


Visual Overview

Abstract
Understanding small interfering RNA (siRNA) fraction unbound (fu) in relevant physiologic compartments is critical for establishing pharmacokinetic-pharmacodynamic relationships for this emerging modality. In our attempts to isolate the equilibrium free fraction of N-acetylgalactosamine–conjugated siRNA using classic small-molecule in vitro techniques, we found that the hydrodynamic radius was critical in determining the size exclusion limit requirements for fu isolation, largely validating the siRNA “rigid rod” hypothesis. With this knowledge, we developed an orthogonally validated 50 kDa molecular-mass cutoff ultrafiltration assay to quantify fu in biologic matrices including human, nonhuman primate, rat, and mouse plasma, and human liver homogenate. To enhance understanding of the siRNA-plasma interaction landscape, we examined the effects of various common oligonucleotide therapeutic modifications to the ribose and helix backbone on siRNA fu in plasma (fu,plasma) and found that chemical modifications can alter plasma protein binding by at least 20%. Finally, to gain insight into which specific plasma proteins bind to siRNA, we developed a qualitative screen to identify binding “hits” across a panel of select purified human plasma proteins.
Introduction
Fraction unbound (fu) is a measure of free drug at equilibrium in a biologic matrix of interest. In this paper, we describe methods to quantify small interfering RNA (siRNA) fu in plasma [fu,plasma; commonly referred as plasma protein binding (PPB)] and liver tissue homogenate (fu,liver). Fraction unbound is routinely quantified for small-molecule therapeutic candidates according to the free drug hypothesis, wherein only the unbound fraction of drug is available to exhibit pharmacologic effects (Rowland et al., 2011). However, the role, if any, of fu on the pharmacokinetic-pharmacodynamic (PK-PD) relationship has yet to be established for therapeutic siRNA.
siRNA is a rapidly emerging therapeutic modality, with the first US Food and Drug Administration (FDA) approval granted in August 2018 (Hoy, 2018). Although oligonucleotide therapeutics such as siRNA are generally treated as small molecules for regulatory filings, the FDA has not issued any specific guidance around the reporting of in vitro absorption, distribution, metabolism, and excretion properties (e.g., PPB) on this modality to date. While fu,plasma has been described for antisense oligonucleotide (ASO) therapeutics using 30 kDa molecular-weight cutoff (MWCO) ultrafiltration, a corresponding assay has not yet been described for siRNA (Watanabe et al., 2006). For small molecules, fu,plasma is typically measured via ultracentrifugation, ultrafiltration, or equilibrium dialysis. In these assays, the molecular size, shape, mass, and/or density of the small molecule relative to the plasma protein milieu largely determine its differential partitioning based on sedimentation velocity (ultracentrifugation) or porous membrane exclusion limits (ultrafiltration and equilibrium dialysis). For siRNA fu isolation, the larger size of siRNA [approximately 15 kDa with triantennary N-acetylgalactosamine (GalNAc) conjugation] needed to be taken into consideration.
In this publication, we report an orthogonally validated 50 kDa MWCO ultrafiltration assay to quantify fu,plasma and fu,liver of a therapeutic siRNA in human matrices at clinically relevant concentrations and across relevant preclinical species (fu,plasma only). The 21-mer double-stranded siRNA used throughout the study, referred to as siRNA-X, is chemically modified RNA with phosphorothioate (PS) bonds, 2′O-methyl (2′-OMe) and 2′deoxy 2′-fluoro (2′-F) ribose modifications, and GalNAc conjugation. PS bonds replace specific phosphodiester bonds to increase exonuclease resistance (Braasch et al., 2004), 2′-OMe and 2′-F enhance both stability and RNA-induced silencing complex (RISC) interactions (Allerson et al., 2005; Choung et al., 2006), and GalNAc enables targeted hepatocyte uptake via the asialoglycoprotein receptor (ASGPR) (Foster et al., 2018; Janas et al., 2018; Springer and Dowdy, 2018). siRNA-X is highly efficacious, eliciting greater than 80% target mRNA and protein knockdown over at least three months after a single 3-mg/kg dose in nonhuman primates (manuscript in preparation). Numerous chemical modifications and ligands used in the current generation of oligonucleotide therapeutics—namely, ASO therapeutics—have been demonstrated to alter the extent of protein binding (Wilce et al., 2012; Geary et al., 2015; Bhandare and Ramaswamy, 2016; Juliano, 2016; Schirle et al., 2016; Bailey et al., 2017; Gaus et al., 2018). To understand how RNA modifications and ligand conjugation affect siRNA PPB specifically, we investigated the effects of PS, 2′-OMe, 2′-F, GalNAc, and biotin on fu,plasma.
Protein-siRNA interactions may affect siRNA tissue clearance, macroscopic (tissue-level) and microscopic (cell-level) distribution, and/or pharmacological activity; conversely, binding of siRNA to certain proteins may change the function or fate of those proteins. Taken together, these works, addressing both total siRNA-matrix interactions to inform the former and screening for specific siRNA-protein interactions to inform the latter, provide a set of complementary tools to begin to establish the role and the relevance of protein binding for this emerging modality. Furthermore, while the extent of total PPB at equilibrium is of interest from a PK-PD modeling perspective, knowledge of interactions between therapeutic siRNA and specific plasma proteins may help identify potential off-target protein-binding liabilities, drug-drug interactions, and aid the design of next-generation siRNA molecules.
