transformed using thionyl chloride into the corresponding acyl chloride, further reacted into the target according to an already described procedure (Wright and Corbett, 1993). To a solution of 8-oxo-8-(phenylamino)octanoic acid (0.30 g, 1.2 mmol) in dry DCM (3 ml) thionyl chloride (330 µl, 4.6 mmol) was slowly added at room temperature. The solution was heated to reflux for 3 hour and later a stream of nitrogen was used to remove the solvent and the excess of thionyl chloride. To the crude product was added a solution of concentrated aqueous ammonia solution (1.03 ml, 26.4 mmol) and NH4Cl (0.22 g, 4.1 mmol) in 1 ml of water and the mixture was stirred at 20°C for 8 hour. The reaction was quenched with NaBH4 (23 mg, 0.6 mmol) and after 1 hour at room temperature HCl 2 N aqueous solution was added dropwise to pH∼2. The precipitate was collected to give DS116 as white powder (0.24 g, 80%). Rf = 0.30 (DCM/Et2O/MeOH/, 9.5:1:0.5); Retention time for HPLC was 9.18 minute; mp = 161–163°C; 1H-NMR (400 MHz, DMSO-d6): δ 1.22–1.34 (m, 4H), 1.44–1.52 (m, 2H), 1.63–1.53 (m, 2H), 2.03 (t, J = 7.4 Hz, 2H), 2.29 (t, J = 7.4 Hz, 2H), 6.68 (br s, 1H), 7.01 (t, J = 7.4, 1H), 7.22 (br s, 1H), 7.28 (t, J = 7.8 Hz, 2H), 7.58 (d, J = 7.8 Hz, 2H), 9.85 (s, 1H); LRMS (ESI) m/z 247.1 [M-H]−.
Synthesis of N-Hydroxyphenacetin
N-hydroxyphenacetin was prepared using a previously reported procedure with minor modifications (Hinson and Mitchell, 1976). 4-Nitrophenetole (2.0 g, 11.96 mmol) and NH4Cl (0.64 g, 11.96 mmol) were dissolved in 40 ml of a C2H5OH/H2O mixture (4:1, v/v) at room temperature. After the addition of Zn dust (3.2 g, 48.94 mmol) the reaction mixture was stirred for 10 minutes. The Zn dust was filtered and washed with 40 ml (C2H5)2O. The ethereal phase was washed with 50 ml brine and separated from the aqueous phase. NaHCO3 (1.6 g, 19.04 mmol) was suspended in 5 ml H2O at 0°C and the N-hydroxyphenetidine containing ethereal phase was added. 500 μl of 2.5% acetyl chloride in (C2H5)2O were slowly added over 1 hour. The formation of the product was TLC controlled. N-hydroxyphenacetin gave a red, N-hydroxyphenetidine a blue spot with FeCl3. After disappearance of the blue spot, 20 ml of H2O was added and twice extracted with (C2H5)2O. The combined layers were washed with H2O and twice with brine. The product was extracted with 100 ml of cold 0.2 M NH3-solution. After neutralization to pH 7.0 with NaHCO3 the product was extracted in 300 ml (C2H5)2O, dried over anhydrous Na2SO4 and the solvent was evaporated to dryness. N-hydroxyphenacetin was recrystallized with (C2H5)2O/hexane, 15% yield from 4-nitrophenetole. 1H-NMR (300 MHz, DMSO-d6): δ 10.48 (s, 1H, hydroxyl), 7.45 (d, J = 8.7 Hz, aromatic, 2H), 6.91 (d, J = 9.1 Hz, aromatic, 2H), 4.01 (q, J = 7.0 Hz, 2H, H3CH2-), 2.14 (s, 3H, CH3CO−), 1.31 (t, J = 7.0 Hz, 3H, CH3CH2-); LC-MS (ESI), m/z 196 [M+H]+, 178, 150.
Protein Sources
Subcellular porcine tissue fractions were purified from female pigs as described earlier (Ott et al., 2014). Expression and purification of human mARC1 (reference sequence NP_073583) and mARC2 (reference sequence NP_060368), CYB5B (reference sequence NP_085056) and CYB5R3 (reference sequence NP_000389) was carried out in Escherichia coli as described by Wahl et al. (2010). Protein content was determined using (BCA) protein assay kit (Pierce, Rockford, IL) according to the manufacturer’s protocol. Heme content in CYB5B was determined according to the method of Estabrook by recording the difference spectrum of oxidized and NADH-reduced protein (Estabrook and Werringloer, 1978). FAD-content in CYB5R3 was measured at 450 nm according to Whitby after sample preparation by heating at 100°C for 10 minute and centrifugation at 22,000g for 5 minute at room temperature (Whitby, 1953). For the quantification of FAD a calibration curve (0.01–0.1 mM) was used.
