


Abstract
17α-Ethynylestradiol (EE), a major component of many oral contraceptives, affects the activities of a number of the human cytochrome P450 (P450) enzymes. Here, we characterized the effect of EE on CYP2J2, a major human P450 isoform that participates in metabolism of arachidonic acid. EE inactivated the hydroxyebastine carboxylation activity of CYP2J2 in a reconstituted system. The loss of activity is time and concentration dependent and requires NADPH. The KI and kinact values for the inactivation were 3.6 μM and 0.08 minute−1, respectively. Inactivation of CYP2J2 by EE was due to formation of a heme adduct as well as an apoprotein adduct. Mass spectral analysis of CYP2J2 partially inactivated by EE showed two distinct protein masses in the deconvoluted spectrum that exhibited a mass difference of approximately 312 Da, which is equivalent to the sum of the mass of EE and one oxygen atom. Liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis revealed a heme adduct with MH+ ion at m/z 875.5, corresponding to alkylation of an iron-depleted prosthetic heme by EE plus one oxygen atom. The reactive intermediate responsible for covalently modifying both the prosthetic heme and apoprotein was characterized by trapping with glutathione (GSH). LC-MS/MS analysis revealed two GSH conjugate isomers with MH+ ions at m/z 620, which were formed by reaction between GSH and EE with the oxygen being added to either the internal or terminal carbon of the ethynyl moiety. High-pressure liquid chromatography analysis revealed that three other major metabolites were formed during EE metabolism by CYP2J2.
Introduction
17α-Ethynylestradiol (EE) (Fig. 1), an orally bioactive component of many oral contraceptive formulations, is metabolized by cytochrome P450 (P450) enzymes in various animal species and by humans (Bolt et al., 1977; Maggs et al., 1983; Guengerich, 1990; Kent et al., 2002; Wang et al., 2004; Lin and Hollenberg, 2007). EE has been used by more than 70 million women worldwide for more than 50 years and its interactions with clinically used drugs including rifampicin, bupropion, ritonavir, nevirapine, selegiline, mephenytoin, omeprazole, and tizanidine have been studied extensively (Bolt et al., 1977; Ouellet et al., 1998; Laine et al., 2000; Mildvan et al., 2002; Palovaara et al., 2003; Granfors et al., 2005; Zhang et al., 2007). In some cases, an association between hypertension or cardiovascular disease and oral contraceptives containing EE has been reported (Ahluwalia et al., 1977; Godsland et al., 1995; Olatunji and Soladoye, 2006). CYP3A4 and CYP2C9 are the two major P450 enzymes contributing to the 2-hydroxylation of EE, and the formation of 2-OH-EE has also been observed for CYP1A1, 1A2, 2B6, 2C8, 2C19, and 3A5 using human liver microsomes or recombinant P450 reconstituted systems (Guengerich, 1990; Kent et al., 2002; Wang et al., 2004; Lin and Hollenberg 2007). EE exhibits an inhibitory effect on various human P450s including CYP1A1, 1B1, 2B6, 2C8, 2C9, 2C19, 2J2, 3A4, and 3A5 (Lin et al., 2002; Lin and Hollenberg 2007; Chang et al., 2009).

Chemical structure of EE. Some of the carbon atoms are numbered.
CYP2J2 is primarily expressed in the human cardiovascular system and it is also expressed in the intestines, lungs, livers, kidney, brain, and tumor tissues (Wu et al., 1996; Zeldin et al., 1996, 1997; Dutheil et al., 2009; Jiang et al., 2009). A major endogenous substrate of CYP2J2 is arachidonic acid, which is converted to four biologically active regioisomeric epoxyeicosatrienoic acids (EETs) (Wu et al., 1996; Jenkins et al., 2009). These four EETs have biologic effects on numerous processes including cellular proliferation, inflammatory responses, and hormone release (Zeldin et al., 1996, 1997; Node et al., 1999). They also play critical roles in cellular signaling pathways responsible for cardiovascular functions, including control of blood pressure, and in the protection of endothelial cells from hypoxia-reoxygenation injury (Wu et al., 1996; Yang et al., 2001; Fleming and Busse, 2006). Since it is now well recognized that alterations in the EET pathway contribute to the pathophysiology of the cardiovascular system, the interest in research on human CYP2J2 has grown significantly in the last 10 years. Current areas of investigation include: 1) identification of potent and selective CYP2J2 inhibitors (Lafite et al., 2006; Chen et al., 2009; Lee et al., 2012; Ren et al., 2013); 2) identification of additional drugs currently in use that are metabolized by CYP2J2 (Lee et al., 2010; Wu et al., 2013); 3) mechanism(s) by which EETs protect the myocardium following ischemia/reperfusion injury (Seubert et al., 2007); and 4) mechanisms by which overexpression of CYP2J2 or EET administration protect rat hearts against injury by tumor necrosis factor-α (Zhao et al., 2012). More recent studies have demonstrated elevated levels of CYP2J2 mRNA and protein in human-derived cancer cells and human cancer tissues, raising the possibility of a role for CYP2J2 in some cancers (Jiang et al., 2005, 2009; Nithipatikom et al., 2010). Thus, the detection of CYP2J2 overexpression is currently used as a biomarker for cancer cells, and selective inhibitors of CYP2J2-mediated EET biosynthesis may provide a promising therapeutic strategy for the treatment of some human cancers.
CYP2J2 plays an important role in sequential metabolism of ebastine (EB), a second-generation antihistamine shown to be effective in the treatment of both seasonal and perennial allergies with a low potential for inducing drowsiness. The hydroxylation of EB to the intermediate metabolite hydroxyebastine (OHEB) followed by conversion of OHEB to carebastine (CEB), the pharmacologically active metabolite, is preferentially catalyzed by CYP2J2 (Hashizume et al., 2002; Liu et al., 2006). Initially, EB was used to characterize the catalytic activity and kinetics of purified CYP2J2 in the reconstituted system with NADPH-P450 reductase. However, it was found that 10% of OHEB formed from EB was further metabolized to CEB after 5-minute incubation with CYP2J2 (Hashizume et al., 2002; Matsumoto et al., 2002). To avoid the possibility of subsequent metabolism confounding the measurement of catalytic activity and the kinetic studies, OHEB is used here as the substrate probe to characterize the mechanism-based inactivation of CYP2J2 by EE (Lin et al., 2017). During the inactivation of CYP2J2 by EE, the formation of both heme and apoprotein adducts was identified by mass spectrometry.