Materials and Methods
Materials
All oligonucleotides were synthesized in house using commercially available reagents or purchased from Integrated DNA Technologies (Skokie, IL). Human plasma protein [albumin (#A3782), α-1-acid glycoprotein (#G9885), α-2-macroglobulin (#SRP6314), fibronectin (#F2006), fibrinogen (#F3879), haptoglobin (#372022), IgG Fc fragment (#AG714)] buffer reagents, and Amicon Ultra 0.5-ml centrifugal filters [30 kDa MWCO (#UFC503008) and 50 kDa MWCO (#UFC505008)] were obtained from Millipore Sigma (Burlington, MA). Slide-A-Lyzer 0.5-ml MINI Dialysis Devices (20 kDa MWCO; #88402) were obtained from Thermo Fisher (Waltham, MA). α-thrombin was obtained from MP Biomedicals (#02194918; Santa Ana, CA). All plasma (K2 EDTA treated) and liver tissues were obtained from BioIVT (Westbury, NY). All plasma samples were from frozen pooled, mixed-gender donors from CD-1 mice (MSEPLEDTA2; 300 donors), cynomolgus monkeys (CYNPLEDTA2; 22 donors), Sprague-Dawley rats (RATPLEDTA2; 60 donors), and humans (HMPLEDTA2; 69 donors). Human liver tissue was from three pooled, female donors (aged 30–40 years) and homogenized (from frozen) in Tissue Extraction Reagent I (#FNN071; Invitrogen, Carlsbad, CA). Blocking reagents were sourced as follows: heparin (#84020), bovine serum albumin (#A9418), gelatin (#9000-70-8), 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate (CHAPS; #C3023), and spermidine (#S2626) from Sigma-Aldrich (St. Louis, MO); I-Block (#T2015) from Invitrogen; and Tween-20 (#85113), Blocking Reagent (#11096176001), and Triton X-100 (#28314) from Thermo Fisher. siRNA-specific rabbit polyclonal antibody (pAb) was generated by Lampire Biologic Laboratories (Encinitas, CA) by immunization of rabbits with siRNA-X.
Methods
Time to Equilibrium of siRNA Binding to Human Plasma by Biolayer Interferometry.
Biotinylated siRNA (biotin conjugated to the 3′ terminus of the sense strand; bn-siRNA) in buffer A [10 mM Tris (pH 7.4), 150 mM NaCl, 1 mM CaCl2, 1% gelatin (w/v), 0.13% Triton X-100 (w/v)] was loaded onto pre-equilibrated high-precision streptavidin biosensors (ForteBio LLC, Fremont, CA) to obtain a response of ∼1 nm over 1000 seconds. bn-siRNA–loaded tips were rinsed in buffer A for 1 minute, followed by exposure to a five-point (plus two blanks), 3-fold dilution series of human plasma in buffer A [top concentration: 3.3% human plasma in buffer A (v/v)] over 10 minutes. Data were collected on a ForteBio Octet-384 instrument, and analysis and fitting were performed using the ForteBio software (version 10.0).
fu,plasma and fu,liver Determination by Ultrafiltration.
To prepare the Ultracel regenerated cellulose filters for use, residual glycerin was removed by twice adding 0.5 ml of phosphate-buffered saline [PBS; 137 mM NaCl, 2.7 mM CaCl2, 10 mM Na2HPO4, 1.8 mM KH2PO4 (pH 7.4)] and spinning in a bench-top centrifuge for 10 minutes at 3000g. The remaining PBS was removed before adding 0.5 ml of PBS with 0.1% Tween-20 (w/v; PBST) and repeating the spin to prevent nonspecific binding of drug to the filter. PBST was removed from the filter and collection tube immediately prior to sample addition (take care to avoid drying the filter). Samples were prepared by spiking known siRNA or small-molecule concentrations into neat plasma or tissue homogenate (pre-equilibrated to 37°C) and incubated at 37°C for 30 minutes, shaking at 500 rpm. A 500-μl sample was transferred into prepared filters and spun at 1500g until no more than 20% of the volume had passed through the filter. Only a small volume of ultrafiltrate should be collected, as the protein concentration in the upper reservoir rises during the filtration process (Zeitlinger et al., 2011). To mitigate matrix effects, after ultrafiltration, all donor samples were pretreated with an equivalent volume of PBST, and all ultrafiltrate receiver samples were pretreated with an equivalent volume of plasma/homogenate (standard curves were treated the same way). To measure recovery, drug-spiked PBST controls were performed at every concentration tested. During method development, we set an arbitrary cutoff of 80% recovery in buffer at each concentration tested to ensure that the corresponding fu readout in plasma or homogenate was representative of the majority of siRNA in the sample. Ultracentrifugation and equilibrium dialysis methods are provided in the Supplemental Methods.
siRNA Detection and Quantitation via 96-Well Plate-Based Hybridization Assay.
Sheep anti-digoxigenin polyclonal antibody (#11222089001; Roche) was conjugated with a ruthenium label using the MSD GOLD Sulfo-Tag NHS-Ester conjugation kit (#R31AA-1; Meso Scale Diagnostics, Rockville, MD). Standards and sample buffer were made as follows: 10 mM Tris-HCl (pH 8.0) and 1 mM EDTA. Hybridization buffer was made as follows: 60 mM Na2PO4 (pH 7.0, dibasic), 1 M NaCl, 5 mM EDTA, and 0.02% Tween-20. Lyophilized oligonucleotide probes were custom synthesized from Qiagen Inc. (Hilden, Germany). Sequence-specific capture and detection probes were conjugated to biotin and digoxigenin, respectively. All chemicals and reagents were analytical grade or higher, if not specified.
