


Abstract
Circulating trimethylamine N-oxide (TMAO) predicts poor cardiovascular outcomes in patients with chronic kidney disease (CKD). Accumulation of serum TMAO has been observed in CKD patients; however, the mechanisms contributing to this finding have been inadequately explored. The purpose of this study was to investigate the mechanisms responsible for TMAO accumulation in the setting of decreased kidney function using a CKD mouse model. Mice were fed a diet supplemented with 0.2% adenine to induce CKD, which resulted in increased serum TMAO concentrations (females: CKD 29.4 ± 32.1 μM vs. non-CKD 6.9 ± 6.1 μM, P < 0.05; males: CKD 18.5 ± 13.1 μM vs. non-CKD 1.0 ± 0.5 μM, P < 0.001). As anticipated, accumulation of circulating TMAO was accompanied by a decrease in renal clearance (females: CKD 5.2 ± 3.8 μl/min vs. non-CKD 90.4 ± 78.1 μl/min, P < 0.01; males: CKD 10.4 ± 8.1 μl/min vs. non-CKD 260.4 ± 134.5 μl/min; P < 0.001) and fractional excretion of TMAO. Additionally, CKD animals exhibited an increase in hepatic flavin monooxygenase (FMO)–mediated formation of TMAO (females: CKD 125920 ± 2181 pmol/mg per 60 minutes vs. non-CKD 110299 ± 4196 pmol/mg per 60 minutes, P < 0.001; males: CKD 131286 ± 2776 pmol/mg per 60 minutes vs. non-CKD 74269 ± 1558 pmol/mg per 60 minutes, P < 0.001), which likely resulted from increased FMO3 expression in CKD mice. The current study provides evidence that both decreased renal clearance and increased hepatic production of TMAO may contribute to increments in serum TMAO in the setting of CKD. Hepatic FMO activity may represent a novel therapeutic target for lowering circulating TMAO in CKD patients.
Introduction
Patients with chronic kidney disease (CKD) exhibit a substantial burden of cardiovascular disease (CVD), which is disproportionate to that observed in patients with comparable comorbidities and normal kidney function (Foley et al., 1998; Go et al., 2004; Khalique et al., 2007). It is clear that this increased burden of CVD in CKD patients is not solely driven by a higher prevalence of traditional risk factors for CVD development, such as diabetes and hypertension (Gansevoort et al., 2013). Emerging evidence suggests that a number of metabolic byproducts produced by intestinal bacteria may serve as cardiovascular toxins and nontraditional risk factors for accelerated CVD progression in CKD (Ramezani et al., 2016).
Trimethylamine N-oxide (TMAO) is an organic compound that is a downstream byproduct produced by bacterial metabolism of dietary L-carnitine and choline derivatives. Following ingestion, these dietary constituents are converted to trimethylamine (TMA), a gas that is rapidly absorbed from the intestinal lumen and oxidized to form TMAO by the hepatic flavin monooxygenase (FMO) enzymes. FMO3 and FMO1 are the predominate FMO isoforms responsible for the conversion of TMA to TMAO; however, FMO3 exhibits increased specificity for TMA compared with the FMO1 isoform. When expressed, it is estimated that FMO1 accounts for 11%–12% of TMAO formation (Veeravalli et al., 2018). In mice, testosterone is a negative regulator of FMO3 expression; therefore, compared with females, male mice have reduced FMO3 expression and FMO1 serves a more prominent role in TMAO production (Ripp et al., 1999; Bennett et al., 2013).
Prior studies in genetically modified mice predisposed to atherosclerosis development have demonstrated that TMAO directly exacerbates atherosclerosis formation (Wang et al., 2011; Koeth et al., 2013). Moreover, in patients with both normal and decreased kidney function, high serum TMAO concentrations independently predict adverse CVD outcomes (Wang et al., 2011; Tang et al., 2015; Stubbs et al., 2016). Taken together, these data support TMAO as a non-traditional risk factor for the development of CVD.
CKD patients exhibit serum TMAO concentrations that far exceed those observed in patients with normal kidney function, with serum concentrations often 30- to 100-fold higher in patients with end-stage renal disease (Kaysen et al., 2015; Stubbs et al., 2016; Shafi et al., 2017). Existing data support that TMAO clearance from the bloodstream occurs primarily through urinary excretion (Al-Waiz et al., 1987); however, formal studies that have investigated the mechanisms responsible for TMAO accumulation in CKD are lacking. In the current study, we used CKD mice to investigate how decreases in kidney function alter both renal excretion and hepatic production of TMAO.