Materials and Methods
Chemicals.
NADPH, glutathione (GSH), EE, chloramphenicol, and catalase were purchased from Sigma-Aldrich (St. Louis, MO). EB, OHEB, and CEB were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). 16β-Hydroxytestosterone was purchased from Steraloids Inc. (Newport, RI). All other chemicals and solvents were of the highest purity available from commercial sources.
Enzyme Purification.
CYP2J2 was expressed using the modifications as described previously (Smith et al., 2008). The CYP2J2 cDNA construct was a gift from Dr. Rheem Totah at the University of Washington (Seattle, WA). Briefly, the CYP2J2 plasmid was transformed into Escherichia coli C41(DE3) competent cells and the CYP2J2 protein was expressed as previously described (Lin et al., 2017). CYP2J2 purification was performed as described previously for purification of other members of the CYP2B family (Scott et al., 2001). CYP2B1, CYP3A4, and NADPH-P450 reductase were expressed in E. coli TOPP3 cells and purified according to previously published procedures (Scott et al., 2001; Lin et al., 2002).
Catalytic Activity and Inactivation of the P450s.
To measure the catalytic activity of CYP2J2 in the reconstituted system, the assays were performed as previously described with OHEB as the substrate (Lin et al., 2017). The data were analyzed for the time- and concentration-dependent loss of activity. The calculations of the kinetic values for mechanism-based inactivation were performed as previously described (Kent et al., 2002; Lin et al., 2013). Each experiment was repeated at least twice and done in triplicate with the replicates differing by no more than 10%. The goodness of fit for all experiments in which regression analysis was used had the value of r2 > 0.95.
Determination of the Partition Ratio.
EE at various concentrations ranging from 1 to 200 μM was added to the reconstituted system containing CYP2J2 and incubated as described by Lin et al. (2017). The reactions were initiated by the addition of 1 mM NADPH and incubated for 30 minutes to allow the inactivation to go to completion (Silverman, 1966). Aliquots were removed and assayed for residual catalytic activity as described previously and the data were analyzed as described previously (Lin et al., 2002).
Analysis of the Prosthetic Heme.
Aliquots containing 100 pmol of control (−NADPH) or inactivated (+NADPH) CYP2J2 that had been incubated with 50 μM EE for 10 minutes were analyzed for prosthetic heme and heme adducts as previously described (Lin et al., 2017). Under the same conditions, 100 pmol of CYP2B1 and CYP3A4 was incubated with EE to characterize the changes in the prosthetic heme and the formation of heme adducts to compare them to that observed with CYP2J2.
Electrospray Ionization (ESI)-Liquid Chromatography (LC)-Mass Spectrometry (MS) Analysis of the Apoprotein.
Control and inactivated samples incubated with 50 μM EE for 10 minutes were prepared as described previously. Aliquots containing 50 pmol of P450 were analyzed by ESI-LC-MS as described previously (Kent et al., 2006; Lin and Hollenberg, 2007; Lin et al., 2017).
Liquid Chromatography–Tandem Mass Spectrometry (LC-MS/MS) Analysis of the EE-Heme Adduct.
For CYP2J2, the major heme adduct eluted from the C4 reverse-phase column and was manually collected from a 200 pmol primary reaction mixture during the high-pressure liquid chromatography (HPLC) analysis. The samples were concentrated to 100 μl by blowing down with N2 gas and subjected to LC-MS/MS analysis using a C18 reverse-phase column (Jupiter, 5 μm, 2.0 × 100 mm; Phenomenex, Torrance, CA). Solvent A was 0.1% trifluoroacetic acid (TFA) in H2O and solvent B was 0.1% TFA in acetonitrile. The initial gradient was from 40% to 60% B for 5 minutes followed by a linear gradient to 80% B over 20 minutes, and then increasing linearly to 95% over 10 minutes at a flow rate of 0.3 ml/min. The column effluent was directed into the ESI source of a LCQ mass spectrometer (Thermo Fisher Scientific, San Jose, CA). The ESI source conditions were: the sheath gas was set at 90 arbitrary units, the auxiliary gas was set at 30 arbitrary units, the spray voltage was 4.2 kV, the tube lens was set at 25 V, and the capillary temperature was 200°C. Data were acquired in positive ion mode using Xcalibur software (Thermo Fisher Scientific) with one full scan followed by two data-dependent scans of the most intense and the second most intense ions.
For CYP2B1, an aliquot (25 pmol) of the primary reaction mixture inactivated by incubation with EE was injected onto a reverse-phase HPLC column (XTerra MS C18, 2.1 × 150 mm; Waters Corporation, Milford, MA) equilibrated with 65% solvent A (0.05% TFA/0.05% formic acid in water) and 35% solvent B (0.05% TFA/0.05% formic acid in acetonitrile) at a flow rate of 0.3 ml/min. The column was held at 35% solvent B for 5 minutes, increased linearly to 80% solvent B over 40 minutes, and then ramped to 95% solvent B over 10 minutes. The effluent from the HPLC was directed into a Thermo Fisher Scientific LTQ linear ion trap mass spectrometer. The ESI source was tuned with the heme moiety from horse heart myoglobin to optimize the conditions. The source parameters were 4 kV for the spray voltage, 300°C for the capillary temperature, 47 V for the capillary voltage, 100 V for the tube lens, 30 arbitrary units for the sheath gas, and five arbitrary units for the auxiliary gas. The ion trap was operated in the positive ion mode. Full-scan data were collected from m/z 300 to 2000 and the MS/MS spectra were acquired in a data-dependent mode on the six most abundant ions in the survey scan. The normalized collision energy was set to 35%.