Test article standard curves were prepared in PBS or plasma at a concentration range of 2.6–166 pM (0.04–2500 ng/ml). The standard curves and samples were diluted 1:10 in sample buffer and added to a 96-well polymerase chain reaction plate to a final volume of 50 µl. Both sequence-specific capture and detection probes were added to hybridization buffer to a final concentration of 50 nM, and 50 µl was added to the samples in buffer. Sample and probes were hybridized in a thermal cycler under the following conditions: 90°C for 5 minutes, 35°C for 30 minutes, and a final hold at 12°C. After hybridization, samples were transferred to an MSD GOLD 96-well Streptavidin SECTOR PR plate (#L13SA; Meso Scale Diagnostics) for 30 minutes at room temperature. Plates were washed and incubated for 1 hour with 50 μl of 2 μg/ml ruthenium-labeled anti-digoxigenin antibody in SuperBlock T20 TBS Blocking Buffer (#37536; Thermo Fisher). After a final wash, 150 μl of MSD Read Buffer T (#R92TD; Meso Scale Diagnostics) was added, and the plate was read in an MSD Sector S 600 instrument (Meso Scale Diagnostics).
Quantitation was performed against standard curves by nonlinear four parameter logistic regression using GraphPad Prism (version 7.04). For ultrafiltration and equilibrium dialysis, percentage recovery (% recovery) in buffer was calculated using eq. 1: (1)
(1)
where [donor] is the concentration of siRNA in buffer before addition to the apparatus, and [receiver] is the concentration of siRNA recovered in the ultrafiltrate or on the other side of the dialysis membrane, respectively. The fu was calculated using eq. 2: (2)
(2)
Dilution of human liver tissue in homogenization buffer was accounted for using eq. 3 (Kalvass et al., 2007): (3)
(3)
where D is the dilution factor.
Comparative Liquid Chromatography–Tandem Mass Spectrometry Analysis of Small-Molecule fu,plasma and fu,liver via Ultracentrifugation and Ultrafiltration.
Liquid chromatography–tandem mass spectrometry was used to quantify post-50 kDa MWCO ultrafiltration and ultracentrifugation of warfarin, antipyrine, and timolol in human plasma and rosuvastatin in human liver homogenate (see Supplemental Methods for ultracentrifugation fu,plasma and fu,liver method). Samples were quenched with 40% acetonitrile in water and spun for 10 minutes at 4000g before injection on a Kinetex C18 column (2.6 µm, 50 × 2.1 mm; Phenomenex) using a Shimadzu ultrafast liquid chromatography system coupled to an AB Sciex Qtrap 4500 mass spectrometer with a source temperature of 550°C and an ion spray voltage of 4500 V. The mobile phases consisted of 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in acetonitrile (mobile phase B) using a flow rate of 1 ml/min and a gradient as follows: 5% B for 0.8 minutes, 99% B for 0.5 minutes, and return to 5% B to 1.5 minutes. Analytes were detected in positive ion mode using multiple reaction monitoring (Q1→Q3; collision energy, V): warfarin (309.0→163.0 m/z; 20 V), timolol (317.0→261.1 m/z; 25 V), antipyrine (189.1→161.1 m/z; 25 V), rosuvastatin (482.4→258.1 m/z; 45 V), and internal standard tolbutamide (309.0→163.0 m/z; 25 V). Compound peak areas were integrated using Analyst 1.6.2 software (Sciex) and normalized to the internal standard.
Binding of Select Purified Human Plasma Proteins to Biotinylated siRNA-X ± GalNAc.
Binary siRNA–protein interactions were measured by biolayer interferometry (BLI) with biotin-conjugated siRNA immobilized on streptavidin tips. bn-siRNA-X (100 nM in buffer A) with or without GalNAc was loaded onto streptavidin BLI tips over 10 minutes to ∼2 nm. Loaded tips were rinsed in buffer for 1 minute, then introduced to 1:2 titrations of anti-siRNA pAb antibody (positive control), α-2-macroglobulin, α-thrombin, fibrinogen, fibronectin, albumin, α1-acid glycoprotein, α1-antitrypsin, haptoglobin, and IgG Fc fragment, starting at 1 μM (except the anti-siRNA pAb control at 0.2 μM, and α-2-macroglobulin, α-thrombin, fibrinogen, fibronectin at 0.5 μM). Association and dissociation steps were performed for 10 minutes each.
Results
Time to Equilibrium and Quasi-Kinetic Binding Analysis Using Biolayer Interferometry.
Determination of fu requires that the system be at equilibrium (Schmidt et al., 2010). BLI was used to measure the time taken for biotin-conjugated siRNA-X loaded onto streptavidin tips to reach steady state (Fig. 1). We observed that time to steady state decreased with increasing plasma concentrations, and at 3.3% plasma the system approached equilibrium in 10 minutes. Based on this result and liquid chromatography–mass spectrometry evidence that siRNA-X is stable in plasma for more than 30 minutes (Supplemental Fig. 1), we determined that 30 minutes was sufficient for accurate fu determination in neat plasma. It is important to recognize that, for all PPB experiments, small molecules included, a compromise must be made to balance time to equilibrium with metabolic stability. Given that the rate of equilibration depends on the ligand-protein complex half-life (t1/2; Corzo, 2006), the sensorgrams do not appear to be approaching zero in the dissociation phase, and that plasma is a highly heterogeneous mixture of proteins, it is possible that a population of plasma proteins that bind siRNA tightly have not reached equilibrium in 30 minutes. Therefore, to ensure reproducibility, it is essential to perform the experiment with strict adherence to the 30-minute equilibration time and temperature (37°C).