In Vitro N-Reductive Activity Assay
In vitro biotransformation assay was carried out at 37°C in a shaking water bath. Incubation mixture consisted of 100 µg of porcine subcellular fractions in 100 mM potassium phosphate buffer, pH 6.0 or in case of recombinantly expressed human proteins of 7.5 µg mARC1 or mARC2, CYB5B resulting in 75 pmol heme and CYB5R3 resulting in 7.5 pmol FAD in 20 mM MES buffer, pH 6.0. Different substrate concentrations were used and for CP544439, vorinostat and bufexamac 4.0%, 4.8% and 8.0% DMSO were added respectively. After 3 minute of pre-incubation, the reaction was started by adding 1 mM NADH, resulting in a total volume of 150 µl. Incubation was stopped after 15 minute by adding 150 µl of cold methanol. Afterward, samples were shaken for 5 minute at room temperature and centrifuged for 5 minute with 9500g at room temperature. Supernatants were analyzed by HPLC. For the determination of kinetic parameters with recombinantly expressed human protein the DMSO concentration was kept at the same level and only the substrate concentration was modified.
HPLC Analysis for Incubated Samples
The flow rate was kept at 1.0 ml/min and the injection volume was 10 µl for all performed HPLC analysis. All benzhydroxamic acid and CP544439 related samples were measured on a Waters e2695 Separation Module with a Waters 2998 Photodiode Array Detector and Waters Empower 2 Build 2154 as integration software.
For the separation of benzhydroxamic acid (rt = 5.0 ± 0.2 minute) benzamide (rt = 8.1 ± 0.2 minute) and benzoic acid (rt = 12.7 ± 0.1 minute) a Phenomenex Gemini NX-C18 (5 µm), 150 × 4.6 mm with a Phenomenex C18 4 × 3.0 mm pre-column was used. The mobile phase consisted of 50 mM KH2PO4, pH 4.6, 10 mM tetramethylammonium chloride and 10% acetonitrile (v/v). Detection wavelength was 210 nm.
For the separation of CP544439 (rt = 4.8 ± 0.3 minute), deoxy CP544439 (rt = 5.9 ± 0.2 minute) and the carboxylic derivative (rt = 7.0 ± 0.2 minute) a Waters Sunfire C18, 3.5 µm, 150 × 4.6 mm with a Phenomenex C18 4 × 3.0 mm pre-column was used. Solvent A (0.2% formic acid in H2O (v/v)) and Solvent B (0.2% formic acid in acetonitrile (v/v)) were used. Starting with 60% A, the gradient changed linearly from 3 to 7 minute to 10% A. At 11 minute A was set to 60% over 0.5 minute. Total runtime was 16 minute. Detection wavelength was 247 nm. The column temperature was maintained at 25°C and sample storage temperature at 18°C.
All vorinostat, bufexamac, N-hydroxyphenacetin and benzamidoxime related samples were measured on a Waters HPLC system consisting of a Waters 717 autosampler, a Waters 1525 pump and a Waters 2487 dual absorbance detector at room temperature. A Phenomenex Gemini NX-C18 (5 µm), 150 × 4.6 mm with a Phenomenex C18 4 × 3.0 mm pre-column was used with exception for benzamidoxime where a LiChrospher 60 RP-select B (5 µm), 250 × 4 mm column combined with a Lichrospher 60 RP-select pre-column was used.
For separation of vorinostat (rt = 6.7 ± 0.0 minute), DS116 (rt = 8.9 ± 0.1 minute) and DS92 (rt = 18.9 ± 0.2 minute) the mobile phase consisted of 100 mM KH2PO4 and 23% acetonitrile (v/v). The detection wavelength was set to 254 nm.
For separation of bufexamac (rt = 16.9 ± 0.1 minute), deoxy bufexamac (rt = 20.9 ± 0.1 minute) and the carboxylic derivative (rt = 28.5 ± 0.2 minute) the mobile phase consisted of 50 mM KH2PO4, pH 4.4, 10 mM tetramethylammonium chloride and 45% methanol (v/v). Detection wavelength was set to 228 nm.
For separation of N-hydroxyphenacetin (rt = 7.8 ± 0.2 minute) and phenacetin (rt = 9.1 ± 0.0 minute) the mobile phase consisted of 1% formic acid in H2O (v/v) and 22.5% acetonitrile (v/v). Detection wavelength was 245 nm.