Materials and Methods
Animal Preparation and Study Protocol.
All mice were maintained in accordance with the recommendations in the “Guide for Care and Use of Laboratory Animals,” from the Institute on Laboratory Animal Resources, National Research Council, and all animal protocols were reviewed and approved by the University of Kansas Medical Center Institutional Animal Care and Use Committee prior to the commencement of this research. Kidney disease was induced by feeding C57BL/6J mice a standard diet supplemented with 0.2% adenine (TD.160072; Harlan Teklad, Madison, WI) starting at 6 weeks of age. Non-CKD mice were maintained on the same diet but lacking adenine supplementation (TD.120786; Harlan-Teklad). Due to differences in kidney disease progression between sexes consuming an adenine diet, females were maintained on the diet for 12 weeks, whereas males were provided the diet for 10 weeks (Diwan et al., 2014). At the time of sacrifice, mice were placed in metabolic cages to obtain a fasting urine collection (12 hours). Prior to this fasting urine collection, mice were housed in the metabolic cages 8 hours/day for 3 days for environmental acclimation and provided a 1% agar diet to promote hydration. At sacrifice, harvested tissue was either fixed for histology or snap frozen, and then stored at −80°C until further processing.
Serum and Urine Biochemistries.
Serum blood urea nitrogen and creatinine were measured using an Integra 400 Plus Bioanalyzer (Roche Diagnostics, Indianapolis, IN). Urine creatinine was measured by colorimetric assay (Caymen Chemical, Ann Arbor, MI), relying on the Jaffe reaction. Serum and urine TMAO levels were quantified by ultra-performance liquid chromatography–tandem mass spectrometry as previously reported (Ocque et al., 2015). Renal clearance and fractional excretion of TMAO (FETMAO) levels were calculated using eqs. 1 and 2, respectively:
 (1)
(1) (2)
(2)Kidney Histology.
Kidneys were fixed in 4% paraformaldehyde for 24 hours, and following paraffin embedding 5 μm sections were obtained. Midsagittal kidney sections were stained by the periodic acid-Schiff method (Sigma, St. Louis, MO).
Assessment of Liver FMO Expression and Activity.
For quantitative real-time polymerase chain reaction assessment of liver FMO3 and FMO1 gene expression, total RNA was isolated following homogenization of liver tissue using TRI-Reagent (Molecular Research Center, Cincinnati, OH). First strand cDNA was synthesized from 1 μg RNA using iScript (Bio-Rad Laboratories, Hercules, CA). The polymerase chain reactions contained 40 ng cDNA, 150 nM of each primer, and 1X iQ SYBR Green Supermix (Bio-Rad Laboratories) in 20 μl. The threshold cycle (Ct) of each gene product was normalized to the Ct for HPRT and gene expression was calculated using the Pfaffl method (Pfaffl, 2001). Gene expression for CKD animals was expressed as fold change relative to the gene expression observed in non-CKD mice of the same sex. The primer sequences used were: FMO3 forward: 5′-CTCAGGCTGTGACATTGCTG-3′, FMO3 reverse: 5′-CGACTCATCACCCAAGAACC-3′; FMO1 forward: 5′-TCGAGAGGAGCAGTGACCTG-3′, FMO1 reverse: 5′-CCACTGTCCAG AGACAGCAA-3′; and HPRT forward: 5′-TGATAGATCCATTCCTATGACTGTAGA-3′, HPRT reverse: 5′-AAGACATTCTTTCCAGTTAAAGTTGAG-3′.
Hepatic FMO protein expression and activity was assessed in isolated liver microsomes. Microsomes were isolated from frozen liver tissue by differential ultracentrifugation, as previously described with slight modification (Alshogran et al., 2015). Briefly, mouse livers from the same group were pooled together in sets of three and homogenized in ice-cold buffer (50 mM Tris-HCl buffer, 150 mM KCl, 1 mM EDTA, 0.1 mM dithiothreitol, and 20% glycerol, pH 7.4) containing 0.1 mM phenylmethylsulfonyl fluoride and 0.113 mM butylated hydroxytoluene as protease inhibitors. The homogenate was centrifuged at 20,000g for 30 minutes at 4°C. The supernatant was then centrifuged at 140,000g for 30 minutes at 4°C. The microsomal pellet was resuspended in 0.154 M KCl and was then centrifuged at 140,000g for 30 minutes at 4°C. The microsomal pellet was then finally resuspended in 20 mM Tris-HCl (pH 7.4) containing 0.25 M sucrose. Protein concentrations were determined using a Bio-Rad protein assay (Bio-Rad Laboratories).