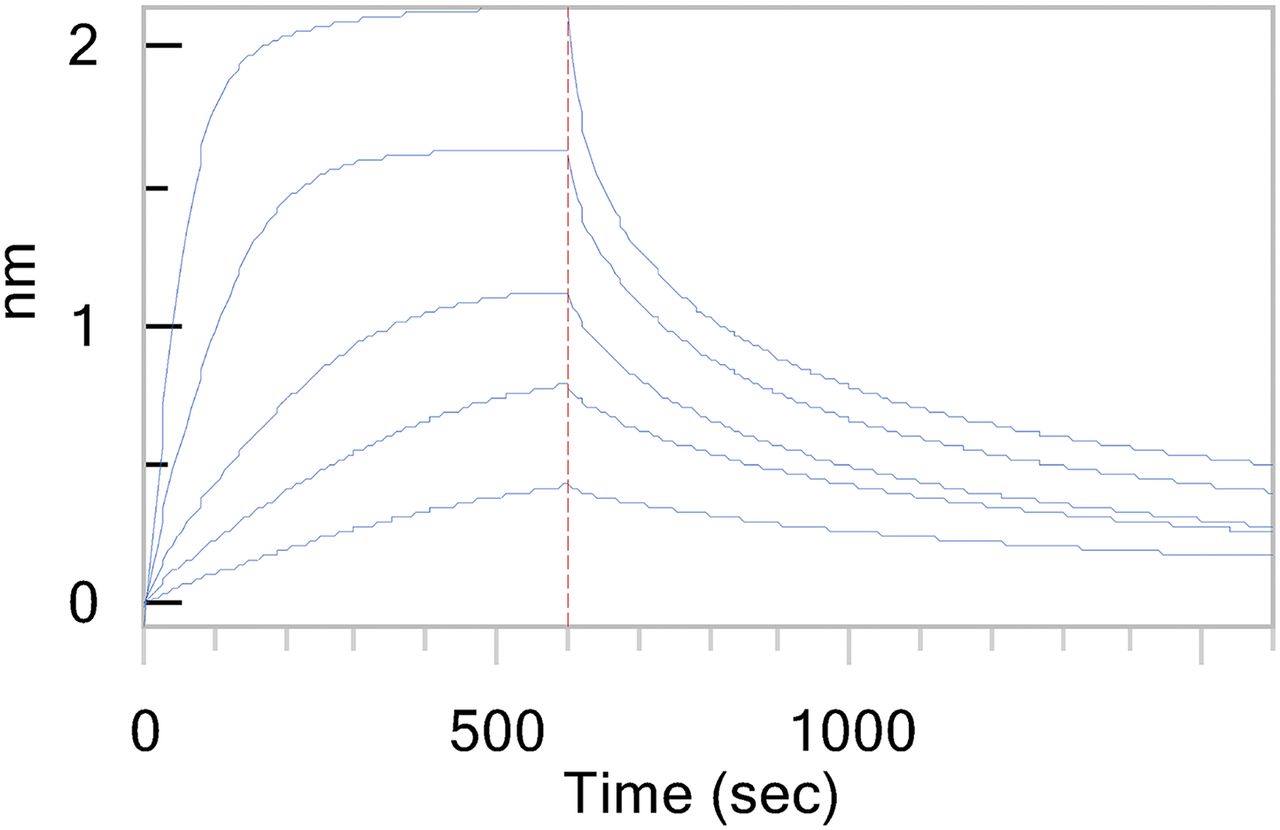
Representative time to equilibrium of siRNA binding to total human plasma. Reference tip-subtracted sensorgrams depicting a titration of total human plasma interacting with biotinylated siRNA on streptavidin tips. Top plasma concentration is 3.3% (v/v) in buffer A followed by a 1:2 dilution series.
Comparison of Classic Small-Molecule PPB Methods to Determine the Unbound Fraction of siRNA in Plasma.
At roughly 15 kDa, GalNAc-conjugated siRNA is significantly larger than a typical small molecule. Consequently, isolation of the unbound fraction by a semipermeable physical barrier (ultrafiltration and equilibrium dialysis) or by differential sedimentation (ultracentrifugation) requires some adaption from small-molecule fu isolation methods. Initially, we tested ultrafiltration and equilibrium dialysis devices with 30 and 20 kDa MWCO exclusion limits, respectively, based on commercial availability of devices with MWCOs close to, but greater than, 15 kDa. For ultracentrifugation, owing to complexities of achieving differential sedimentation of similarly sized macromolecular species (Hughes et al., 1938), we elected to test our existing ultracentrifugation small-molecule protocol without modification. We ran initial tests in protein-free medium to ensure that siRNA could freely diffuse across the semipermeable membrane (ultrafiltration and equilibrium dialysis) or would remain in the supernatant after spinning (ultracentrifugation).
Table 1 shows representative percentage recoveries of 1 μM siRNA-X in PBST in the receiver compartment (ultrafiltration and equilibrium dialysis) or supernatant (ultracentrifugation) for the three techniques. The recoveries—0.0062%, 5.5%, and 24.8% for ultracentrifugation (small-molecule method), equilibrium dialysis (20 kDa MWCO), and ultrafiltration (30 kDa MWCO), respectively—were too low for use in siRNA fu determination. At first, we hypothesized that the poor recovery was due to nonspecific surface binding to filters, dialysis membranes, and ultracentrifugation tubes. We explored a range of potential blocking reagents, including heparin, alternative siRNA molecules, bovine serum albumin, gelatin, I-Block, spermidine, Blocking Reagent, and Triton X-100. None of the blocking agents tested improved recovery over PBST, and several of them, including heparin, alternative siRNA, and spermidine, significantly reduced the sensitivity of the hybridization detection assay, potentially due to charge-based competition for the capture and detection probes. We subsequently discovered that the pore size of the permeable membrane was the largest determinant for recovery, as we recovered 92% of 1 μM siRNA-X in the receiver compartment of a filtration device with a 50 kDa MWCO in PBST. While this recovery represented a great improvement compared with where we started, recovery can be further improved with optimization of blocking reagents, filter-blocking routines, and centrifugation speeds. We did investigate the effect of replacing PBST with PBS with 0.1% CHAPS (w/v) and found that it significantly improved recovery of asymmetric [larger hydrodynamic radius (Rh)] GalNAc-siRNA molecules (Supplemental Fig. 2). It is important to note that Watanabe et al. (2006) reported over 90% recovery of a fully phosphorothioated 20-mer DNA ASO using a 30 kDa MWCO ultrafiltration method, highlighting that single-stranded DNA and double-stranded RNA (dsRNA) likely differ significantly in their structural conformations.
