


Visual Overview

Abstract
The identification of nonopioid alternatives to treat chronic pain has received a great deal of interest in recent years. Recently, the engineering of a series of Nav1.7 inhibitory peptide-antibody conjugates has been reported, and herein, the preclinical efforts to identify novel approaches to characterize the pharmacokinetic properties of the peptide conjugates are described. A cryopreserved plated mouse hepatocyte assay was designed to measure the depletion of the peptide-antibody conjugates from the media, with a correlation being observed between percentage remaining in the media and in vivo clearance (Pearson r = −0.5525). Physicochemical (charge and hydrophobicity), receptor-binding [neonatal Fc receptor (FcRn)], and in vivo pharmacokinetic data were generated and compared with the results from our in vitro hepatocyte assay, which was hypothesized to encompass all of the aforementioned properties. Correlations were observed among hydrophobicity; FcRn binding; depletion rates from the hepatocyte assay; and ultimately, in vivo clearance. Subsequent studies identified potential roles for the low-density lipoprotein and mannose/galactose receptors in the association of the Nav1.7 peptide conjugates with mouse hepatocytes, although in vivo studies suggested that FcRn was still the primary receptor involved in determining the pharmacokinetics of the peptide conjugates. Ultimately, the use of the cryopreserved hepatocyte assay along with FcRn binding and hydrophobic interaction chromatography provided an efficient and integrated approach to rapidly triage molecules for advancement while reducing the number of in vivo pharmacokinetic studies.
SIGNIFICANCE STATEMENT Although multiple in vitro and in silico tools are available in small-molecule drug discovery, pharmacokinetic characterization of protein therapeutics is still highly dependent upon the use of in vivo studies in preclinical species. The current work demonstrates the combined use of cryopreserved hepatocytes, hydrophobic interaction chromatography, and neonatal Fc receptor binding to characterize a series of Nav1.7 peptide-antibody conjugates prior to conducting in vivo studies, thus providing a means to rapidly evaluate novel protein therapeutic platforms while concomitantly reducing the number of in vivo studies conducted in preclinical species.
Introduction
It has been estimated that over half a million people in the United States alone died from drug overdoses between 2001 and 2015, and the majority of these deaths were attributed to the growing opioid crisis (Humphreys, 2017). As such, the identification of nonopioid alternatives to treat chronic pain has become a rapidly emerging area of research in recent years. The Nav1.7 voltage-gated sodium ion channel (SCN9A) represents one key pharmacological target given the high degree of human genetic validation for its role in modulating pain states (Dib-Hajj et al., 2013; Emery et al., 2016; Vetter et al., 2017). Multiple examples of small-molecule, peptide, and antibody approaches to inhibit Nav1.7 activity have been reported in the literature. Recently, we described the engineering of two series of inhibitory Nav1.7 peptide-antibody conjugates, linking peptides based on the GpTx-1 or JzTx-V toxins found in tarantula venom to nontargeting antibodies to increase the half-life and stability of the inhibitory peptides (Biswas et al., 2017; Moyer et al., 2018; Wu et al., 2018; Murray et al., 2019).
As with monoclonal antibodies, it is likely that the pharmacokinetics of peptide-antibody conjugates are also affected by both physicochemical properties, target-mediated drug disposition, and receptor-mediated interactions (Datta-Mannan et al., 2015a). Key physicochemical properties that have been shown to contribute to pharmacokinetics of antibody therapeutics include molecular charge, isoelectric point, hydrodynamic radius, size, valency, and post-translational modifications (Boswell et al., 2010; Bumbaca et al., 2012; Liu, 2015; Schoch et al., 2015). Interactions with the neonatal Fc receptor (FcRn) are perhaps the most well studied protein-receptor interactions for therapeutic proteins, as they facilitate intracellular recycling, resulting in the long serum half-life of IgG (Roopenian and Akilesh, 2007; Giragossian et al., 2013). Antibodies are pinocytosed into intracellular endosomes where the Fc region binds to FcRn under acidic conditions (pH ∼5.5–6). The complex is protected from proteolytic degradation and recycled to the cell surface where the antibody is released at neutral pH (Antohe et al., 2001; Roopenian and Akilesh, 2007). Importantly, it has been shown that modification to the non-Fc portion of an antibody (such as drug conjugation) can alter FcRn binding (Wang et al., 2011). In addition to FcRn, there are other cell surface receptors, many expressed in the liver, that recognize post-translational modifications or aberrant proteins and thus may also function as determinants of recycling or elimination for protein therapeutics. Examples include the Stab/Scarb1 family of scavenger receptors, the mannose and galactose receptors, the low-density lipoprotein (LDL) receptor, and the asialoglycoprotein receptor 1 (Ashwell and Morell, 1974; Ashwell and Harford, 1982; Inamoto and Brown, 1991; Stahl, 1992; Terpstra et al., 2000; Allavena et al., 2004; Goetze et al., 2011).
While small-molecule drug discovery has optimized many approaches over the years to predict pharmacokinetics prior to conducting in vivo studies, the characterization of pharmacokinetic properties for protein therapeutics is still highly dependent upon the use of preclinical species, with available in vitro and in silico tools only now beginning to emerge (Jaramillo et al., 2017; Avery et al., 2018). Further complicating the development of such tools is the fact that they are often based on the processes that underwrite the catabolism and disposition of monoclonal antibodies, although protein therapeutic developmental efforts now favor highly engineered protein platforms where in vivo disposition may not be governed by the same mechanisms as monoclonal antibodies (Boehm et al., 1999; Vugmeyster et al., 2012; Ueda, 2014; Ramsland et al., 2015).
To that end, various in vitro and in silico tools are currently being developed to understand protein therapeutic disposition, including surface plasmon resonance to measure FcRn affinity, hydrophobic interaction chromatography to compare relative hydrophobicity, and computational approaches that use either sequence-based or three-dimensional structural modeling to identify drug candidates (Dostalek et al., 2017). More recently, studies of cellular-based approaches to assess nonspecific clearance pathways have been reported. Nonspecific binding to HEK293 or CHO cells has been correlated to high in vivo clearance (Datta-Mannan et al., 2012, 2015b; Xu et al., 2013), and association with liver sinusoidal endothelial cells has been implicated in the rapid clearance of a series of IgG–extracellular domain and IgG-scFv bispecific antibodies (Datta-Mannan et al., 2016).
In this study, several of the aforementioned assays were retrospectively applied to a series of Nav1.7 peptide-antibody conjugates based on observations from biodistribution studies that suggested Nav1.7 peptide properties altered tissue distribution of the conjugate relative to the parent antibody. Experiments were designed to test the hypothesis that hepatocytes may play a role in the distribution and elimination of engineered proteins, such as these Nav1.7 peptide-antibody conjugates. To this end, an in vitro approach utilizing cryopreserved plated mouse hepatocytes was implemented to measure the depletion of the peptide-antibody conjugates from the media, similar to the approach commonly used in small-molecule stability assays. Physicochemical, receptor-binding, and in vivo pharmacokinetic data were generated and compared with the results from the hepatocyte assay. Finally, receptor phenotyping and in vivo knockout studies were conducted to identify receptor-mediated pathways beyond FcRn which may contribute to antibody binding in vitro and, subsequently, to pharmacokinetic profiles in vivo.
Materials and Methods
Materials.
Biotinylated murine anti-human IgG Fc monoclonal antibody (clone no. 1.35.1) and all antibodies or peptide-antibody conjugates were prepared and purified at Amgen, Inc. (Thousand Oaks, CA) as previously described (Murray et al., 2019). Stable labeled peptides used as internal standards were obtained from Mid-West Biotech, Inc. (Fishers, IN). Cryopreserved mouse hepatocytes and FcRn knockout mouse hepatocytes were purchased from Triangle Research Laboratories (Durham, NC). CM4 surface plasmon resonance chips were purchased from GE Healthcare Life Sciences (Pittsburgh, PA). Acetyl–low-density lipoprotein, Dulbecco’s phosphate-buffered saline, HALT Protease Inhibitor Cocktail (100×), Streptavidin Dynal M-280 magnetic beads, and and trypsin (N-tosyl-L-phenylalanine chloromethyl ketone treated) were were purchased from Thermo Fisher Scientific (Waltham, MA). Oasis HLB 96-well µElution Plates were obtained from Waters Corporation (Milford, MA). All other reagents were from Sigma-Aldrich (St. Louis, MO) and were of the highest grade possible.