LC-MS/MS Analysis of the GSH Conjugates of EE.
Samples containing 250 pmol of CYP2J2 in 250 μl of reconstituted system reaction mixtures were prepared as described previously. The reaction mixtures were incubated with 50 μΜ EE and 2 mM GSH in the absence or presence of 1 mM NADPH at 37°C for 30 minutes and the reactions were terminated by the addition of 1 ml of acetonitrile and analyzed by ESI-LC-MS as previously described (Lin et al., 2017).
Metabolism of EE.
CYP2J2 or CYP3A4 (0.5 nmol) was incubated with 100 μM EE in the reconstituted system at 37°C for 30 minutes. The samples were then extracted with 4 ml of ethyl acetate and dried under N2 gas. The samples were dissolved in 50% methanol (110 μl) to be analyzed by HPLC on a Microsorb-MV C18 column (5 μm; 4.6 × 250 mm; Agilent Technologies, Santa Clara, CA) using a system consisting of solvent A (0.1% acetic acid in water) and solvent B (0.1% acetic acid in 29.9% methanol and 70% acetonitrile). A linear gradient from 40% to 50% B was used for 10 minutes followed by a linear gradient to 55% B over 20 minutes, and then increasing linearly to 95% for 10 minutes at a flow rate of 1 ml/min. The eluate was monitored at 280 nm on a Waters HPLC with a 490E multi-wavelength detector (Waters Corporation).
Results
Kinetic Values for the Inactivation of CYP2J2 by EE.
The inactivation of the CYP2J2-catalyzed metabolism of OHEB by EE was measured using various concentrations (1–200 μM) of EE and at various time points for each concentration. Representative HPLC elution profiles showing the internal standard 16β-hydroxytestosterone, CEB, and OHEB at time 0 and the loss of CEB formation following incubation of the reaction mixtures with EE and NADPH for 6 and 12 minutes are shown in Fig. 2A. As shown in Fig. 2B, the inactivation of CYP2J2 was time and concentration dependent. Linear regression analysis of the time course data was used to estimate the initial rate constants (kobs) for the inactivation of CYP2J2 by the various concentrations of EE. The kinetic values were obtained by fitting the kobs values at the various concentrations of EE to the Michaelis-Menten equation (GraphPad Prism 5, San Diego, CA). The KI, kinact, and t1/2 values were determined to be 3.6 μM, 0.08 minute−1, and 9 minutes, respectively.

Calculation of the kinetic values and the partition ratio for the inactivation of CYP2J2 by EE. (A) Representative HPLC metabolite profile showing the time-dependent loss in the formation of CEB from OHEB after the reaction mixtures were incubated with 20 μM EE and 1 mM NADPH for 6 and 12 minutes. (B) The time- and concentration-dependent inactivation of CYP2J2 by EE. The reconstituted system was incubated with 0 (●), 1 (○), 5 (▴), 20 (△), 50 (▪), and 200 μM (□) EE, and aliquots were removed at the times indicated and assayed for residual OHEB metabolism activity as described in Materials and Methods. The catalytic activity at time zero was used as the 100% control to calculate the initial rate constants for the inactivation (kobs) for each concentration of EE. The inset shows the fitting of the initial rate constants to the Michaelis-Menten equation as a function of the EE concentrations. The KI and kinact values were determined to be 3.6 μM and 0.08 minute−1. (C) Determination of the partition ratio for the inactivation of CYP2J2 by EE. The percentage of catalytic activity remaining was determined as a function of the molar ratio of EE to CYP2J2 as described in Materials and Methods. The partition ratio was estimated from the intercept of the linear regression line from the lower ratios of EE to CYP2J2 and the straight line obtained from the higher ratios of EE to CYP2J2. The data represent the average of two separate experiments done in duplicate and that did not differ by >10%.
Partition Ratio for the Inactivation of CYP2J2 by EE.
CYP2J2 was incubated with various concentrations of EE for 30 minutes in order for the inactivation to reach completion. The percentage of activity remaining was plotted as a function of the molar ratio of inactivator to P450 (Fig. 2C). The partition ratio was estimated from the intercept of the linear regression line obtained from the lower ratios of EE to P450 with the straight line derived from higher ratios of EE to P450. Using this method, the partition ratio was estimated to be ∼20 for the inactivation of CYP2J2 by EE (Fig. 2C).
Heme Modification Following the Inactivation of P450s by EE.
Reaction mixtures containing CYP2J2, CYP2B1, and CYP3A4 were incubated with EE in the absence or presence of NADPH and analyzed by HPLC. The absorbance of the eluate was monitored at 400 nm. Initially, the change in the elution profile of the prosthetic heme moiety was characterized for CYP2J2. When a heme adduct was observed for CYP2J2 that had been inactivated by EE, we then compared this result with that for two other major mammalian P450s, which had previously been shown to be inactivated by EE (Kent et al., 2002; Lin et al., 2002). Figure 3A shows the HPLC elution profiles for the native heme of 2J2, 2B1, and 3A4 in the absence of NADPH. Figure 3B shows the HPLC elution profiles for the native heme and heme adducts in the presence of NADPH. The amounts of native heme remaining for CYP2J2, CYP2B1, and CYP3A4 following inactivation by EE were ∼40%, ∼20%, and ∼50%, respectively. The heme adduct observed for CYP2J2 was designated as “a.” The heme adducts observed for CYP2B1 were designated as “b” and “c.” The heme adducts observed for CYP3A4 were designated as “d” and “e.” The photodiode-array absorption spectra for all five heme adducts (a, b, c, d, and e) were essentially identical and the maximum was at ∼409 nm, as displayed in Fig. 3C. The maximum in the photodiode-array absorption spectrum of native heme was at ∼398 nm (data not shown). The red shift of the maximum in the absorption spectrum suggests that the prosthetic heme has been modified by a reactive intermediate of EE (Kuo et al., 1999).