For separation of benzamidoxime (rt = 9.1 ± 0.3 minute) and benzamidine (rt = 15.9 ± 0.1 minute) the mobile phase consisted of 10 mM sodium 1-octanesulfonate and 20% acetonitrile. Detection wavelength was 229 nm.
Cell Culture
HEK-293 cells, derived from a female human source, were maintained in minimum essential medium supplemented with 10% FBS, 2 mM L-glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, and 1.5 g/l sodium bicarbonate. The cell line was incubated at 37°C in 5% CO2.
siRNA Transfection and Design of Knockdown Experiments
HEK-293 cells were reverse transfected and mARC-protein down-regulated according to the previous described procedure (Plitzko et al., 2015).
N-Reductive Metabolism of Benzhydroxamic Acid in HEK 293 Cells
For N-reduction studies in HEK-293 the culture medium was removed, and cells were carefully washed and pre-incubated with substrate-free incubation buffer (Hanks’ balanced salt solution containing 10 mM HEPES, pH 7.4) at 37°C for 10 minute. After removing the substrate-free incubation buffer, the vital cells were then incubated with benzhydroxamic acid-containing incubation buffer (3 mM, 0.5% (v/v) DMSO) at 37°C for 180 minute. After the designated time, the culture supernatant was carefully removed, centrifuged to eliminate cellular debris and analyzed by HPLC as described above.
Total Cellular Protein Extraction
Cellular protein was harvested and protein contents determined as previously described (Plitzko et al., 2015).
Western Blot Analysis
SDS-PAGE and Western Blot analysis to verify down-regulation of mARC-protein in HEK-293 cells was carried out as described previously (Plitzko et al., 2015).
Statistical Analysis
Statistical analyses were carried out using the SigmaPlot 11 software (Systat Software Inc., Erkrath, Germany). The significance of observed differences was evaluated by Bonferroni test. A probability less than 5% was considered to be significant. All experimental values are given as means ± S.D.
Results
From all analyzed subcellular fractions, N-reductive activity in mitochondria was found to be enhanced compared with other tissue fractions for all N-unsubstituted hydroxamic acids (Fig. 3). These findings reflect the enrichment of mARC depending enzyme activities in this fraction as published earlier (Krompholz et al., 2012). In the microsomal and cytosolic fractions only minor/no N-reductive activity was detectable. By contrast, no reduction of N-hydroxyphenacetin could be detected after incubation with the mitochondrial fraction and only minor reduction rates were detected within the cytosolic fraction (data not shown). Besides reduction to the amide, hydrolysis to the corresponding carboxylic acids was monitored and was found to be more pronounced in microsomes compared with other fractions; this indicates the involvement of a microsomal enzymatic system or a non-enzymatic reaction (Fig. 3, A and D). In the case of bufexamac and CP544439 the microsomal fraction was the only fraction where hydrolysis could be detected to a small extent (data not shown).

N-reduction of benzhydroxamic acid, bufexamac, CP544439 and vorinostat in hepatic subcellular fractions. Biotransformation assay consisted of 100 µg protein, 1 mM NADH and either 1.0 mM vorinostat and 4.8% DMSO, 3.0 mM benzhydroxamic acid, 0.5 mM CP544439 and 4.0% DMSO or 1.0 mM bufexamac and 8.0% DMSO. Incubation was carried out for 15 minute and stopped by addition of methanol. Activities are means ± S.D. of two biologic determinations. (A) benzhydroxamic acid, (B) bufexamac, (C) CP544439, (D) vorinostat, *under limit of quantification, n.d. not detectable.