Mouse Pharmacokinetics and Biodistribution Studies.
Animals were cared for in accordance with the Guide for the Care and Use of Laboratory Animals, 8th Edition (National Research Council, 2011). All in vivo pharmacokinetic and biodistribution studies were conducted at an AAALAC International–accredited facility and were approved by the Amgen Institutional Animal Care and Use Committee (Thousand Oaks, CA). Mice were maintained on a 12:12-hour light:dark cycle, and ambient temperature and humidity were kept between 68 and 79°F and 30%–70%, respectively. Animals were allowed access to pelleted feed (Teklad, 2920X irradiated; Envigo, Indianapolis, IN) and reverse osmosis–purified water ad libitum.
Pharmacokinetic Studies.
Studies to characterize the intravenous clearance of a set of peptide-antibody conjugates were performed in male CD-1 mice (8–12 weeks old; Charles River Laboratories, Hollister, CA). Peptide-antibody conjugates were formulated in 10 mM sodium acetate with 9% sucrose (pH 5.2) at 1 mg/ml. Dosing (5 mg/kg) was via intravenous bolus tail vein injection. Blood samples were collected at predetermined time points up to 168 hours postdose by submandibular venipuncture. Whole blood was collected and placed into Sarstedt Microvette 500 plasma tubes containing K2-EDTA and inverted several times prior to centrifugation at 14,000g for 5 minutes. The resulting plasma was stored at −70°C until analysis. The pharmacokinetics of Activity-dependent neuroprotective protein and conjugate 4 were also evaluated in cynomolgus monkeys following a 3-mg/kg intravenous dose (10 mM sodium acetate with 9% sucrose, pH 5.2). Blood samples were collected through 672 hours postdose and processed as noted earlier.
In Vivo Biodistribution Studies.
Representative anti-DNP antibodies were radiolabeled with 64Cu as previously described (Murray et al., 2019). In brief, antibodies were modified via conjugation to 1,4,7-triazacyclononane,1-glutaric acid-4,7-acetic acid (NODA-GA) and incubated with 64CuCl2 (1 to 2 mCi in 5–10 μl, 0.1 M HCl) for 1 hour at 37°C. The reaction was quenched with 10 mM EDTA for 5 minutes to remove any unchelated 64Cu. Thin-layer chromatography with radiochemical detection was used to determine labeling efficiency as previously noted, with all conjugates exhibiting greater than 95% purity.
Mice (male, CD-1) were administered intravenous bolus tail vein injections with 100 μCi of radiolabeled conjugate in 100–125 μl of saline for a total dose of 30–40 mg/kg. Mice were anesthetized using 1% to 2% isoflurane delivered in 100% oxygen gas, provided with supplemental heating, and were monitored for physical well being over the course of the positron emission tomography scan acquisition. All images were acquired using a Siemens Inveon scanner (Siemens, Erlangen, Germany) at 3 hours postdose. Data analysis incorporated Siemens Inveon Acquisition Workplace and Inveon Research Workplace software applications.
Hepatocyte Depletion Assay.
An in vitro cellular assay using cryopreserved, plated CD-1 mouse or cynomolgus monkey hepatocytes was designed to monitor the depletion of the antibody-peptide conjugates from the hepatocyte media. In brief, hepatocytes (Triangle Research Laboratories) were plated at 1 million cells per well, allowed to attach for 2 hours, and subsequently washed with Dulbecco’s modified Eagle’s medium (DMEM) buffer (Sigma-Aldrich) containing 5% fetal bovine serum. The peptide-antibody conjugates (diluted in DMEM buffer containing 5% fetal bovine serum) were added to the hepatocytes (20 µg/ml, final concentration) and incubated for up to 3 hours at 37°C under 5% CO2. Aliquots were removed at 0, 30, 60, 90, 120, and 150 minutes and immediately frozen for subsequent analysis. Following the 150-minute time point, the hepatocytes were washed three times with DMEM buffer containing 5% fetal bovine serum and lysed by addition of 0.5 ml of lysis buffer. Lysed contents were collected and frozen for subsequent ELISA analysis. In general, cellular viability was >90% over the course of the incubation.
FcRn Binding.
The affinity of the peptide-antibody conjugates toward FcRn was measured by surface plasmon resonance using FcRn conjugated to a CM4 chip via an amine tether (1000–2000 resonance units level of immobilization) on a Biacore T200 (GE Healthcare). Affinity measurements were carried out at 25°C using a flow rate of 50 µl/min; a peptide-antibody concentration between 0.1 and 1 µM; and association and dissociation phases of 1.5 and 10 minutes, respectively. Responses (RU) were monitored at pH 5.5 and pH 7.4 using phosphate-buffered saline (0.005% P20) and 10 mM sodium acetate/150 mM NaCl/0.005% P20 (pH 5.5 or pH 7.4) as running and sample buffers. Binding affinity is expressed as RU values as determined in the Biacore evaluation software.
Hydrophobic Interaction Chromatography.
Peptide-antibody conjugates were analyzed by hydrophobic interaction chromatography (HIC) using a Dionex ProPac HIC-10, 5 μm, 300A, 2.1 × 100–mm column attached to an Agilent analytical high-performance liquid chromatography system by eluting with buffer A [1.0 M (NH4)2SO4, 20 mM NaOAc, pH 5] and buffer B (20 mM NaOAc, 10% CH3CN, pH 5, at a 0.4 ml/min flow rate). The gradient was held at 0% B from 0 to 2 minutes, ramped 0%–100% B from 2 to 12 minutes, held at 100% B from 12 to 13 minutes, ramped 100%–0% B from 13 to 14 minutes, and re-equilibrated at 0% B from 14 to 18 minutes. Samples were diluted 10-fold in buffer A prior to injection, and 5–30 μg of material was injected.
Receptor Phenotyping Experiments.
To evaluate the role of specific cell surface receptors in the depletion of the peptide-antibody conjugates, the unconjugated control antibody (aDNP) or conjugate 4 was incubated in the aforementioned hepatocyte assay in the presence or absence of selective inhibitors. Acetyl–low-density lipoprotein (Scarb/Stab1; 20 µg/ml), ovalbumin (mannose and galactose receptors; 1 mg/ml), chondroitin (class A scavengers; 1 mg/ml), or IgG (FcRn; 500 µg/ml) was added to the plated hepatocytes and allowed to incubate for 5 minutes prior to adding aDNP or conjugate 4 (20 µg/ml; final concentration). Incubations were carried out for 2.5 hours at 37°C under 5% CO2, at which point an aliquot of medium was sampled and frozen for subsequent ELISA. Based on preliminary data, acetylated LDL and ovalbumin were selected for follow-up studies. A range of inhibitor concentrations (acetylated LDL, 0.02–0.5 mg/ml; ovalbumin, 0.3–10 mg/ml) was used to more definitively assess the contribution of these two scavenger pathways to the disposition of conjugate 4.
In Vivo Knockout Studies.
Pharmacokinetic studies were also carried out in knockout mouse models to further evaluate the potential mechanisms responsible for in vivo clearance. FcRn (B6.129P2-B2mtm1Unc/J, stock number 002087), low-density lipoprotein receptor (Ldlrtm1Her/J, stock number 002207), and mannose receptor (B6.129P2-Mrc1tm1Mnz/J, stock number 007620) knockout models were obtained from the Jackson Laboratory (Bar Harbor, ME), whereas asialoglycoprotein receptor 1 (ASGR1) knockout mice (B6.129S4-Asgr1tm1Sau/SaubJxmJ, stock number 009105) were obtained from Charles River Laboratories (Wilmington, MA). Knockout or wild-type mice received a 2-mg/kg intravenous bolus dose of either aDNP or conjugate 4 (0.4 mg/ml in 10 mM sodium acetate with 9% sucrose, pH 5.2) via tail vein injection. Blood sampling and processing details were similar to those described earlier.