Formation of a heme adduct after incubating CYP2J2 with EE and comparison with heme adducts formed with two other major mammalian P450s under the same experimental conditions. (A) HPLC elution profiles monitored at 400 nm for prosthetic heme in the absence of NADPH. (B) HPLC elution profiles showing formation of heme adducts in the presence of NADPH. The heme adducts are labeled as “a” for CYP2J2, “b” and “c” for CYP2B1, and “d” and “e” for CYP3A4. (C) Photodiode-array spectrum of the heme adduct “a” shown for CYP2J2 in (B). The experimental procedures are described in Materials and Methods.
LC-MS/MS Analysis of the CYP2J2 Heme Adduct Formed during Inactivation by EE.
The mass of the heme adduct was determined by collecting the heme adduct from a C4 reverse-phase column during HPLC analysis and then reinjecting it onto a C18 reverse-phase column for LC-MS/MS analysis in the LCQ mass spectrometer. The extracted ion chromatogram (XIC) for heme adduct “a” with MH+ ion at m/z 875.5 is equivalent to alkylation of the Fe-depleted prosthetic heme by oxygenated EE (Fig. 4A). The full mass and MS/MS spectrum are displayed in Fig. 4, B and C, respectively. The proposed structure with oxygen being inserted into the internal carbon of ethynyl moiety is shown in Fig. 4D (Ortiz de Montellano and Komives, 1985; Chan et al., 1993). The fragment ion at m/z 563 is Fe-depleted heme and the fragment ion at m/z 576 results from the cleavage of the C–C bond of ethynyl moiety.

Fe-depleted heme adduct analyzed by LC-MS/MS following inactivation of CYP2J2 by EE. (A) The XIC of the ion at m/z 875.5. (B) The full mass spectrum for the peak having a MH+ ion at m/z 875.5. (C) MS/MS spectrum for the MH+ ion at m/z 875.5. (D) Structure proposed for the heme adduct formed by alkylation of the Fe-depleted heme by EE with the activated oxygen added to the internal carbon of the ethynyl moiety. The dashed lines indicate the sites of fragmentation. The MS/MS spectra were obtained in the positive mode and analyzed using the Xcalibur software package (Thermo Fisher Scientific).
LC-MS/MS Analysis of the CYP2B1 Heme Adduct Formed during Inactivation by EE.
Following the inactivation of CYP2B1 by EE and NADPH, the masses of the heme adducts formed were determined by direct injection of the reaction mixture onto the LTQ linear ion trap mass spectrometer. Figure 5A shows the HPLC elution profile for the native heme remaining and the presence of heme adducts I, II, and III monitored at 400 nm. Figure 5B shows the following: 1) the XICs for peaks I and III with the MH+ ions at m/z 875.5 correspond to alkylation of Fe-depleted prosthetic heme by EE and one oxygen atom; 2) the XIC for peak II with the MH+ ion at m/z 928.7 corresponds to the alkylation of the Fe-containing prosthetic heme by EE and one oxygen atom; and 3) the full mass spectrum for each of the heme adducts. The MS/MS spectrum for the MH+ ion at m/z 875.5 with CYP2B1 is similar to the one observed with CYP2J2 (data not shown). The MS/MS spectrum of the ion at m/z 928.7 and the proposed structure of the Fe-containing heme adduct are displayed in Fig. 5C. The formation of the fragment ions is indicated by dashed lines.

LC-MS/MS analysis of CYP2B1 heme adducts after inactivation by incubation of the reaction mixture with EE. The reaction mixture was injected directly onto a reverse-phase HPLC column and the effluent was analyzed using a photodiode-array detector and a Thermo Fisher Scientific LTQ linear ion trap mass spectrometer. (A) HPLC elution profile for native heme and heme adducts I, II, and III monitored at 400 nm. (B) The XICs for the heme adducts observed in the HPLC elution profile and the full mass spectra of the heme adducts having the MH+ ion at m/z 875.5 or 928.7. (C) The MS/MS spectrum of the MH+ ion at m/z 928.7 and the structure proposed for the Fe-containing heme adduct formed by alkylation of the prosthetic heme by EE with the activated oxygen added to the internal carbon of ethynyl moiety. The dashed lines indicate the sites of fragmentation.
Characterization of the Apoprotein Adduct.
The reaction mixtures were incubated with EE in the absence or presence of NADPH, and then analyzed by ESI-LC-MS. The m/z spectra were deconvoluted to get the masses of the unmodified and modified apoproteins. Figure 6A shows the unmodified CYP2J2 apoprotein with a mass of 54,956 ± 3 Da, the mass expected based on the amino acid sequence of the protein. Figure 6B shows the EE-inactivated CYP2J2 apoprotein with two masses of 54,955 ± 5 and 55,268 ± 9 Da. The difference in mass of 313 ± 3 Da between the unmodified and modified apoprotein is consistent with the addition of the mass of EE plus one oxygen atom.

Detection of the EE-adducted CYP2J2 apoprotein by ESI-LC-MS analysis. The incubation conditions and MS analysis conditions are described in Materials and Methods. (A) Representative deconvoluted mass spectrum of CYP2J2 incubated with EE in the absence of NADPH. (B) Representative deconvoluted mass spectrum of CYP2J2 incubated with EE in the presence of NADPH.
ESI-LC-MS/MS Analysis of GSH Conjugates of EE Having Molecular Ions at m/z 620.