Comparison of percentage of siRNA-X recovery using different fu isolation techniques
Akin to ultracentrifugation approaches to measure small-molecule fu, the ultrafiltration technique described here is likely subject to minor equilibrium perturbation effects as a consequence of the extent and duration of the centrifugal force applied. siRNA-protein interactions most affected by this are weak binders. To ensure reproducible and comparable results using this technique, we recommend using the centrifugation speeds and times described here.
Validation of siRNA Rh via Calculation from Literature Values and siRNA-X Crystal Structure.
After observing that a 50 kDa MWCO exclusion limit was required for adequate recovery of siRNA-X across an ultrafiltration apparatus in buffer (Table 1), we realized that Rh, and not molecular weight (MW), governs filter selection (these filters are typically designed for protein-based applications).
siRNA Rh can be calculated using helical rise per base pair from literature values describing dsRNA A-form helix dimensions (Supplemental Calculations 1; Taylor et al., 1985; Baeyens et al., 1995). Initially, we were concerned that backbone and ribose modification of siRNA might cause deviations from the A-form helix structure; however, a crystal structure of siRNA-X confirmed the “rigid rod” linear geometry of siRNA, as well as the A-form helix (helical rise: 0.26–0.29 nm/base pair; Rh: ∼2.7–3 nm; Fig. 2B; manuscript in preparation). Consequently, we used literature values of known protein Rh versus protein MW to establish a correction factor to determine siRNA-protein MW equivalence (Fig. 6Rh calculations shown in Supplemental Calculations 2). The 21-mer siRNA-X is roughly equivalent to a 48 kDa protein, which is why it requires a 50 kDa MWCO filter.

(A) Workflow of determination of siRNA fu via ultrafiltration. Step 1: pretreat filter with detergent-containing buffer [we found that PBST (Table 1) and PBS+CHAPS (Supplemental Fig. 2) provided good recovery with a 50 kDa MWCO filter]. Step 2: add pre-equilibrated siRNA-spiked matrix into donor compartment of filter and centrifuge. Step 3: collect flow-through and quantify siRNA fu. (B) Depiction of siRNA-X Rh based on the crystal structure.
Orthogonal Validation of a 50 kDa MWCO Ultrafiltration Method for siRNA-X fu Determination Using Electrophoretic Mobility Shift Assay and Liquid Chromatography–Mass Spectrometry.
Increasing the ultrafiltration MWCO from 30 to 50 kDa resulted in a significant buffer recovery increase, leading us to realize the importance of using of siRNA hydrodynamic radius (Rh) rather than MW in determining which device to use for fu determination (Table 1). However, given that human plasma consists of a highly heterogeneous mixture of proteins, many of which are 50 kDa or less, it was important to test whether the “unbound fraction” of siRNA in the receiver compartment of the ultrafiltration device was the true fu or a mixture of unbound siRNA and siRNA bound to small proteins. We ran an electrophoretic mobility shift assay on the ultrafiltrate of 1 μM siRNA-X spiked into human plasma and observed two bands—one consistent with siRNA-X, the other consistent with albumin (Supplemental Fig. 3A). Further testing of the albumin band in plasma without siRNA-X showed that albumin, or some compound that comigrates with albumin, stains with SYBR Gold (Supplemental Fig. 3B); we later demonstrate that albumin does not bind siRNA-X using BLI (Supplemental Fig. 6). Together, these findings suggest that the clear majority of siRNA-X that passes through the 50 kDa MWCO filter is unbound.
Next, to determine whether the 50 kDa MWCO ultrafiltration method was a reliable measure of fu, we tested it on a panel of small molecules with well established fu,plasma and fu,liver values ranging from majority bound (warfarin) to majority unbound (antipyrine). Table 2 provides a comparison of the reference fu values obtained using classic PPB methods compared with the 50 kDa MWCO ultrafiltration method. Warfarin fu,plasma was overestimated approximately 4-fold. This is likely because warfarin binds to albumin, and we showed with electrophoretic mobility shift assay that some fraction of albumin is recovered in the receiver compartment using the 50 kDa MWCO filter (Supplemental Fig. 3); the manufacturer also only reports >95% bovine serum albumin retention on these devices (http://www.emdmillipore.com/US/en/life-science-research/protein-sample-preparation/protein-concentration/amicon-ultra-centrifugal-filters/http://www.emdmillipore.com/US/en/product/Amicon-Ultra-0.5mL-Centrifugal-Filters-for-DNA-and-Protein-Purification-and-Concentration,MM_NF-C82301). Recovery of some quantity of albumin is expected given its high abundance,.Its MW (∼66 kDa) is sufficiently close to 50 kDa, the filter pore sizes represent a distribution at best, and albumin is not a perfect sphere. Rosuvastatin fu,liver (fu,liver = 0.14) was underestimated relative to the literature values (fu,liver = 0.23), which were generated using equilibrium dialysis (Pfeifer et al., 2013). This is likely because applying centrifugal force to homogenate results in blocking of the filtration pores to some extent. Lowering the centrifugation speed could minimize this effect, or this problem could be avoided entirely with equilibrium dialysis.
Validation of 50 kDa MWMCO ultrafiltration method for determination of fu,plasma and fu,liver using well characterized small molecules
Cross-Species Comparison of siRNA PPB.