Measurement of Test Articles from In Vitro Studies.
Peptide-antibody conjugate levels from in vitro studies were detected and quantitated by ELISA. A capture plate was prepared by coating a high binding Costar ELISA plate (Sigma-Aldrich) with an anti-DNP 3A4 monoclonal antibody (2 µg/ml, final concentration) in phosphate-buffered saline. Capture plates were washed (3×) and sample aliquots from in vitro studies (100 µl) were added and incubated for 1 hour at room temperature, followed by a second wash step (5×) prior to the addition of a detection antibody. Analytes were detected by addition of a horse radish peroxide–conjugated mouse anti-human Fc monoclonal antibody (7.4 µg/ml, final concentration; 1-hour incubation at room temperature). Detection plates were washed (5×), followed by addition of 3, 3′, 5, 5′-Tetramethylbenzidine peroxidase substrate and TMB peroxidase substrate solution B (SeraCare, Milford, MA). Plates were incubated for 20 minutes and quenched with 2 N sulfuric acid. Luminescence was read at 450 nm (reference wavelength, 650 nm) on a SpectraMax M2 plate reader (Molecular Devices, Sunnyvale, CA). Standard curves were prepared from 2.4 to 2500 ng/ml, and test article concentrations were extrapolated from the standard curve response.
Measurement of Test Articles in Serum from Pharmacokinetic Studies.
Total and intact peptide-antibody conjugates from pharmacokinetic studies were evaluated using immunocapture followed by tandem mass spectrometry as previously described (Murray et al., 2019). In brief, an aliquot of each plasma or serum sample was diluted 1:1 (v/v) with Dulbecco’s phosphate-buffered saline (1×) with 2× HALT Protease Inhibitor Cocktail. Biotinylated murine anti-human IgG Fc monoclonal antibody (clone number 1.35.1) immobilized on Streptavidin Dynal M-280 magnetic beads was added as immunoaffinity capture reagent to a 96-well-plate format. Samples were vortex-mixed for 1 hour at room temperature, and the beads subsequently washed with Tris-HCl buffer (pH 7.5). Reduction of the samples was accomplished by addition of tris-(2-carboxyethyl)-phosphine) solution (7.5 mM in 250 mM Tris-HCl, 8 M urea, pH 7.5) containing the relevant internal standard as noted below. Samples were alkylated with iodoacetamide for 45 minutes at 60°C and subsequently digested with trypsin (500 µg/ml) in a microwave set to 30°C/400 W for 10 minutes. Samples were quenched with 20% formic acid and centrifuged at 4500 rpm for 10 minutes prior to analysis by liquid chromatography–tandem mass spectrometry.
Liquid chromatography–tandem mass spectrometry analysis of the peptide analytes was achieved using an Acquity ultra-high performance liquid chromatography (Waters Corporation) coupled to a Sciex QTRAP 5500 mass spectrometer (AB Sciex, Foster City, CA) that was operated in the electrospray positive ion multiple reaction monitoring mode. A Phenomenex Aeris C18 column (2.1 × 100 mm, 1.7 µm; Phenomenex, Carlsbad, CA) held at 70°C was used for chromatographic separation of the peptide analytes. Total test article (antibody or peptide conjugate) was quantitated using a universal surrogate peptide (peptide sequence: VVSVLTVLHQDWLNGK) corresponding to the Fc region of human IgG1 monoclonal antibodies together with the corresponding stable label isotopic standard ([13C6,15N]-leucine; peptide sequence: VVSV*LTVLHQDW*LNGK). Specific peptide sequences (Supplemental Table 1) were used to quantitate intact peptide-antibody conjugates.
Total and intact analyte concentrations were calculated by comparing peak area ratios to calibration curves prepared in a similar fashion using a 1/x2 weighted linear least-squares regression. Pharmacokinetic parameters were estimated by noncompartmental analysis using Watson LIMS.
Results
Pharmacokinetic studies in wild-type mice were carried out in support of lead candidate identification efforts for a series of Nav1.7 inhibitory peptide-antibody conjugates with varying peptide identities and formal charges, linker lengths and architecture, or antibody attachment sites. Following a 5-mg/kg intravenous dose, the in vivo clearance of the unconjugated aDNP antibody was 0.0006 l/h/kg. Clearance values for the conjugated antibodies ranged from 0.0013 l/h/kg for conjugates 12 and 13 to 0.079 l/h/kg for conjugate 2 (Table 1). The ratio of intact area under the curve (AUC) to total AUC for the peptide conjugates ranged from <0.1 for conjugate 2 to 0.76 for conjugate 5. The highest clearance values and lowest AUCintact/AUCtotal ratios were observed for conjugates 1 and 2, the two peptide conjugates with a peptide charge of +6. Lower clearance values and higher AUCintact/AUCtotal ratios were observed for peptide conjugates with a peptide charge of +1 or +2 as compared with those with a +6 charge, although no statistically significant difference was observed in clearance or AUCintact/AUCtotal between peptide conjugates with a peptide charge of +1 versus a peptide charge of +2 (Fig. 1A). The stability and pharmacokinetic properties of antibodies are also known to be affected by the global physicochemical properties of the molecules. To evaluate the correlation between the hydrophobicity of the Nav1.7 peptide-conjugates and in vivo clearance or depletion from the in vitro hepatocyte assay, hydrophobic interaction chromatography analyses were performed with the peptide conjugates. A significant correlation (Pearson r = 0.8005, N = 12, P = 0.0018) was observed between the HIC retention time and the clearance measured from in vivo pharmacokinetic studies (Fig. 1B).
List of peptide conjugates
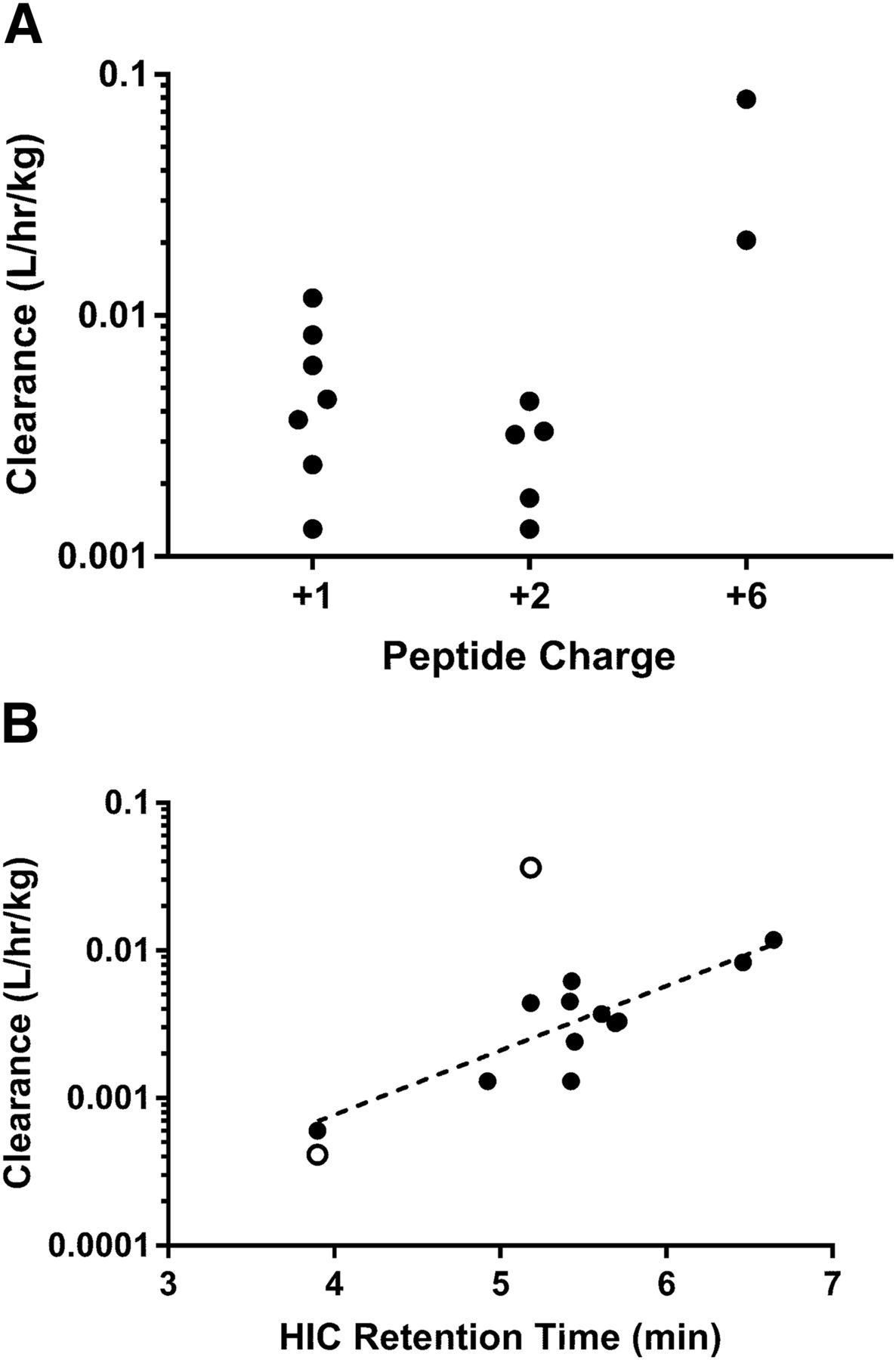
Effect of charge (A) and hydrophobicity (B) on Nav1.7 peptide-antibody conjugate pharmacokinetics. A reduction in clearance was observed for peptide-antibody conjugates with a peptide charge of +1 or +2 as compared with a peptide charge of +6, whereas a Pearson correlation (r = 0.8005) was observed between the HIC retention time and in vivo clearance. Mouse data are denoted with closed circles, whereas cynomolgus monkey data are denoted by open circles.