To identify the masses and determine the structures of reactive intermediate(s), the formation of GSH conjugates was characterized by LC-MS/MS analysis. The XICs of GSH conjugates eluting at 21.9 and 26.2 minutes having precursor ions at m/z 620 are shown in Fig. 7A. The molecular masses of the GSH conjugates correspond to the sum of the mass of GSH plus EE and one oxygen atom. The MS/MS spectra of the ions eluting at 21.9 and 26.2 minutes are displayed in Fig. 7, B and C, respectively. The fragmentation patterns are very different, indicating that they have different structures. Ortiz de Montellano and Komives (1985) and Chan et al. (1993) have proposed that if oxygen is added to the internal carbon of the ethynyl moiety, the heme will be alkylated; whereas, if oxygen is added to the terminal carbon of ethynyl moiety, the apoprotein will be modified. Thus, the two structures are tentatively proposed and displayed in the right-hand side panels of Fig. 7, B and C with the addition of oxygen to the external and internal carbons, respectively.

Structural identification of the two GSH conjugates of EE with precursor ions at m/z 620 analyzed by LC-MS/MS. (A) The XICs of the GSH conjugates having the molecular ions at m/z 620 eluting at 21.9 and 26.2 minutes. (B) MS/MS spectrum of the GSH conjugate eluting at 21.9 minutes. (C) MS/MS spectrum of the GSH conjugate eluting at 26.2 minutes. The proposed structures of the GSH conjugates are displayed in the panel on the right-hand side. The dashed lines indicate the sites of fragmentation.
For the GSH conjugate eluting at 21.9 minutes, the precursor ion at 620 undergoes a loss of Gly and Glu to produce ions at m/z 545 and 491, respectively (Baillie and Davis, 1993). The loss of water from the precursor ion at m/z 620 and the ion at m/z 491 give rise to fragment ions at m/z 602 and 473, respectively. The fragment ion at m/z 345 results from the cleavage of the C–S bond within the GSH moiety and the fragment ion at m/z 308 is due to the protonated GSH moiety.
For the GSH conjugate eluting at 26.2 minutes, the formation of the fragment ions at m/z 602, 545, 491, and 308 are the same as the GSH conjugate eluting at 21.9 minutes. The ions at m/z 527 and 455 are from the loss of water from the ions at m/z 545 and 473, respectively. The combination of the losses of Gly, Glu, and water from the precursor ion at m/z 620 produces the ion at m/z 398 and the combination loss of Gly, Glu, and CO from the precursor ion at m/z 620 produces the ion at m/z 388. The cleavage between the sterol moiety and the 17α-CO side chain will produce fragments at m/z 271 and 350. The further loss of water from the ion at m/z 271 will form the ion at m/z 253, and the further loss of Glu and water from the ion at m/z 350 will form an ion at m/z 203.