To measure protein expression, 30 μg of pooled liver microsomal protein was diluted in NuPAGE reducing buffer (Life Technologies, Carlsbad, CA), incubated at 70°C for 10 minutes, and resolved on 4%–12% Bis-Tris gels (Life Technologies). Proteins were transferred onto polyvinylidene difluoride membranes using semidry transfer. Membranes were stained with Ponceau S, blocked in 5% milk, and then incubated with either primary FMO3 (PA5-27916; Thermo Fischer Scientific, Waltham, MA) or FMO1 (sc-376924; Santa Cruz Biotechnology, Inc., Dallas, TX) antibody. Blots were incubated with horseradish peroxidase–conjugated secondary antibodies and an enhanced chemiluminescent substrate was used to generate luminescence (GE Healthcare, Piscataway, NJ). Blots were visualized using GE Amersham Imager 600 (GE Healthcare), and densitometry analysis was performed using Image J software (National Institutes of Health, Bethesda, MD). Protein quantification was normalized to total protein content after Ponceau S staining of the membrane.
To measure FMO-mediated formation of TMAO from TMA, incubations were conducted for each pooled group using 0.75 mg/ml of microsomal protein in 20 mM Tris-HCl buffer (pH 7.4) containing 5 mM MgCl2 and 1 mM NADPH to assess FMO-mediated conversion of TMA to TMAO. Prior to adding the substrates, the microsomal incubations in the presence of NADPH were prewarmed for 3 minutes at 37°C. Reactions were started by the addition of 3 μl of various concentrations of TMA-HCl (5, 10, 25, 50, 100, 200, 500, 1000, 2500, and 10,000 μM) and incubated for 60 minutes at 37°C. Each concentration of substrate was conducted in triplicate. The final reaction volume was 300 μl. Experimental controls omitting substrate and NADPH were included in each experiment. Incubations were stopped by adding 300 μl ice-cold MeOH. Final TMAO concentrations were quantified as described previously.
Data and Statistical Analysis.
Comparisons between non-CKD and CKD animals were evaluated by Student’s t test, or Mann-Whitney test for data exhibiting a non-normal distribution. TMAO formation rates were calculated from their measured concentrations. A nonlinear regression Michaelis-Menten kinetic model was used to fit the formation of TMAO and to estimate the maximum velocity (Vmax) and the affinity constant (Km) values, which were incorporated into Eadie-Hofstee plots. Michaelis-Menten parameters were compared between groups by extra-sum-of-squares F tests. All calculations were performed using Prism 7 (GraphPad Software, San Diego, CA) and are presented as mean ± S.D. unless otherwise specified.
Results
Assessment of Kidney Function and Serum TMAO in CKD and Control Mice.
Consumption of a 0.2% adenine diet led to substantial kidney injury in both female and male mice, as demonstrated by kidney histology revealing tubular injury and dilation, along with marked infiltration of immune cells (Fig. 1A). Additionally, CKD mice exhibited elevated serum blood urea nitrogen concentrations (Fig. 1B; females: CKD 117.2 ± 35.3 mg/dl vs. non-CKD 26.1 ± 4.9 mg/dl, P < 0.001; males: CKD 120.5 ± 32.5 mg/dl vs. non-CKD 28.5 ± 3.1 mg/dl, P < 0.001). At baseline, there was a slight elevation in serum TMAO in female mice compared with male mice (Fig. 1C; females 6.9 ± 6.1 μM vs. males 1.0 ± 0.5 μM, P = 0.07). Serum TMAO concentrations in all CKD mice were considerably elevated compared with non-CKD controls (Fig. 1C; females: CKD 29.4 ± 32.1 μM vs. non-CKD 6.9 ± 6.1 μM, P < 0.05; males: CKD 18.5 ± 13.1 μM vs. non-CKD 1.0 ± 0.5 μM, P < 0.001).