To characterize the large differences observed for in vivo clearance between the unconjugated aDNP antibody and the corresponding peptide-antibody conjugates, image-based biodistribution studies were carried out using 64Cu-NODA–labeled antibodies. 64Cu-NODA imaging data suggested that at 3 hours postdose, both the unconjugated aDNP antibody (aDNP) and a representative symmetrically conjugated divalent peptide-antibody conjugate (P2S-aDNP; peptide charge: +6) molecule were constrained primarily to the vasculature, with the bladder being the main secondary site of accumulation (Fig. 2). Conversely, a representative asymmetrically conjugated divalent peptide-antibody conjugate containing two peptides on a single monoclonal antibody heavy chain (P2A-aDNP; peptide charge: +6) or symmetrically conjugated tetravalent peptide-antibody conjugate containing two peptides on each heavy chain (P4S-aDNP; peptide charge: +6) demonstrated rapid distribution out of the central compartment and accumulation predominantly into the liver at 1 hour postinjection (Fig. 2).
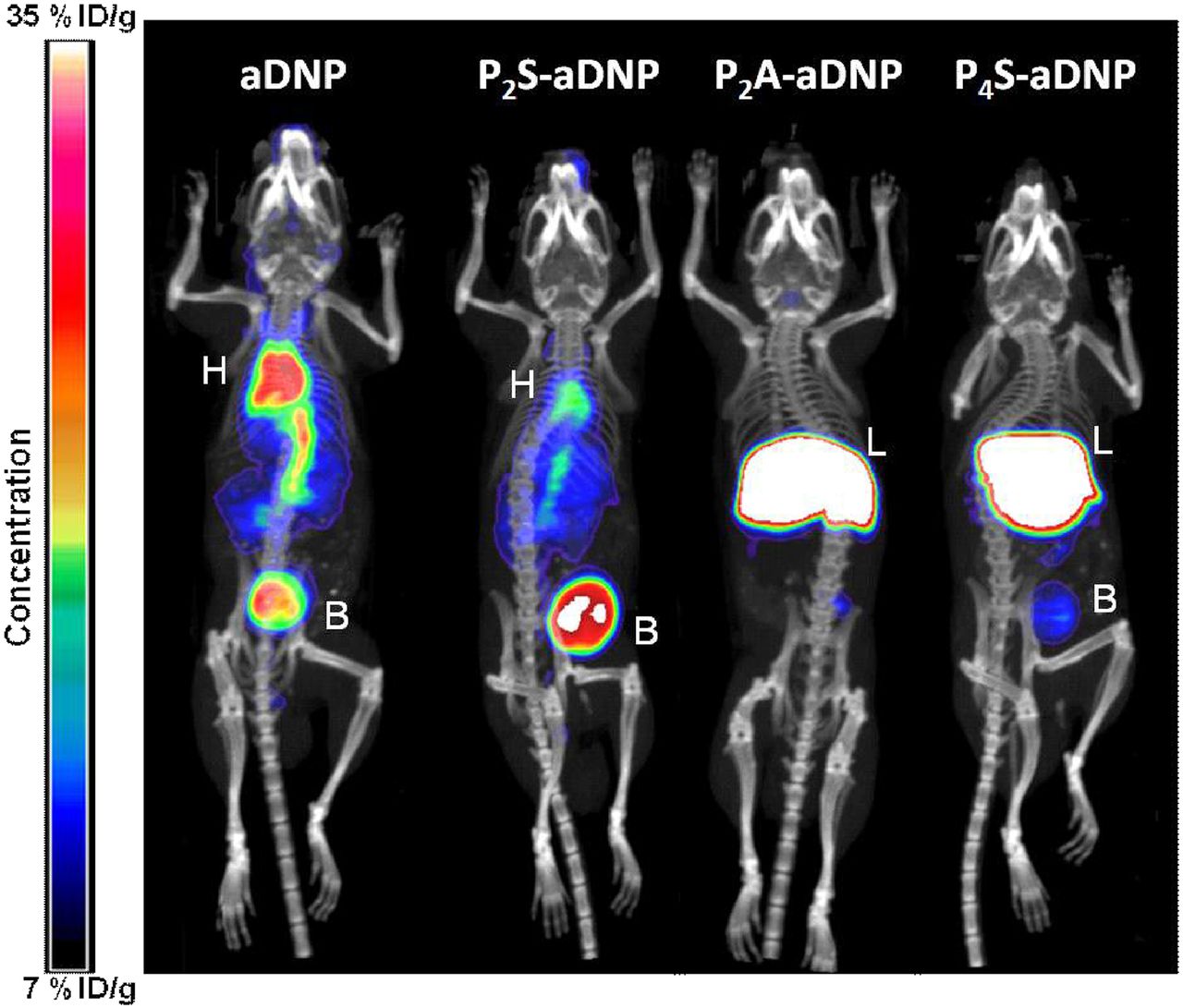
64Cu-NOTA–labeled imaging studies of the unconjugated antibody (aDNP) and three peptide-antibody conjugates. At 3 hours postdose, aDNP and a representative symmetrical divalent conjugate (P2S-aDNP) were confined primarily to the central compartment, while the asymmetric divalent conjugate (P2A-aDNP) and a tetravalent peptide-antibody conjugate (P4S-aDNP) distributed rapidly to the liver. B, bladder; H, heart; %ID/g, percent injected dose per gram; L, liver.