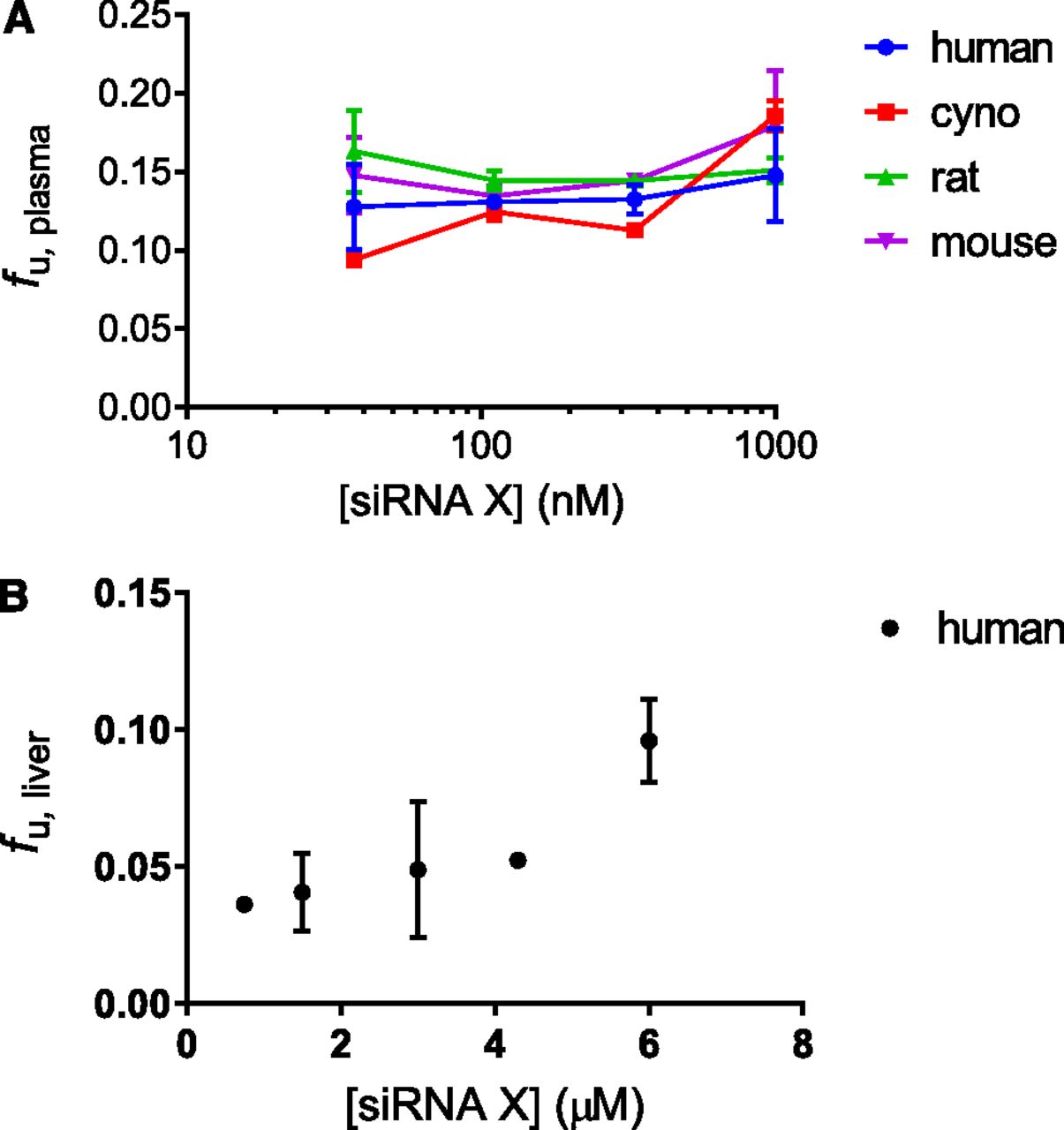
As the development of therapeutic molecules necessitates testing in multiple preclinical species, it is important to understand if PPB properties are consistent across relevant species—in this case, mouse, rat, and cynomolgus monkeys—as well as in humans over a range of therapeutically relevant concentrations. Cross-species PPB comparisons could help at least partly explain any observed differences in PK profiles. A two-way analysis of variance (ANOVA) of the data in Fig. 3A indicated that there was no significant distinction for siRNA-X fu,plasma across the species tested (P > 0.05), but that there was an increase in the fraction unbound with increasing concentration from 0.037 to 1 μM (P < 0.01). The latter is consistent with PPB data reported across multiple modalities (Schmidt et al., 2010). However, given the limited concentration range, it cannot be determined if PPB is concentration-independent (linear) or concentration-dependent (nonlinear), as both cases have been reported (Deitchman et al., 2018). It should be noted that Gaus et al. (2018) recently observed that plasma binding of a 50% phosphorothioated DNA/RNA duplex was ∼19-fold higher in monkey plasma compared with humans (mouse and rat were intermediate). This remains an area of active research.

PPB and liver protein binding of siRNA-X. (A) Cross-species comparison of fu,plasma across a range of therapeutically relevant siRNA concentrations. There was a significant increase in fu,plasma with concentration (P < 0.01) that was not dependent on species (determined by two-way ANOVA, GraphPad Prism). (B) siRNA-X fu,liver across a range of therapeutically relevant concentrations. Plasma measurements were performed in triplicate; liver measurements were performed in duplicate.
Determination of siRNA-X fu,liver in Human Liver Tissue Homogenate.