Adenine consumption led to impaired kidney function and an increase in serum TMAO concentrations. CKD was induced by consumption of a diet containing 0.2% adenine, as described in Materials and Methods. (A) Periodic acid-Schiff staining of midsagittal kidney sections revealed intense immune infiltration and tubular dilation in both female and male CKD mice (scale bar, 200 μm; original magnification, 40× ) The induction of CKD was accompanied by a rise in serum blood urea nitrogen (BUN) (B) and serum TMAO (C). *P < 0.05; ***P < 0.001 vs. non-CKD controls.
Urinary Excretion of TMAO.
There was a sex difference in the renal clearance of TMAO in non-CKD mice (females 90.4 ± 78.1 μl/min vs. males 260.4 ± 134.5 μl/min, P < 0.05). In both female and male mice, CKD resulted in a substantial decrease in the renal clearance of TMAO (Fig. 2; females: CKD 5.2 ± 3.8 μl/min vs. non-CKD 90.4 ± 78.1 μl/min, P < 0.01; males: CKD 10.4 ± 8.1 μl/min vs. non-CKD 260.4 ± 134.5 μl/min; P < 0.001). In addition to decreased renal clearance, there was a decrease in the FETMAO levels in both sexes with kidney injury (Fig. 3; females: CKD 52.5% ± 19.6% vs. non-CKD 178.3% ± 157.3%; P < 0.05; males: CKD 64.8% ± 21.2% vs. non-CKD 240.9% ± 158.9%, P < 0.01).
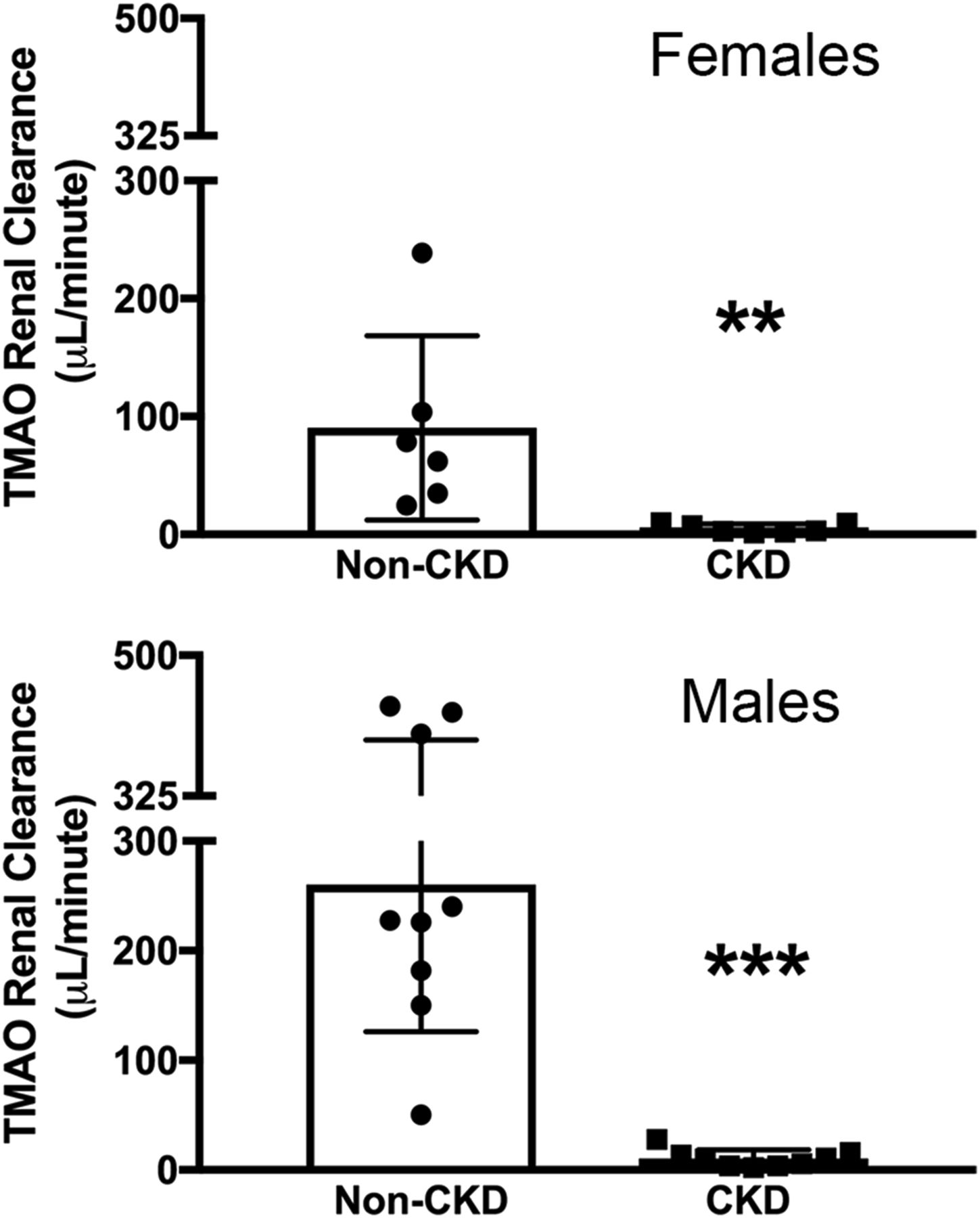
Kidney damage resulted in decreased renal clearance of TMAO. In both female and male CKD mice, there was a substantial decrease in the renal clearance of TMAO compared with non-CKD mice. **P < 0.01; ***P < 0.001 vs. non-CKD controls.