ESI-LC-MS/MS Analysis of GSH Conjugates of EE Having Molecular Ions at m/z 618.
In addition, two GSH conjugates having MH+ ions at m/z 618 were observed following incubation of CYP2J2 with EE. The molecular masses of these GSH conjugates correspond to the mass of GSH plus EE and one oxygen atom minus two mass units. The XICs of these two GSH conjugates, eluting at 22.0 and 24.7 minutes, are shown in Fig. 8A. The MS/MS spectra of these two GSH conjugates are similar, and the one eluting at 22.0 minutes is shown in Fig. 8B. By using rat and human liver microsomes, it has previously been demonstrated that GSH can bind to either the C1 or C4 position of chemically reactive quinone or semiquinones derived from 2-OH-EE (Maggs et al., 1983). Thus, the structure proposed is displayed in Fig. 8C showing that GSH may bind at either the C1 or C4 position. The neutral loss of the Gly and Glu moieties from the precursor ion will produce the ions at m/z 543 and 489, respectively. The ions at m/z 600 and 471 are produced from the loss of water from the precursor ion at m/z 618 and the ion at m/z 489, respectively. The combination of the losses of Gly, Glu, and CO will form the ion at m/z 386. The ion at m/z 343 is from the binding of sulfur to the 2-OH-EE at the C1 or C4 position.

Structural identification of the two GSH conjugates of EE with precursor ions at m/z 618 analyzed by LC-MS/MS. (A) The XICs of the GSH conjugates having molecular ions at m/z 618 eluting at 22.0 and 24.7 minutes. (B) MS/MS spectrum of GSH conjugate eluting at 22.0 minutes. (C) The proposed structure of the GSH conjugate. The dashed lines indicate the sites of fragmentation.
Metabolism of EE.
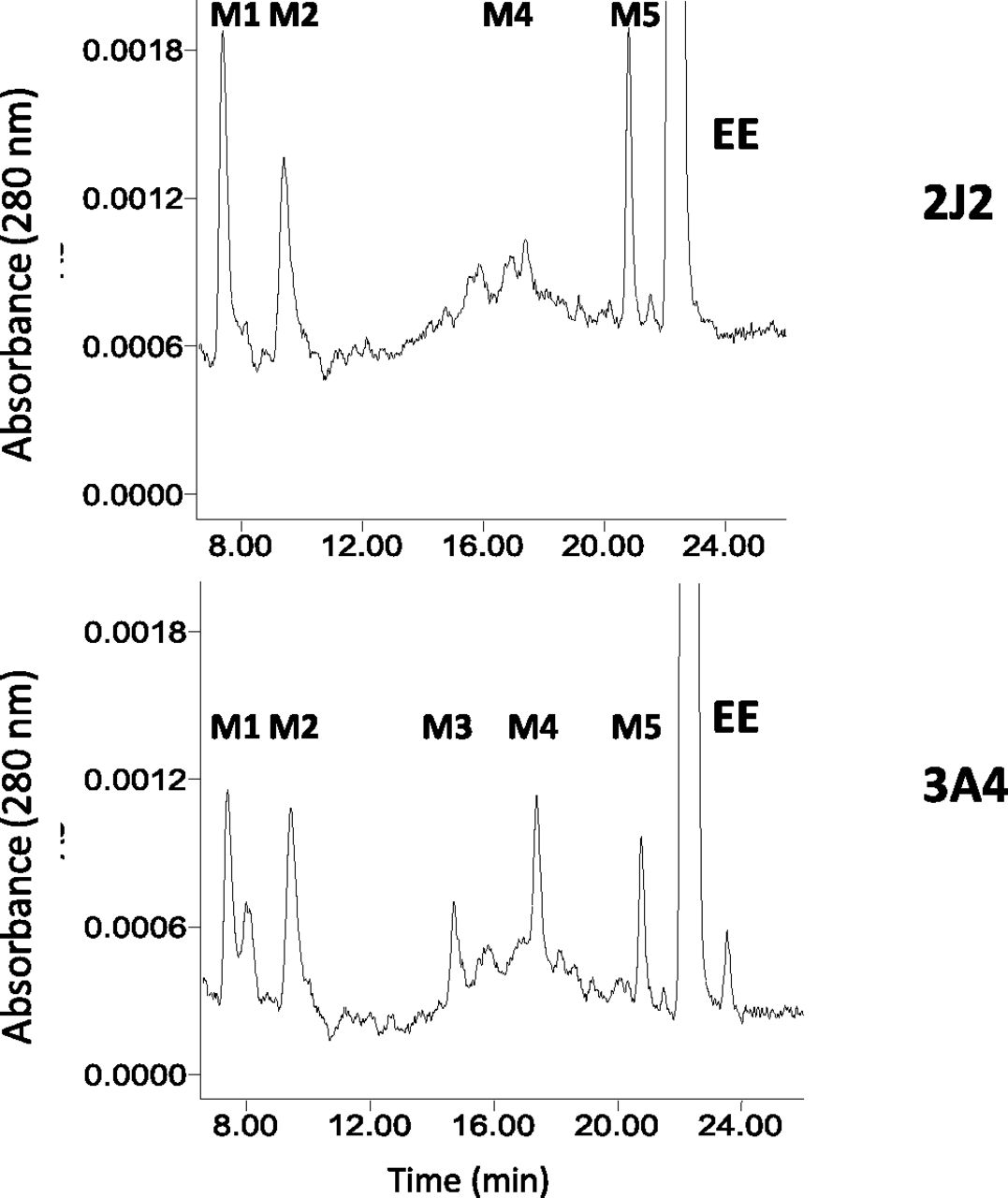
The metabolism of EE by CYP2J2 was determined by HPLC analysis (Fig. 9). For comparison, we also show the metabolites generated by CYP3A4. CYP3A4 generated five major metabolites M1–M5), whereas CYP2J2 generated three major metabolites (M1, M2, and M5) with a small amount of M4. On the basis of the previous reports on EE metabolism by CYP2B1, CYP3A4, and CYP3A5 (Kent et al., 2002; Lin et al., 2002; Lin and Hollenberg, 2007), M1 is estriol, M2 is unknown (but the mass is 312 Da and it is likely that it may be a stable product related to the reactive electrophilic species responsible for the inactivation), M3 is 4-OH-EE, M4 is 2-OH-EE, and M5 is estrone.
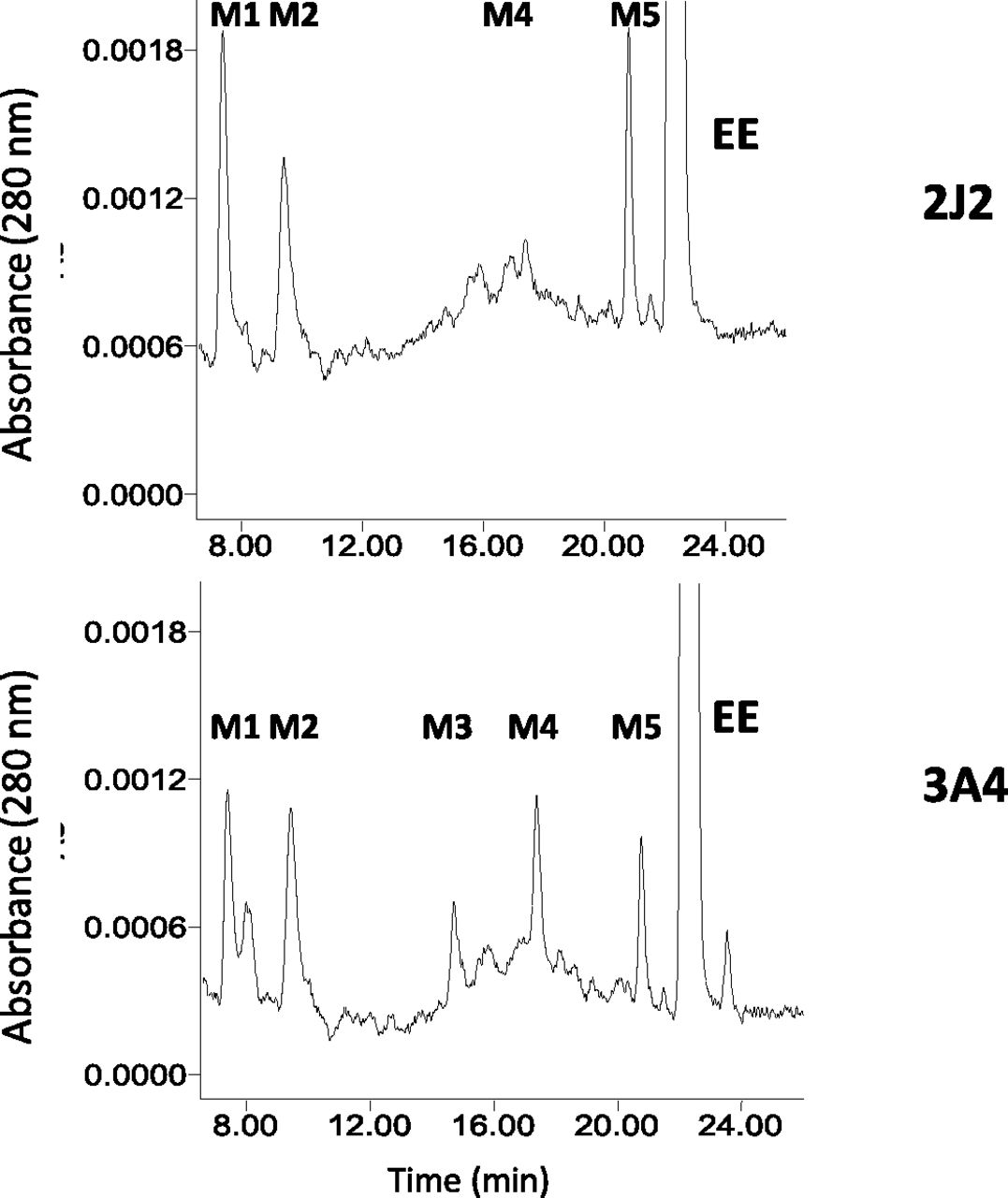
HPLC separation of the major metabolites of EE formed, catalyzed by CYP2J2 and CYP3A4. Reaction mixtures containing CYP2J2 (top panel) and CYP3A4 (bottom panel) in the reconstituted system were incubated with EE and NADPH at 37°C for 30 minutes, and then were extracted with ethyl acetate and analyzed by HPLC as described in Materials and Methods.