Many siRNA molecules currently under investigation as therapeutics, including siRNA-X, are targeted to the liver via GalNAc conjugation, which enables delivery via ASGPR-mediated uptake. It was therefore of interest to measure the unbound fraction in the liver. For small molecule, fu,liver is typically measured by equilibrium dialysis (Pfeifer et al., 2013); however, in the absence of commercially available devices with ∼50 kDa MWMCO for siRNA, we adapted the plasma ultrafiltration method described earlier to human liver homogenate (Fig. 3B). For siRNA-X, fu,liver ranged from 0.018 to 0.051 over a therapeutically relevant concentration range (0.375–6 μM), indicating that it is mostly bound in human liver tissue, and that binding was higher in liver homogenate compared with plasma. Over this concentration range, the data did not appear to be strictly linear.
The Effect of Chemical Modifications and Ligand Conjugates on siRNA PPB.
To investigate the effect of different chemical modifications on siRNA PPB, we measured fu,plasma on constructs with the same siRNA-X sequence that had been modified to be entirely 2′-OMe, 2′-F, or PS modified (Fig. 4). Consistent with the literature, we observed that PS increases PPB, and 2′-OMe decreases PPB relative to the siRNA-X control (Braasch et al., 2004; Allerson et al., 2005; Choung et al., 2006; Gaus et al., 2018). There was no statistically significant difference between siRNA-X and fully 2′-F siRNA fu,plasma. We could not find evidence in the literature discussing the role of 2′-F in siRNA PPB differences; however, in certain ASO cases, 2′-F appears to confer increased specificity and/or affinity to select cytosolic proteins (Shen et al., 2015; Vickers and Crooke, 2016). At 1 μM siRNA, the effects of PS and 2′-OMe appear informative from an siRNA therapeutic design perspective because alterations in the numbers of these modifications could significantly affect the PK profile of siRNA in the blood.

Effect of chemical modifications on fu,plasma at 1 μM siRNA concentration. (A) Constructs tested with the sense strand depicted at the top (5′-3′) and the complementary antisense strand at the bottom. The RNA base sequence was constant across constructs, with only the conjugated ligand(s) (GalNAc and biotin), the ribose (2′-OMe, 2′-F), and the backbone (PS or phosphodiester) changing. (B) fu,plasma for each of the constructs in plasma; siRNA-X was also measured in serum. Results were compared using an ordinary one-way ANOVA with multiple comparisons in GraphPad Prism. Significant differences between the test article, siRNA-X in plasma, and the other constructs are reported as *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001.
We also looked at the effect of siRNA ligand conjugation on PPB and found that removal of GalNAc increased protein binding, and biotin had no statistically significant effect compared with siRNA-X. The implications of these findings are that conjugating biotin to siRNA as a functional handle does not affect PPB, and that GalNAc may be important for reducing PPB interactions. Furthermore, while siRNA-X fu,plasma was slightly less than fu,serum, this result was not statistically significant, indicating that the role of clotting factors in total PPB is minimal. In addition, we ran the same PPB assay on a panel of four other therapeutic candidate siRNA molecules with different sequences and similar modification patterns, and found that at 1 μM siRNA, fu,plasma varied between approximately 0.08 and 0.15, which was significantly less than the effects observed with more extreme modification patterns here (data not shown).
Qualitative Determination of Specific Interactions between siRNA-X and Select Human Plasma Proteins.
We used BLI to gain further insight into which specific plasma proteins bound to siRNA-X. We elected to compare binding in the presence and absence of GalNAc due to the observation of different trends in total plasma protein binding when GalNAc was present or absent in various constructs (Supplemental Figs. 4 and 5), and because siRNA-X PPB significantly increased when GalNAc was removed (Fig. 4). Selection criteria for panel inclusion was based on plasma abundance [albumin, IgG Fc fragment, fibrinogen, α-2-macroglobulin, α-1-antitrypsin, and haptoglobin (Anderson and Anderson, 2002)], prior evidence of prominent small-molecule or ASO drug binding [albumin, α-1-acid glycoprotein, α-2-macroglobulin (Cossum et al., 1993; Brown et al., 1994; Srinivasan et al., 1995; Watanabe et al., 2006)], or RNA aptamer binding precedence [fibronectin (Ulrich et al., 2002) and α-thrombin (Long et al., 2008)]. An siRNA-specific rabbit pAb was used as a positive control.
α-2-Macroglobulin and α-thrombin bound to both siRNA-X constructs, while fibronectin and fibrinogen bound to bn-siRNA-X (-Gal) only (Fig. 5), and no binding was observed with albumin, α1-acid glycoprotein, α1-antitrypsin, haptoglobin, and IgG Fc (negative results provided in Supplemental Fig. 6) (Fig. 6).

BLI sensorgrams of positive screening hits for select human plasma proteins binding to biotinylated siRNA-X ± GalNAc. A side-by-side comparison of anti-siRNA pAb (positive control), α-2-macroglobulin, α-thrombin, fibrinogen, and fibronectin binding to bn-siRNA-X with (left column) or without (right column) GalNAc conjugation. Titrations were 1:2 dilutions with a top concentration of 0.5 μM (except pAb = 0.2 μM).

The relationship between Rh and MW for globular proteins does not hold for the dsRNA linear polymer according to Rh prediction based on dsRNA helical rise and the “siRNA rigid rod” assumption. siRNA is marked in red. GalNAc was not included in the Rh calculation. Linear regression was performed on the protein subset to estimation of the MW for a protein with an equivalent Rh to siRNA. The equation of the line obtained (with siRNA omitted) was y = 0.03509x + 1.233, and this was used to calculate the siRNA-protein MW equivalence value of 48 kDa (GraphPad Prism; calculations provided in Supplemental Calculations 2).