Impaired kidney function resulted in a decrease in FETMAO. The FETMAO levels in both female and male non-CKD mice were greater than 100%, suggesting a significant contribution of tubular secretion to renal clearance of TMAO in non-CKD animals. The FETMAO levels in CKD animals were markedly decreased compared with controls. *P < 0.05; **P < 0.01 vs. non-CKD controls.
Liver FMO Expression and Activity.
Quantitative polymerase chain reaction was performed on liver tissue to assess the impact of CKD on hepatic FMO3 and FMO1 gene expression. With chronic kidney injury, there was a 13-fold increase in gene expression in females (Fig. 4A; CKD mean Ct value 20.3 vs. non-CKD Ct value 25.0; P < 0.01) and an 18-fold increase in males (Fig. 4A; CKD mean Ct value 21.2 vs. non-CKD Ct value 29.6; P < 0.001). There was no observed difference in FMO1 gene expression in CKD mice compared with non-CKD mice (Fig. 4B). Immunoblotting analysis revealed increased FMO3 protein expression in microsomes isolated from CKD males versus those isolated from non-CKD male mice; however, there was no observed difference in FMO3 protein expression in female mice with kidney injury (Fig. 4, C and D). Similar to the gene expression data, there was no obvious change in FMO1 protein expression in female and male mice with kidney injury (Fig. 4, C and E).

Hepatic FMO gene and protein analysis. Quantitative polymerase chain reaction analysis revealed an increase in liver FMO3 gene expression (A) and no change in FMO1 gene expression (B). (C) Immunoblotting analysis revealed increased FMO3 protein expression in males (D) and no changes in FMO1 protein expression (E). **P < 0.01; ***P < 0.001 vs. non-CKD controls.
To validate the functional significance of these gene and protein expression changes observed in CKD mice, we performed ex vivo studies on hepatic FMO activity (Fig. 5). At baseline, wild-type (non-CKD) females exhibited significantly higher FMO activity compared with males (females: 110,299 ± 4196 pmol/mg per 60 minutes vs. males: 74,269 ± 1558 pmol/mg per 60 minutes; P < 0.001). Similar to our observations with FMO3 gene expression, CKD mice exhibited enhanced hepatic FMO activity in both sexes (Fig. 5A; females: CKD 125,920 ± 2181 pmol/mg per 60 minutes vs. non-CKD 110,299 ± 4196 pmol/mg per 60 minutes, P < 0.001; males: CKD 131,286 ± 2776 pmol/mg per 60 minutes vs. non-CKD 74,269 ± 1558 pmol/mg per 60 minutes, P < 0.001).

Increased hepatic FMO activity in animals with impaired kidney function. (A) A nonlinear regression Michaelis-Menten kinetic model was used to fit the formation of TMAO and estimation of Vmax and Km. (B) Eadie-Hofstee transformation of the TMAO formation was performed to examine whether one or two enzymes are responsible for TMAO production. All activity data are presented as mean ± S.E.M. ***P < 0.001 vs. non-CKD controls.