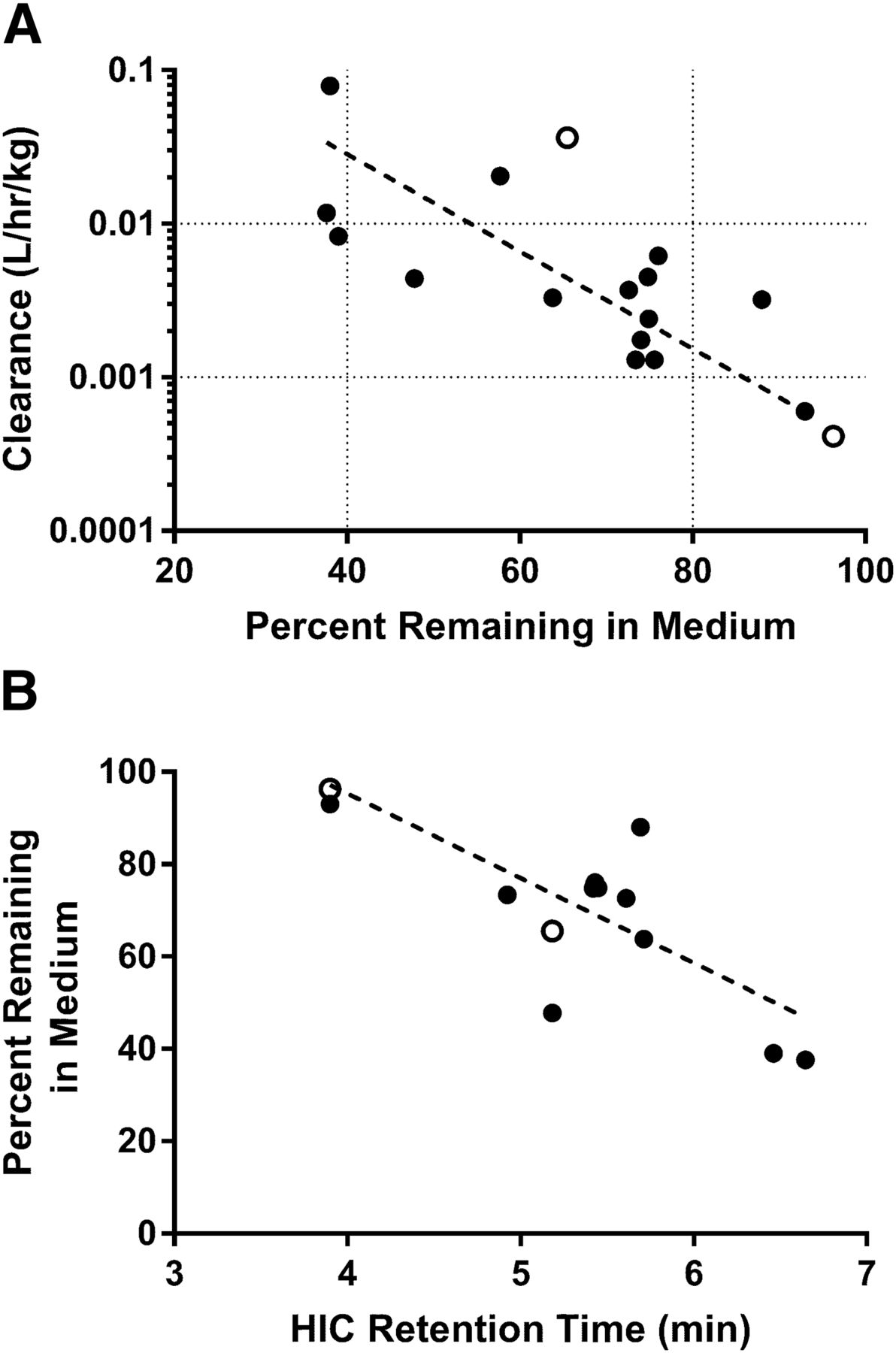
Based on the results from the biodistribution studies, it was hypothesized that binding and/or uptake into hepatocytes may contribute to the elimination of the peptide-antibody conjugates with high clearance values in vivo. To test this hypothesis, a diverse chemical series of peptide-antibody conjugates were initially incubated with suspended mouse hepatocytes, and the percentage remaining in the media was monitored over time. Exploratory experiments (data not shown) suggested a limited dynamic range for the assay, and as such, subsequent experiments were carried out with cryopreserved plated mouse hepatocytes, and the percentage remaining antibody versus time in the media was quantitated by ELISA. After incubation of the unconjugated or conjugated antibodies for up to 3 hours in the plated hepatocyte assay, 38%–93% of the starting antibody concentration remained in the media (Table 1). The percentage remaining in the media exhibited a significant correlation with the observed in vivo clearance (Pearson r = −0.5525, N = 15, P = 0.0327), with more rapid depletion from the media correlating with higher in vivo clearance (Fig. 3A). A significant correlation was also observed between HIC retention time and percentage remaining in the media from the hepatocyte assay (Pearson r = −0.7185, N = 15, P = 0.0085; Fig. 3B).
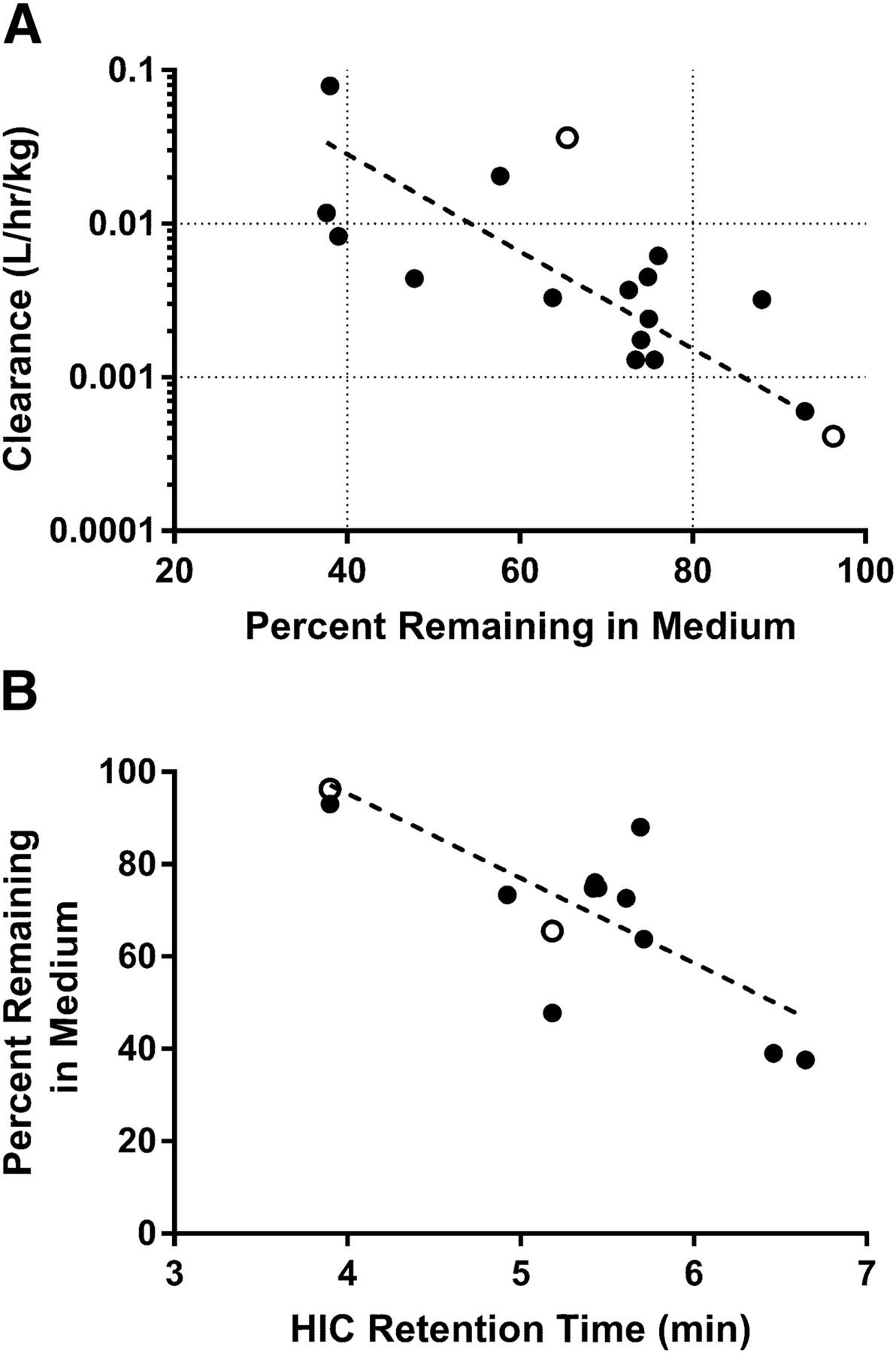
Percentage of peptide-antibody conjugates remaining in the medium after 2.5 hours in plated mouse hepatocyte assays versus in vivo clearance from intravenous pharmacokinetic studies (A) or HIC retention time (B). Peptide conjugates exhibiting extensive association with hepatocytes (lower percentage remaining in media) demonstrated higher in vivo clearance values than those with decreased association (higher percentage remaining in media; Pearson r = −0.5525). A correlation was also observed between HIC retention time and percentage remaining in the hepatocyte media (Pearson r = −0.7185). Mouse data are denoted with closed circles, whereas cynomolgus monkey data are denoted by open circles.