Discussion
CYP2J2, the only CYP2J isoform in humans, is predominantly expressed in the cardiovascular system and other extrahepatic tissues including the intestines, kidneys, and lungs (Wu et al., 1996; Zeldin et al., 1996, 1997). CYP2J2 converts arachidonic acid to four biologically active EETs in the heart, thus CYP2J2-related research has attracted increased attention during the past 20 years (Wu et al., 1996; Chaudhary et al., 2009; Michaud et al., 2010; Sato et al., 2011; Xu et al., 2013). EETs play a pivotal role in the regulation and maintenance of cardiovascular homeostasis and they display potent anti-inflammatory actions (Wu et al., 1996; Seubert et al., 2004, 2007; Chaudhary et al., 2009). However, recent studies have demonstrated that increased expression of 2J2 is associated with increased tumor growth and lung metastasis and that CYP2J2 is upregulated in human carcinoma cell lines (Jiang et al., 2005, 2009; Nithipatikom et al., 2010).
Because liver microsomes express low levels of CYP2J2, the activity of this enzyme and its potential role in deleterious drug-drug interactions has not been routinely investigated. EB and astemizole are antihistamine prodrugs and their pharmacologically active metabolites are CEB and desmethylastemizole, respectively (Hashizume et al., 2002; Matsumoto et al., 2002). Studies have identified that the hydroxylation of EB and the demethylation of astemizole are catalyzed in the small intestine primarily by CYP2J2 leading to their bioactivation (Hashizume et al., 2002; Matsumoto et al., 2002; Liu et al., 2006). Moreover, amiodarone 4-hydroxylation and albendazole hydroxylation are catalyzed primarily by CYP2J2 (Lee et al., 2012; Wu et al., 2013). Because of the recognition of the increasingly important role of CYP2J2 in drug metabolism and its broad spectrum of clinically relevant substrates (Lee et al., 2010; Xu et al., 2013), it is essential that we expand our knowledge of CYP2J2 and investigate the potential of mechanism-based inactivation of this P450 by therapeutic drugs leading to drug-drug interactions.
EE is the major synthetic estrogen component of many oral contraceptives. Although the incorporation of the acetylenic moiety increases oral bioavailability of EE, metabolism of this acetylenic moiety by P450 enzymes can lead to mechanism-based inactivation (Kunze et al., 1983; Ortiz de Montellano and Komives, 1985; Chan et al., 1993). For those P450 enzymes that have been tested thus far, EE is a mechanism-based inactivator of CYP2B1, 2B6, 3A4, and 3A5, but not of 2B2, 2B4, or 2E1 (Kent et al., 2002; Lin et al., 2002; Lin and Hollenberg, 2007). Although the IC50 shifts for EE in the absence of preincubation and following preincubation with EE were ∼2 for CYP2J2-catalyzed hydroxylation of terfenadine, suggestive of mechanism-based inactivation, the kinetic parameters describing the mechanism-based inactivation were not determined (Chang et al., 2009). Here, we report that the loss of the OHEB carboxylation activity of CYP2J2 following incubation with EE is time and concentration dependent. The apparent KI, kinact, and kinact/KI (efficiency) values and partition ratio of CYP2J2 from this study are summarized in Table 1 and compared with results previously obtained with CYP3A4 (Lin et al., 2002). As shown in Table 1, the mechanism-based inactivation of CYP2J2 is much more efficient than that of CYP3A4. Therefore, the effect of EE on CYP2J2-catalyzed drug metabolism merits further investigation.
The kinetic constants for the inactivation of CYP2J2 and CYP3A4 by EE
The data presented for CYP2J2 are calculated from Fig. 3, B and C in this study and those for CYP3A4 are from a previous study (Lin et al., 2002).
Following the inactivation of CYP2J2 by EE, HPLC analysis revealed heme destruction with the concomitant generation of one major heme adduct. This heme adduct is much more abundant than that previously observed during the inactivation of CYP3A4 or CYP3A5 by EE (Lin et al., 2002; Lin and Hollenberg, 2007). Because of the recent availability of a high-resolution C4 reverse-phase column, heme modification of two major mammalian P450 enzymes, CYP2B1 and CYP3A4, was reevaluated in the present study. Two heme adducts with maximal absorbance at ∼408 nm were observed, suggesting that the reactive intermediate of EE alkylates more than one pyrrole ring of the tetrapyrrolic skeleton (Kunze et al., 1983). The relative amounts of heme adducts formed by CYP3A4 were much less than those formed by CYP2B1 or CYP2J2. When the heme adducts were further analyzed by LC-MS/MS, two species with MH+ ions at m/z 875.5 and 927.5, which correspond to Fe-depleted and Fe-containing heme adducts, respectively, were identified. The mass of the reactive intermediate of EE adducted to the heme is 312 Da, which corresponds to the mass of EE plus one oxygen atom. To our knowledge, this is the first report describing the masses of the precursor ions of heme adducts formed by mechanism-based inactivation using LC-MS/MS analysis in which both the Fe-depleted and Fe-containing heme adducts are observed from P450s inactivated by therapeutic drugs.