Eadie-Hofstee analysis was performed to explore two-enzyme contribution to TMAO formation (Fig. 5B). The analysis revealed the presence of a two-enzyme kinetic profile in non-CKD females, suggesting both FMO1 and FMO3 contribute to TMAO formation in this group. On the other hand, in non-CKD males, only a single enzyme appears to contribute with a higher Km value, suggesting that FMO1 may primarily contribute to the reaction. The CKD females and males have a less apparent two-enzyme kinetic profile with similar lower Km values, suggesting that the high-affinity FMO3 primarily contributes to the reactions.
Discussion
Based on data from both human and animal studies, TMAO has been independently linked to atherosclerosis formation and CVD progression (Wang et al., 2011; Koeth et al., 2013). Additional investigations have demonstrated a substantial elevation of circulating TMAO concentrations in patients with impaired kidney function (Wang et al., 2011; Tang et al., 2015; Stubbs et al., 2016); thus, TMAO may be an example of a nontraditional CVD risk factor that is of particular importance in this setting. While it has been widely speculated that increments in circulating TMAO in CKD primarily result from decreased urinary clearance of this compound, no formal studies have been conducted to rigorously test this hypothesis. Therefore, the current study was conducted to test how alterations to both urinary excretion and hepatic formation of TMAO may be altered in CKD using translational science approaches in mice.
Our first notable observation of discrepant serum TMAO concentrations between female and male wild-type (non-CKD) mice corroborates recent findings in wild-type mice (Veeravalli et al., 2018) (Fig. 1C). While the difference in TMAO concentrations between healthy female and male mice was not statistically significant, mean TMAO concentrations were approximately 6-fold higher in the non-CKD females. Interestingly, female wild-type animals exhibited both decreased renal clearance (Fig. 2) and increased FMO activity compared with wild-type males (Fig. 5). The observed difference in TMAO formation between sexes could be explained by the established function of testosterone to serve as a negative regulator of FMO3 expression (Ripp et al., 1999; Bennett et al., 2013). As for the apparent sex difference in renal clearance of TMAO, studies evaluating tubular transport in rat kidneys have previously demonstrated a decrease of organic cation transporter (OCT)-2 expression in female animals (Urakami et al., 1999). Since OCT-1 and OCT-2 are believed to be the primary regulators of TMAO secretion by tubular epithelial cells (Miyake et al., 2017), it is plausible that decreased renal TMAO clearance in wild-type females is a direct result of lower transporter abundance.
Similar to humans with CKD, mice with adenine-induced CKD exhibit substantially elevated circulating concentrations of TMAO. As anticipated, renal clearance of TMAO in CKD mice of both sexes is dramatically decreased (Fig. 2). As depicted in Fig. 3, the FETMAO level in the setting of normal kidney function is well above 100%, indicating that tubular secretion plays a predominant role in urinary elimination of this compound. Prior studies on tubular transport mechanisms in rats with adenine-induced CKD have found a dramatic decrease in tubular expression of both OCT-1 and OCT-2 (Komazawa et al., 2013), the suspected regulators of TMAO transport by renal tubules. Thus, we hypothesize that a decrease in both glomerular filtration and tubular secretion of TMAO actively contribute to its systemic accumulation in CKD.
The most noteworthy finding of our current investigation was an increase in hepatic FMO activity in both male and female CKD animals compared with non-CKD controls (Fig. 5A). The Eadie-Hofstee analysis (Fig. 5B) revealed non-CKD females to have a two-enzyme kinetic profile, corroborating previous findings that FMO1 and FMO3 contribute to metabolic reactions (Bennett et al., 2013; Veeravalli et al., 2018). This supports our FMO1 and FMO3 expression data in non-CKD females (Fig. 4). Kidney injury in female mice resulted in a 5-fold lower Km value, suggesting an increased role of FMO3 in producing TMAO. Interestingly, while we did observe increased FMO3 gene expression in CKD females compared with non-CKD females, this was not reflected in our protein expression analysis. This finding could support the presence of post-translational modifications that influence enzyme kinetics in CKD females. By contrast, we did not observe a two-enzyme kinetic profile in non-CKD males (Fig. 5), likely because FMO1 is believed to predominately contribute to TMAO formation in non-CKD male mice.