The role of FcRn as a recycling pathway for IgG antibodies is well documented (Roopenian and Akilesh, 2007). Surface plasmon resonance studies were carried out at pH 5.5 and pH 7.4 to assess FcRn binding of our conjugate panel in vitro. Positive correlations were noted between FcRn binding at pH 7.4 (Pearson r = 0.9107, N = 7, P = 0.0044) or pH 5.5 (Pearson r = 0.8088, N = 7, P = 0.0276) and the in vivo clearance for the peptide-antibody conjugates (Fig. 4, A and B). A positive correlation was also observed between FcRn binding at pH 7.4 (Pearson r = 0.7114, N = 6, P = 0.1129) or pH 5.5 (Pearson r = 0.8449, N = 6, P = 0.0342) and the HIC retention time (Fig. 4, C and D), although the correlation at pH 7.4 was not statistically significant. Finally, a strong negative correlation was observed between FcRn binding at pH 7.4 and the percentage remaining in the media from the aforementioned hepatocyte assay (Pearson r = −0.9455, N = 7, P = 0.0013; Fig. 4E). Conversely, no correlation was observed between FcRn binding at pH 5.5 and the percentage remaining in the hepatocyte assay media (Pearson r = −0.3902, N = 7, P = 0.3868; Fig. 4F). A summary of the statistical analysis for the relationships between in vivo clearance, percentage remaining in hepatocyte media, HIC retention time, and FcRn binding at pH 7.4 or 5.5 is shown in Table 2 Initial attempts to assess the translatability of the aforementioned correlations to cynomolgus monkeys are shown in open circles in Figs. 1, 3, and 4, although due to the limited nature of the data set, no statistical analyses were performed.

Observed correlation between FcRn binding affinity of peptide conjugates at pH 7.4 or 5.5 (RUs) and in vivo clearance (A and B), HIC retention time (C and D), or percentage remaining in hepatocyte media (E and F). Peptide conjugates with high affinity for FcRn tended to exhibit higher in vivo clearances, HIC retention times, and depletion rates from hepatocyte media (Pearson r values noted in Table 3). Mouse data are denoted with closed circles, whereas cynomolgus monkey data are denoted by open circles.
In vivo pharmacokinetic parameters following 2-mg/kg intravenous administration of aDNP or conjugate 4 to wild-type mice and receptor knockout mouse models
Summary of Pearson correlation results for in vivo clearance (Cl), percentage remaining in hepatocyte media, HIC retention time, and FcRn binding at pH 7.4 or 5.5
To further explore the role of cell surface receptors in the non-target-mediated binding and/or uptake of the Nav1.7 peptide-antibody conjugates to plated hepatocytes, receptor phenotyping experiments were designed to evaluate the individual contributions of surface receptors known to play a role in binding antibodies and proteins (Ashwell and Harford, 1982; Terpstra et al., 2000). Similar to earlier experiments, aDNP was not depleted from the media of plated hepatocytes, whereas only 33% of conjugate 4 remained in the media after 2.5 hours (Fig. 5). Incubation of conjugate 4 in FcRn knockout hepatocytes resulted in a lack of depletion from the media, similar to what was observed for aDNP in control hepatocytes. Inclusion of acetylated LDL or ovalbumin in the incubation increased the percentage of remaining conjugate 4 to 88% or 69%, respectively, suggesting a role for the Stab/Scarb1 receptors or mannose/galactose receptors in depleting the peptide-antibody conjugates from the media. Follow-up experiments with increasing amounts of acetylated LDL resulted in complete inhibition of conjugate 4 uptake (IC50 = 0.024 mg/ml; Fig. 6), whereas addition of ovalbumin reached a maximum inhibition of 64% of control uptake (estimated IC50 = 1.91 mg/ml; Fig. 6). Inclusion of chondroitin (class A scavengers) or IgG (multiple receptors) had no measurable effect on the depletion of conjugate 4 from the media.

Depletion of unconjugated antibody (aDNP) or conjugate 4 from media of plated mouse hepatocyte assays. aDNP demonstrated limited uptake into mouse hepatocytes, whereas greater than 60% of conjugate 4 was removed from the media over 2.5 hours. Uptake of conjugate 4 into mouse hepatocytes was attenuated in FcRn knockout hepatocytes or by the addition of acetylated LDL (acLDL; Stab/Scarb1) or ovalbumin (mannose/galactose receptors) to wild-type hepatocytes.

IC50 determination for the inhibition of conjugate 4 association with mouse hepatocytes by acetylated LDL (acLDL; Stab/Scarb1 receptor) or ovalbumin (mannose/galactose receptors). Addition of acetylated LDL to wild-type mouse hepatocytes resulted in complete inhibition of conjugate 4 uptake (IC50 = 0.024 mg/ml), whereas addition of ovalbumin reached a maximum inhibition of 64% of control uptake (IC50 = 1.91 mg/ml).
Finally, pharmacokinetic studies were carried out in knockout mouse models to evaluate the in vitro to in vivo translation of the results from hepatocyte experiments. Experiments used FcRn, LDL receptor, and mannose receptor knockout models, as well as a knockout mouse model for ASGR1, a receptor expressed on the surface of hepatocytes that has been shown to be involved in protein uptake (Inamoto and Brown, 1991). The pharmacokinetics of aDNP and conjugate 4 (both total and intact conjugate) were evaluated in each strain. The clearance of aDNP, conjugate 4total and conjugate 4intact increased 15.3-, 49.3-, and 6.81-fold, respectively, in FcRn knockout mice (Fig. 7; Table 2). A modest increase (2.18-fold) in the clearance of conjugate 4total was observed in mannose receptor knockout mice. Clearance and AUC values for all three analytes were within 2-fold of wild-type mice for the LDL receptor and ASGR1 knockout models. A model to evaluate the contribution of the Stab/Scarb1 pathway was not available at the time this manuscript was prepared.

In vivo pharmacokinetics of aDNP and conjugate 4 in wild-type (WT), FcRn knockout , LDL receptor (LDLr) knockout, mannose receptor knockout, or ASGR1 knockout mouse models following 2-mg/kg intravenous administration. Both aDNP and conjugate 4 exhibited higher in vivo clearance values in FcRn knockout mice as opposed to wild-type controls. Conjugate 4 also exhibited higher in vivo clearance values in mannose receptor knockout mice. No difference was observed between wild-type and LDL receptor or ASGR1 knockout mice.
Discussion
The identification of nonopioid alternatives to treat chronic pain is an area of intense scientific research owing in no small part to the addiction risks and societal burdens associated with the current opioid-based standard of care (Dunn et al., 2016; Humphreys, 2017; Kaye et al., 2017). The Nav1.7 voltage-gated sodium ion channel (SCN9A) represents one potential pharmacological target with convincing human genetic validation for the reduction of pain without the use of opioids (Dib-Hajj et al., 2013; Emery et al., 2016; Vetter et al., 2017). Humans expressing loss-of-function mutations in the SCN9A gene have a congenital insensitivity to pain, whereas those with gain-of-function mutations are often diagnosed with primary erythromelalgia or paroxysmal extreme pain disorder (Yang et al., 2004; Cox et al., 2006; Fertleman et al., 2006; Goldberg et al., 2007; Faber et al., 2012). As such, significant efforts have been placed on identifying Nav1.7 inhibitors across a range of therapeutic modalities, including small molecules, peptides, and antibodies.