To prove the involvement of mARC in the reduction of hydroxamic acids, a cell based siRNA experiment was performed. In HEK-293 cells reductive conversion of benzhydroxamic acid to benzamide occurs in a time-dependent (data not shown) and substrate-dependent manner (Fig. 4A) and followed Michaelis Menten kinetics (vmax = 0.06 ± 0.01 nmol benzamide·min−1·mg protein−1). By siRNA-mediated down-regulation of mARC1, benzamide formation decreased dramatically to approximately 30% compared with the negative control (Fig. 4B). Knockdown of mARC2 in HEK-293 cells did not affect the reduction of benzhydroxamic acid in HEK-293 cells. This same behavior is also observed with the model substrate benzamidoxime and is attributed to the low level of mARC2-protein expression in HEK-293 cells (Plitzko et al., 2013). Simultaneous knockdown of both mARC-proteins led to a small, further decrease in N-reductive activity than was observed in the mACR1-only knockout experiments. To further elucidate the differences between mARC1 and mARC2 the kinetic parameters vmax and KM were determined with the in vitro reconstituted recombinant N-reductive system (Fig. 5). All hydroxamic acids were clearly reduced to the corresponding amides with the exception of N-hydroxyphenacetin. These results are consistent with previous observations in subcellular fractions. Benzhydroxamic acid, bufexamac, CP544439 and vorinostat were exclusively reduced to their corresponding amides; none of the corresponding carboxylic acid products were detected. The reductions obey Michaelis-Menten kinetics for both mARC-proteins. The calculated KM and vmax values are presented in Table 1. The conversion rates of hydroxamic acids are lower in comparison with the model compound benzamidoxime (e.g., for vorinostat, vmax is 15 times slower). For both incubation types with subcellular fractions and recombinant expressed proteins, highest vmax values were obtained for the model compound benzhydroxamic acid in comparison with all other studied hydroxamic acids.

N-reductive metabolism of benzhydroxamic acid in HEK-293 cells. N-reductive activities of cells were determined as described in Materials and Methods. Results are presented as means ± S.D. (n = 3). (A) Substrate dependent metabolism. Incubation time was 180 minute. (B) Effect of mARC knockdown on N-reduction. HEK-293 cells were transfected with 20 nM mARC siRNA or non-targeting (NC) siRNA. The siRNA-mediated down-regulations of the proteins of interest were verified by western blot using anti-mARC1, anti-mARC2 or anti-calnexin antibody. Calnexin levels were used as loading control. mARC2-protein could not be detected. N-reductive activities were determined on day 4 after transfection. ***P < 0.001.

Substrate concentration dependence for mARC1 (black) and mARC2 (white). Biotransformation assay consisted of 7.5 µg hmARC, 75 pmol CYB5B and 7.5 pmol CYB5R3. Incubation time was 15 minutes. Quantification was done by HPLC analysis. Activities are means ± S.D. of two biologic determinations. (A) benzhydroxamic acid, (B) bufexamac, (C) CP544439, (D) vorinostat.
N-reduction of vorinostat, benzhydroxamic acid, bufexamac, CP544439, N-hydroxyphenacetin and benzamidoxime by the reconstituted recombinant mARC-system. Biotransformation assays were carried out as described in Materials and Methods
Activities are means ± S.D. of two biologic determinations.
According to the vmax values, CP544439 and vorinostat are reduced to the same amount and smallest conversion rates were detected for bufexamac. Vorinostat, bufexamac and CP544439 required the use of DMSO as a solubilizer. Due to the negative influence of high DMSO concentrations in the incubation mixture (see Supplemental Data), only the minimum amount of DMSO required for solubility was added. In the case of benzhydroxamic acid vmax was about four times higher with mARC2 than with mARC1 but the KM increased sevenfold. Only slight differences of the kinetic parameters for vorinostat and bufexamac were detected for both mARC forms, whereas CP544439 was exclusively reduced by mARC1. In accordance with the results obtained with subcellular liver fractions and RNAi studies all N-unsubstituted hydroxamic acids are reduced by mARC.
Discussion
The mARC-containing three component enzyme system is responsible for the reduction of various N-hydroxylated compounds (Ott et al., 2015) and the results of our recent investigations demonstrate clearly that hydroxamic acids belong to this class of compounds. The hydroxamate moiety exhibits strong cation chelating properties and thereby possesses the ability to affect a variety of enzymes. Hydroxamic acid moieties are found in a multitude of drugs and drug candidates (Halouska et al., 2014; Zhang and Zhong, 2014). In addition, aryl hydroxamic acids have been shown to possess toxic and mutagenic properties (Miller et al., 1961; Vaught et al., 1981). Therefore, the investigation of the metabolic fate of hydroxamic acids is of particular relevance for further drug developments and for the understanding of detoxification pathways. We could demonstrate that hydroxamic acids can serve as substrates for the mARC-system. The model compound benzhydroxamic acid, as