Previously, SDS-PAGE analysis demonstrated that radiolabeled EE was irreversibly bound to the CYP3A4 and CYP3A5 proteins, suggesting that metabolic activation of EE leads to covalent binding to apoprotein (Lin et al., 2002; Lin and Hollenberg, 2007). However, attempts to determine the increase in mass due to adduct formation using mass spectrometry were not successful. Here, Fig. 6 shows the identification of an aproprotein adduct that leads to an increase in mass of approximately 312 Da for the NADPH-treated CYP2J2. Mass spectral analysis of EE-inactivated CYP2B1 has shown an increase in the mass of apoprotein of approximately 313 Da, consistent with the mass of EE plus one oxygen atom (Kent et al., 2006). Therefore, our LC-MS/MS analysis results with CYP2J2 inactivated by EE are in agreement with the mass change observed for the CYP2B1 apoprotein inactivated by EE.
To determine the precise masses and structures of the reactive intermediates, they were trapped with GSH and the masses and structures of EE-GSH conjugates were analyzed. On the basis of the structures of GSH conjugates determined, the potential sites on EE and the mechanisms for its bioactivation can be proposed (Baillie and Davis, 1993). From previous studies on the inactivation of CYP3A5 by EE, it was suggested that the reactive intermediates forming the GSH conjugates with an MH+ ion at m/z 620, but not those forming GSH conjugates having an MH+ ion at m/z 618, contributed to the mechanism-based inactivation of CYP3A5 by EE (Lin and Hollenberg, 2007). The mass increases of the heme adducts as well as the apoprotein adduct of CYP2J2 inactivated by EE is ∼312 Da, which is consistent with the mass of the reactive intermediate forming the GSH conjugate with the MH+ ion at m/z 620 and not at m/z 618. Thus, the reactive intermediate is formed by the oxidation of a carbon-carbon triple bond to give a 17α-oxirene-related species, and the reactive quinone or o-semiquinone derived from oxidation of the a-ring of 2-OH-EE is not involved in the inactivation. The 17α-oxirene-related species can then undergo partitioning of the oxygen between the internal or terminal carbon (Ortiz de Montellano and Komives, 1985; Chan et al., 1993). The same reactive intermediate responsible for forming the GSH conjugates shown in Fig. 8 is also responsible for covalently binding to the heme or apoprotein.
Homology modeling has suggested that the active site volume of CYP2J2 is relatively large, that it is similar to that of CYP3A4, and that there is a strong overlap in substrate recognition by CYP2J2 and CYP3A4 among all of the recently identified CYP2J2 substrates (Lee et al., 2010). In general, CYP3A4 has been shown to metabolize the substrates at multiple sites, whereas substrate metabolism by CYP2J2 is more restricted. Unlike CYP3A4, CYP2J2 exhibited a single site of metabolism for cyclosporine, tamoxifen, thioridazine, and mesoridazine (Lee et al., 2010). Here, we observed that CYP3A4 metabolized EE to give five major metabolites (M1, M2, M3, M4, and M5), whereas CYP2J2 metabolized EE to three major metabolites (M1, M2, and M5) with a small amount of M4 being formed. On the basis of our previous studies on the functional role of threonine-205 in the mechanism-based inactivation of P450 2B1 by two ethnyl substrates (Lin et al., 2004) and the inactivation of P450 3A5 by EE (Lin and Hollenberg, 2007), we suggested that the reactive intermediate leading to the formation of M2 may be responsible for EE-dependent inactivation of P450s. As shown in Fig. 9, the formation of M2 by CYP2J2 appears to be similar to that by CYP3A4. The efficiency of inactivation (kinact/KI) of CYP2J2 is ∼10-fold higher than that of CYP3A4 and the partition ratio of CYP2J2 is significantly lower than that of CYP3A4, suggesting that CYP2J2 is inactivated more efficiently. Therefore, the inactivation of CYP2J2 may occur more readily in vivo than CYP3A4 and may play an important role in the adverse effect or bioavailability of some therapeutic drugs.
In conclusion, CYP2J2 is able to metabolize EE and can oxygenate the acetylene moiety at both the internal and terminal carbons, resulting in the formation of heme and apoprotein adducts, respectively, and ultimately leading to the mechanism-based inactivation. The inactivation of CYP2J2 by EE may significantly alter the metabolism of arachidonic acid in some tissues as well as the metabolism of xenobiotics. Patients taking oral contraceptives as well as drugs primarily or preferentially metabolized by CYP2J2, such as EB and astemizole, may require dose adjustments to prevent adverse drug reactions.
Acknowledgments
We thank Dr. Vyvyca J. Walker for informative discussions. Technical support from Dr. Jaime D’Agostino and Dr. Kathleen R. Noon for the LC-MS/MS analysis of the CYP2J2 and CYP2B1 heme adducts, respectively, is greatly appreciated.
Authorship Contributions
Participated in research design: Lin.
Conducted experiments: Lin.
Contributed new reagents or analytic tools: Lin, Zhang.
Performed data analysis: Lin, Zhang.
Wrote or contributed to the writing of the manuscript: Lin, Zhang, Hollenberg.
Footnotes
- Received February 11, 2018.
- Accepted March 27, 2018.
This work was supported in part by the National Institutes of Health National Cancer Institute [Grant R01 CA-16954] to P.F.H.
Abbreviations
- CEB
- carebastine
- EB
- ebastine
- EE
- 17α-ethynylestradiol
- EET
- epoxyeicosatrienoic acid
- ESI
- electrospray ionization
- GSH
- glutathione
- HPLC
- high-pressure liquid chromatography
- LC
- liquid chromatography
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- MS
- mass spectrometry
- OHEB
- hydroxyebastine
- P450
- cytochrome P450
- TFA
- trifluoroacetic acid
- XIC
- extracted ion chromatogram
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}