Previous reports have described internal efforts to identify potent inhibitory peptides of Nav1.7 based on GpTx-1 and JzTx-V peptide scaffolds that have been identified and isolated from tarantula spider venom (Biswas et al., 2017; Moyer et al., 2018; Wu et al., 2018). Because many therapeutic peptides suffer from poor pharmacokinetic profiles due to proteolysis and/or renal filtration (Diao and Meibohm, 2013; Di, 2015), a half-life extension approach was adopted whereby the inhibitory peptides were conjugated via various linkers to nontargeting antibodies to increase their molecular weight and hydrodynamic radius while concurrently taking advantage of antibody recycling mechanisms (Biswas et al., 2017; Murray et al., 2019). An inherent challenge to this approach is the suboptimal pharmacokinetics often observed with modified antibodies and the subsequent need to conduct numerous in vivo pharmacokinetic studies, given the nascency of in vitro approaches to prioritize protein therapeutics for in vivo studies (Vugmeyster et al., 2012; Jaramillo et al., 2017). To that end, we set out to develop an integrated in vitro strategy to optimize the pharmacokinetic properties of Nav1.7 peptide-antibody conjugates prior to conducting in vivo pharmacokinetic studies, with an eye toward more rapidly identifying a lead candidate to progress into late-stage preclinical studies while concurrently fostering the “3Rs principle” (refine, reduce, replace) with regard to animal studies (Russell, 1995).
In conjunction with lead candidate identification efforts, intravenous pharmacokinetic studies were conducted across a panel of Nav1.7 peptide-antibody conjugates to set a baseline for which to characterize in vitro tools. The in vivo clearance of the unconjugated, nontargeting aDNP antibody (aDNP) was 0.0006 l/h/kg, within the range of what would be expected for a typical antibody in the absence of target-mediated clearance (Table 1). Conjugation of the aDNP antibody with Nav1.7 inhibitory peptides resulted in an approximate 22- to 132-fold increase in its clearance, suggesting that the nature of the peptide and linker had a significant effect on the clearance of the antibody and provided a sufficiently wide range of clearance values to compare with in vitro predictors. In addition to total peptide-antibody conjugate clearance, the ratio of intact to total conjugate (AUCintact/AUCtotal) was also measured, as it has been previously shown that proteolytic cleavage of peptide conjugates can result in a loss of pharmacological activity (Hecht et al., 2012; Hager et al., 2013). The observed AUCintact/AUCtotal ratios ranged from 0.1 to 0.76, with the lowest ratios being attributed to the two peptide-antibody conjugates with a peptide charge of +6 (conjugates 1 and 2). As these two conjugates also exhibited the highest clearance values in vivo, it was hypothesized that the charge of the peptide was a key determinant of the in vivo pharmacokinetic characteristics of the corresponding peptide conjugate.
The charge and resulting isoelectric point of protein therapeutics are well documented determinants of their in vivo pharmacokinetic attributes (Boswell et al., 2010; Bumbaca et al., 2012; Li et al., 2014; Datta-Mannan et al., 2015b). Proteins with net positive charges often tend to interact more readily with negatively charged cell surface components, resulting in a more rapid clearance and increased biodistribution compared with corresponding proteins with less positive charges (Bergmann et al., 1984; Pardridge et al., 1995; Lee and Pardridge, 2003; Hervé et al., 2008). As the initial JzTx-V peptides were highly basic with a positive charge of +6 and isoelectric points between 10.5 and 11, it can be hypothesized that conjugating multiple peptides to the aDNP antibody would result in a substantial increase to the overall pI of the peptide-antibody conjugate and negatively affect the resulting pharmacokinetic properties of the conjugates. Indeed, lower clearance values were observed when the net charge on the peptide was reduced to +1 or +2, although we were unable to further differentiate clearance values when comparing peptide-antibody conjugates with +1- versus +2-charged peptides (Fig. 1A), suggesting that additional assays were needed to efficiently differentiate the conjugates prior to advancement to in vivo studies. Similar results were reported by Murray et al. (2019), where reductions in the positive nature of the conjugated peptide resulted in decreased clearance and increased exposure for Nav1.7 peptide-antibody conjugates. In addition to charge and isoelectric point, the structural stability and pharmacokinetic properties of antibodies are also known to be related to the overall hydrophobicity of the molecule (Deng et al., 2012). When the series of Nav1.7 peptide conjugates were analyzed by hydrophobic interaction chromatography, a trend was observed between longer HIC retention times and increased in vivo clearance (Fig. 1B). In a similar vein, decreases in clearance for a series of anti-CD70 antibody-drug conjugates were observed as modifications were made to the linker region to minimize the overall hydrophobicity of the antibody-drug conjugate (Lyon et al., 2015). As the analytical approach is relatively straightforward and requires a minimal amount of material, it can be readily incorporated into structural optimization efforts prior to progressing molecules to in vivo pharmacokinetic studies.
As mentioned earlier, cationic proteins with higher isoelectric points tend to more readily distribute into tissues as compared with their uncharged or negatively charged counterparts (Bickel et al., 1994; Pardridge et al., 1995, 1998; Khawli et al., 1996; Hong et al., 1999; Kobayashi et al., 1999; Lee and Pardridge, 2003; Boswell et al., 2010). As binding to target was not a biodistribution mechanism for the nontargeting aDNP antibodies, biodistribution studies were carried out using whole-body positron emission tomography with representative 64Cu-NOTA–labeled Nav1.7 peptide antibody conjugates to determine if modifications to the peptide affected the broader biodistribution pattern of the peptide-antibody conjugate. The distribution of the unconjugated antibody or a representative peptide-antibody conjugate with two symmetrically conjugated peptides (P2S-aDNP; peptide charge: +6) following intravenous administration was generally constrained to the vasculature, with a small amount found in the bladder (Fig. 2). However, when the aDNP antibody was conjugated with two peptides in an asymmetric fashion or with four peptides (P2A-aDNP or P4S-aDNP; peptide charge: +6), a rapid distribution to the liver was observed after 3 hours (Fig. 2), suggesting that the molecular asymmetry may shunt the antibody conjugate toward specific pattern-recognition receptors on a given tissue, such as the liver, a finding which may prove useful in general antibody-based protein engineering efforts (Szabo et al., 2006; Mullen et al., 2015). Unfortunately, although hepatic elimination is often considered when evaluating the elimination of small-molecule therapeutics, it is generally overlooked in the pharmacokinetic assessment of protein therapeutics. This approach may be perfectly acceptable when the protein of interest is an unmodified monoclonal antibody; however, as mentioned, engineered platforms such as antibody conjugates, Fc-fusion proteins, and bispecific antibodies may be more readily recognized as modified proteins and thus directed to the liver for elimination through multiple scavenging pathways (Liu, 2018). To this point, Datta-Mannan et al. (2016) recently demonstrated the role of liver sinusoidal endothelial cells in the clearance of modified bispecific antibodies in cynomolgus monkeys. Based on our biodistribution data and the findings from Datta-Mannan and colleagues, we hypothesized that hepatocytes may also play a role in the elimination of the Nav1.7 peptide-antibody conjugates and could serve as a readily available in vitro system with which to rank-order prospective peptide conjugates for in vivo evaluation.