well as three other drugs (vorinostat, bufexamac and CP544439) are reduced to the corresponding amides by porcine mitochondria and the reconstituted recombinant human mARC-system (Table 1). The N-reduction observed in vitro is also evident in intact human cell metabolism and is mARC-dependent as the siRNA-mediated down-regulation leads to a dramatic decrease in N-reductive activity (Fig. 4B). HEK-293 cells were chosen because of the high N-reductive activity in renal tissue (Krompholz et al., 2012). However, the N-reductive conversion of hydroxamic acids is lower compared with the model compound (benzamidoxime). Amidoximes, used as pro-drugs for amidines, are rapidly reduced in vivo (Clement et al., 1992). In the case of hydroxamic acids, reduction leads to inactivation because the amide is not able to form strongly chelating complexes. As metabolism studies for CP544439 reveal, the main metabolism pathways of hydroxamic acids in vivo are glucuronidation, reduction and hydrolysis (Dalvie et al., 2008). The reduction to the amide is of great physiologic relevance, especially in rats, where the amide is the most prevalent metabolite. It has been demonstrated that the aldehyde oxidase is capable of reducing hydroxamic acids to amides (Sugihara et al., 1983a,b; Sugihara and Tatsumi, 1986). Reduction of CP544439 was proposed to be catalyzed by this enzyme as well; studies with human cytosolic liver fractions using the artificial electron donator N-methylnicotinamide resulted in a very low conversion rate (1.6 pmol·min−1·mg protein−1) (Obach, 2004). The aldehyde oxidase is located in the cytosol, but we could not detect any N-reductive activity in the porcine liver cytosol under our tested conditions (Fig. 5C). Our determined conversion rate with the mitochondrial fraction for CP544439 is about 2.5 nmol·min−1·mg protein−1, which is more than 1500 times higher than the previous described cytosolic conversion rate. This finding indicates the important role of mARC in the reduction of hydroxamic acids to the amides. Interestingly, vorinostat has sufficient metabolic stability for therapy and is applied orally, but high doses of 400 mg/day are necessary (Mann et al., 2007). The enhanced lipophilicity of vorinostat compared with benzhydroxamic acid and benzamidoxime could be a pivotal characteristic. The low conversion rates determined for bufexamac, which is a very lipophilic ether compound, support this theory (Fig. 5B). Studies to further elucidate structure activity relationships should be done to evaluate whether lipophilicity is an important feature for enzymatic conversion of hydroxamic acids by the mARC-system and to detect candidates with better stability toward N-reduction.
In the case of N-hydroxyphenacetin no reduction to the amide by mARC was observed. This is the first investigated hydroxamic acid studied by us so far which is not reduced. However, this could be an explanation for the high toxicity of this compound as it needs to be detoxified by other pathways. The most apparent difference between N-hydroxyphenacetin and the other studied hydroxamic acids is the substitution of the nitrogen’s hydrogen with the sterically demanding phenyl group. Due to this steric hindrance mARC might not be able to bind and reduce such kind of substrates. This hypothesis needs to be proved by further structure activity studies.
In conclusion, to properly evaluate the metabolic stability of new hydroxamic acid containing drug candidates, metabolism by the mitochondrial mARC-system must be considered.
Acknowledgments
Thanks to Timothy Zerk for proofreading the article.
Authorship Contributions
Participated in research design: Scheidig, Havemeyer, Clement.
Conducted experiments: Ginsel, Plitzko.
Contributed new reagents or analytical tools: Ginsel, Froriep, Stolfa, Jung.
Expression and purification of recombinant proteins: Kubitza.
Performed data analysis: Ginsel, Plitzko.
Wrote or contributed to the writing of the manuscript: Ginsel, Plitzko, Clement.
Footnotes
- Received May 18, 2018.
- Accepted July 11, 2018.
D.A.S. and M.J. received funding from the European Union’s Seventh Framework Program for research, technological development and demonstration under grant agreements nr. 241865 (SEtTReND).
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ACN
- acetonitrile
- BCA
- bicinchoninic acid
- bs
- broad singlet
- CI
- chemical ionization
- CYB5B
- cytochrome b5 type B
- CYB5R3
- NADH-cytochrome b5 reductase 3
- d
- doublet
- dd
- double doublet
- DMSO
- dimethyl sulfoxide
- DS116
- N’-phenyloctanediamide
- DS92
- 8-oxo-8-(phenylamino)octanoic acid
- dt
- double triplet
- EI
- electron ionization
- ESI
- electrospray ionization
- FBS
- fetal bovine serum
- HDACs
- histone deacetylases
- HPLC
- high performance liquid chromatography
- J
- coupling constant in Hz
- LOQ
- limit of quantification
- m
- multiplet
- mARC
- mitochondrial amidoxime reducing component
- mp
- melting point
- NMR
- nuclear magnetic resonance
- PCI
- positive chemical ionization
- rt
- retention time
- s
- singlet
- t
- triplet
- TLC
- thin layer chromatography
- UV
- ultra violet
- δ
- chemical shift in ppm
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}