It is unclear how and why FMO function would be altered in the setting of impaired kidney function; however, it is possible that the induction of FMO function by CKD could represent a compensatory mechanism to counteract the negative effects of uremia on normal physiologic process. To this end, in fish, high concentrations of urea are thought to stimulate FMO3 activity leading to increased TMAO concentrations (El-Alfy et al., 2002). Moreover, in the in vitro setting, TMAO has been observed to counteract the protein-denaturing effect of urea (Bennion and Daggett, 2004; Meersman et al., 2009). Based on this previously described beneficial function of TMAO, it is plausible that increments in urea, or associated uremic toxins, may stimulate hepatic FMO3 activity in an effort to counteract some of the detrimental effects of uremia. In turn, chronic exposure to high circulating concentrations of TMAO may have the unintended secondary outcome of promoting cardiovascular toxicity.
While studies support a correlation between circulating TMAO concentrations and CVD risk in CKD patients, no studies have confirmed whether lowering TMAO can attenuate CVD progression in this population. If future studies suggest clinical benefit of lowering TMAO concentrations in CKD patients, then it will be important to develop therapeutic strategies to target TMAO production or increase TMAO clearance from the circulation. Dietary interventions to lower the ingestion of TMAO and its precursors would likely alone be insufficient to dramatically lower the high concentrations of TMAO observed in CKD patients. A recently published investigation implied that shifts in the intestinal microbiota observed in CKD patients may facilitate increased production of TMAO (Xu et al., 2017). Thus, one reasonable strategy to lower TMAO in CKD patients could be to promote a shift of the pathogenic bacterial flora to more symbiotic species with less capacity for TMA generation. Unfortunately, existing studies investigating the utility of prebiotics or probiotics for supporting the growth of more symbiotic bacterial flora in CKD patients have largely yielded disappointing results (Poesen et al., 2016). Additionally, in the case of patients with end-stage renal disease, dialysis appears to offer only a transient lowering, with marginal long-term effect on circulating TMAO (Hai et al., 2015; Meyer et al., 2016; Kalim et al., 2018). As with other gut-derived uremic toxins, poor TMAO clearance by dialysis is likely related to the broad tissue distribution of this compound. As demonstrated in a recent post-hoc analysis from the Hemodialysis (HEMO) Study (Meyer et al., 2016), increasing dialysis intensity in end-stage renal disease patients has little impact on chronic TMAO exposure in this population (Kalim et al., 2018). Our study suggests that therapeutic strategies to increase renal clearance of TMAO (in patients with residual kidney function), possibly through increasing the activity of the OCT transporters or inhibiting hepatic FMO activity, may be beneficial in lowering TMAO concentrations in the setting of impaired renal function.
As with any study, the current investigation has both important strengths and limitations. Important strengths of our investigation include: use of a precise and extensively validated method for TMAO quantification, the inclusion of studies to assess hepatic FMO enzyme function, consideration of the impact of gender on the outcomes of interest, performing metabolic studies that included timed urine collections for the assessment of urinary TMAO excretion, and an overall study design that allows for the exclusion of potential confounders that are commonly encountered in human studies (i.e., dietary influences, genetic variability, and the presence of disease comorbidities). Important limitations of our work include: possible differences between human and mouse biology, lack of investigation into specific mechanisms driving the observed alterations in hepatic generation or renal clearance of TMAO, and no assessment of how alterations to the intestinal microbiome may have altered TMA formation rates in CKD mice.
In summary, our observations in CKD mice suggest that both a decrease in renal clearance of TMAO and an increase in hepatic production of TMAO due to increased FMO3 activity contribute to TMAO accumulation in CKD. If future studies confirm our findings in humans, it will be important to further explore the significance of enhanced TMAO formation in CKD and better understand how blockade of FMO3 activity may alter clinical outcomes in this setting.
Authorship Contributions
Participated in research design: Johnson, Prokopienko, West, Nolin, Stubbs.
Conducted experiments: Johnson, Prokopienko, West.
Performed data analysis: Johnson, Prokopienko, West, Nolin, Stubbs.
Wrote or contributed to the writing of the manuscript: Johnson, Prokopienko, West, Nolin, Stubbs.
Footnotes
- Received March 23, 2018.
- Accepted June 7, 2018.
This work was supported in part by an American Heart Association Grant-in-Aid award (#15GRNT25700231) to J.R.S.
Abbreviations
- CKD
- chronic kidney disease
- CVD
- cardiovascular disease
- FETMAO
- fractional excretion of trimethylamine N-oxide
- FMO
- flavin monooxygenase
- OCT
- organic cation transporter
- TMA
- trimethylamine
- TMAO
- trimethylamine N-oxide
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}