The use of cryopreserved hepatocytes to evaluate the metabolic stability of small-molecule therapeutics is commonplace in most absorption, distribution, metabolism, and excretion groups, as hepatocytes contain the full complement of drug-metabolizing enzymes and cofactors involved in the clearance of small molecules (Li, 2001). In addition to small-molecule metabolism, hepatocytes also express numerous intracellular and cell surface receptors, such as FcRn, ASGR1, mannose, galactose, and LDL receptors, in addition to other scavenger receptors, that are known to play a role in protein recycling and elimination (Matsuura et al., 1982; Kindberg et al., 1990; Lagace et al., 2006; Pyzik et al., 2017). It is thus plausible that hepatocytes may represent a holistic system with which to predict the contribution of nonspecific elimination pathways for modified protein therapeutics, such as the Nav1.7 peptide-antibody conjugates. Upon incubating a series of Nav1.7 peptide conjugates with cryopreserved, plated mouse hepatocytes and monitoring depletion from the media, a correlation was observed between the percentage of remaining peptide-antibody conjugate in the media at the end of the incubation and the in vivo clearance, with more rapid depletion from the media correlating with higher in vivo clearance (Fig. 3A). Correspondingly, after washing, lysing, and collecting the remaining cellular content, conjugates with higher in vivo clearance were observed to have more drug-related material bound to or taken up into the hepatocytes as opposed to those with lower in vivo clearance (data not shown). A negative correlation was also observed between the HIC retention time and percentage of remaining peptide-antibody conjugate in the media from the hepatocyte assay, suggesting that hydrophobic interactions may be a key determinant of the depletion of the conjugates from the media (Fig. 3B). Owing to their general ease of use, an similar approach using cryopreserved hepatocytes in suspension was evaluated, but the dynamic range was found to be limited (data not shown). Gene and protein expression patterns in hepatocytes have been shown to be highly dependent on thawing protocols and cellular attachment, and as such, it is possible that plated hepatocytes present a more amenable environment in which to study protein-cellular interactions (Waring et al., 2003).
While the ability of the plated hepatocyte approach to evaluate the in vivo clearance of this series of peptide-antibody conjugates in mice appears robust, the true value (similar to in vitro to in vivo extrapolation approaches with small molecules) will lie in the utility of the system to work with higher-order preclinical species and, ultimately, to predict human pharmacokinetics. To this end, we have extended the current analysis to include pharmacokinetics of aDNP and conjugate 4 in cynomolgus monkeys. The initial results, shown in Figs. 1, 3, and 4, suggest that similar trends may exist in cynomolgus monkey hepatocytes, as was observed with mice, although obviously the data set will need to be significantly expanded before any solid conclusions can be made.
The contribution of the FcRn to the pharmacokinetics of antibody therapeutics is well documented and often a key focus of antibody engineering efforts to optimize pharmacokinetic properties (Roopenian and Akilesh, 2007; Giragossian et al., 2013). To fully take advantage of the FcRn recycling pathway, the binding of the Fc region to FcRn must be balanced in such a way that the antibody rapidly binds to FcRn at pH 5.5 in the endosome but readily dissociates from the receptor at pH 7.4 when the complex is returned to the cell surface. When surface plasmon resonance was used to measure the binding of the Nav1.7 peptide conjugates to mouse FcRn at pH 5.5 or 7.4, a positive correlation was observed between FcRn binding at both pH 5.5 or pH 7.4 and in vivo mouse clearance (Fig. 4, A and B). Of particular interest for the Nav1.7 peptide-antibody conjugates was the similarity of the antibody backbone across the series, suggesting that peptide conjugation to non-Fc regions of the antibody was affecting FcRn binding, similar to what has been previously reported for a set of monoclonal antibodies with identical Fc sequences but varied Fab domains (Wang et al., 2011). A correlation was also observed between the percentage of remaining peptide-antibody conjugate in the media and FcRn binding affinity at pH 7.4 (Fig. 4E) but not pH 5.5, suggesting that the elimination of the peptide conjugates from the hepatocyte media may be a combined function of physicochemical properties and release of the protein from FcRn at the cell surface.
Modifying monoclonal antibodies by conjugating charged peptides to the antibody also opens the possibility that the peptide-antibody conjugate will be recognized as an aberrant protein by other cell surface receptors, as is often the case with scavenger receptors expressed on hepatocytes and liver sinusoidal endothelial cells (Datta-Mannan et al., 2016). To this end, we sought to further characterize other potential receptor-mediated pathways that may be contributing to the observed media depletion in hepatocytes and, subsequently, in vivo clearance. Similar to phenotyping approaches used for small-molecule drug-metabolizing enzymes, inhibitors of various cell surface receptors were added to plated hepatocyte incubations prior to addition of the Nav1.7 peptide-antibody conjugates (Fig. 5). After addition of acetylated LDL or ovalbumin, a decrease in the rate of media depletion for conjugate 4 was observed, suggesting a role for either the Stab/Scarb1 receptor family (acetylated LDL) or the mannose and galactose receptors (ovalbumin) (Smedsrod et al., 1990; Krieger, 2001). As a confirmatory experiment, IC50 curves were generated for the depletion of Nav1.7 peptide-antibody conjugates in the presence of increasing amounts of acetylated LDL or ovalbumin, with acetylated LDL potently inhibiting the depletion of the Nav1.7 peptide-antibody conjugates from the hepatocyte media (Fig. 6). The primary role of the LDL receptor is the endocytosis of cholesterol-bound low-density lipoprotein in the liver, whereas the mannose and galactose receptors are generally involved in the elimination of proteins with complex glycosylation patterns and are expressed on multiple cell types, including those in the liver (Ashwell and Harford, 1982; Goetze et al., 2011; Yang et al., 2015). Conjugate 4 depletion was completely abolished when FcRn knockout hepatocytes were used, further supporting the importance of the FcRn pathway in the disposition of the Nav1.7 peptide conjugates.
Finally, to determine the translatability of the in vitro receptor phenotyping efforts to in vivo outcomes, the pharmacokinetics of aDNP or conjugate 4 were determined in FcRn, LDL receptor, mannose receptor, or ASGR1 knockout mouse models and compared with the pharmacokinetics measured in wild-type mice. As expected, knocking out FcRn had the greatest effect on the pharmacokinetics of aDNP or conjugate 4, with clearance values increasing 15.3-fold or 49.3-fold, respectively, in FcRn knockout mice relative to wild-type mice. Interestingly, a small increase (2.18-fold) in the clearance of conjugate 4total was also observed in mannose receptor knockout mice (Fig. 7). The data suggest that while multiple receptor-mediated pathways may play a role in the pharmacokinetics and distribution of the Nav1.7 peptide-antibody conjugates, interactions with FcRn are still the primary determinant of the observed in vivo pharmacokinetics.
The range of pharmacokinetics of the Nav1.7 peptide-antibody conjugates described in this paper implicates an important role for the inhibitory peptide in determining the in vivo properties of the peptide-antibody conjugate. Taken in their entirety, the data suggest that an integrated strategy combining hydrophobicity, FcRn affinity, and hepatocyte association rates is an efficient way to prioritize Nav1.7 peptide-antibody conjugates for further evaluation and supports the hypothesis that hepatocytes may play a role in the disposition of protein therapeutics. While caution should be exercised in broadly applying such an approach to other engineered proteins, especially when target-mediated disposition may be involved, the data presented herein represent an additional resource to more rapidly triage novel protein therapeutic platforms while concomitantly reducing the number of in vivo studies conducted in preclinical species.
Acknowledgments
The authors thank Dr. Larry C. Wienkers and Dr. Josh T. Pearson for valuable discussions and insights throughout the preparation of this manuscript, as well as Dr. Bill McCarty for assistance with statistical analyses.
Authorship Contributions
Participated in research design: Foti, Biswas, Glaus, Hickman, Murray, Miranda, Moyer, Rock.
Conducted experiments: Foti, Huang, Be, Berry, Conner, Ikotun, Soto.
Contributed new reagents or analytic tools: Murray, Aral, Cheng, Falsey, Herberich, Long, Netirojjanakul, Nixey, Sham, Tegley, Wu, Yin.
Performed data analysis: Foti, Huang, Be, Berry, Conner, Li, Tran.
Wrote or contributed to the writing of the manuscript: Foti, Murray, Rock.
Footnotes
- Received April 26, 2019.
- Accepted July 8, 2019.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- aDNP
- XXX
- ASGR1
- asialoglycoprotein receptor 1
- AUC
- area under the curve
- DMEM
- Dulbecco’s modified Eagle’s medium
- FcRn
- neonatal Fc receptor
- HIC
- hydrophobic interaction chromatography
- LDL
- low-density lipoprotein
- NODA-GA
- 1,4,7-triazacyclononane,1-glutaric acid-4,7-acetic acid
- SCN9A
- Nav1.7 voltage-gated sodium ion channel
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}