Discussion
We have described and validated an assay to measure the free fraction of GalNAc siRNA in plasma and liver homogenate at equilibrium. The rationale for developing the assay was to gain insight into the protein-binding properties of therapeutic siRNA molecules to support PK-PD modeling efforts and to pre-empt any regulatory filing requests. Under the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use and FDA guidelines, therapeutic siRNA is generally treated as a small molecule, as there is no specific guidance for this emerging modality. Small-molecule regulatory filings typically require in vitro PPB data for preclinical animals and humans [S3A Guidance on Toxicokinetics (https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM519697.pdf) and M3(R2) Guidance on Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals (https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073246.pdf)]. To our knowledge, whereas fu methods have been described for heavily phosphorothioated single-stranded DNA–based ASOs, this is the first description of an siRNA assay (Cossum et al., 1993; Braasch et al., 2004; Watanabe et al., 2006).
siRNA Rh Dictates fu Separation Requirements.
A major finding of this work was that MWCO exclusion limits, which are estimated from approximately spherical globular proteins, cannot be directly applied to siRNA for fu experiments because siRNA Rh is ∼2-fold greater than a protein of equivalent MW . Therefore, to ensure siRNA can diffuse freely across a porous membrane for fu separation, filters must be selected based on Rh and not MW.
siRNA is hypothesized to exist as a “rigid rod” in solution due to the geometry of the dsRNA A-form helix (Kornyshev and Leikin, 2013; Kozielski et al., 2013; Dandekar et al., 2015). We recently obtained a crystal structure of siRNA-X confirming its linear geometry (Rh = ∼2.7–3 nm; manuscript in preparation), consistent with literature descriptions of unmodified dsRNA (∼0.28-nm helical rise/base pair or Rh ∼2.9 nm for 21-mer siRNA). As it tumbles freely in solution, 21-mer siRNA Rh is roughly equivalent to a 48 kDa protein (Kok and Rudin, 1981; Wilkins et al., 1999). This conservative estimate does not account for terminally positioned triantennary GalNAc or cation concentration and identity [(shown for DNA (Fujimoto et al., 1994)]. 2′-F, 2′-OMe, and PS modifications likely do not alter the A-form helical properties (Smith and Nikonowicz, 2000; Liu et al., 2011). Consequently, our recovery data (Table 1) support the siRNA rigid rod hypothesis.
An important implication of siRNA Rh elucidation is that certain organs in the body, including the kidney and lymph nodes, use filtration as a means of sorting molecules. Based on our findings, we recommend using Rh-corrected MW to aid predictions of how these tissues process siRNA.
Application of Protein-Binding Data to Interpret PK-PD Profiles.
PPB and liver protein-binding fu values for siRNA-X at equilibrium are not particularly informative in isolation, as they do not address interaction dynamics and binding affinities in whole blood or at the site of action (e.g., liver). The true value of fu measurements lies in how they can be applied to interpret the corresponding in vivo PK and PD data. Typical serum half-lives for GalNAc siRNA range from 2 to 8 hours; in contrast, target mRNA knockdown can last from weeks to months (Nair et al., 2017). Such rapid clearance from blood would suggest that a majority of the observed 85%–95% siRNA-X bound to proteins at equilibrium is only transiently bound with rapid dissociation rates (koff). Consequently, if siRNA-X exhibits high affinity for any plasma proteins at all, these hypothetical proteins could only exist at low concentrations (<<37 nM was the lowest PPB concentration tested). An important implication of this scenario is that tightly bound siRNA-X could compete with endogenous ligands or otherwise interfere with physiologic processes of low-abundance plasma proteins.
As the GalNAc-siRNA chemistry repertoire continues to evolve, we advocate that the relationship between PPB and blood PK continue to be monitored, as changes in the identities of GalNAc-siRNA protein-binding partners and/or their affinities could lead to alterations in the distribution and exposure of the molecule, or adverse effects due to interference with endogenous processes. Moreover, understanding the blood distribution, including protein-binding partners, may be beneficial in defining strategies for extrahepatic delivery.
High-affinity ASO binding to specific plasma proteins (nanomolar; via fluorescence polarization) has directly impacted PD in knockout (α-2-macroglobulin) or knockdown (histidine-2-glycoprotein) mice (Shemesh et al., 2016; Gaus et al., 2018). In both cases, protein removal from circulation resulted in a 2-fold ASO activity increase, suggesting protein binding can modulate shunting to unproductive pathways. Although outside the scope of this paper, this experimental approach will aid understanding of the impact of protein binding on siRNA PD.
Given that GalNAc siRNA is delivered to the liver via rapid ASGPR uptake, understanding siRNA-protein interactions at the site of action may aid understanding of the long duration of response. Our findings indicate siRNA is highly bound in the liver at equilibrium. If the binding turns out to be high-affinity, this could confirm the existence of a protein-bound “depot”—with gradual release of siRNA to RISC. Other prevailing theories suggest that the “depot” is a subcellular organelle like the endosome (Dominska and Dykxhoorn, 2010; Juliano and Carver, 2015) or a consequence of RISC-mediated RNA interference being a catalytic process with a long-lived Ago2-siRNA or Ago2-antisense complex (Okamura et al., 2004; Wang et al., 2009; Nakanishi, 2016). Consequently, the contribution of liver protein binding to GalNAc siRNA remains an open question.
Toward siRNA PPB Engineering.
Aligned with other published oligonucleotide data, our structure-activity relationship data suggest that siRNA PPB is “tunable” (Cossum et al., 1993; Braasch et al., 2004; Watanabe et al., 2006) (Fig. 4). In our limited study looking at the effect of various common chemical modifications, we established that fu,plasma is manipulatable from 0.01 to 0.21 fu at minimum. Whether binding modulation alters pharmacologic outcome remains to be seen. Modulation strategies might include minimizing binding to toxicity-related proteins, reducing drug-drug interaction liabilities, or targeting binding to specific proteins to facilitate siRNA delivery. For example, diacyl-conjugated siRNA displaying albumin binding demonstrated an increased circulation half-life compared with nonconjugated siRNA (Sarett et al., 2017). More generally, therapeutics directly conjugated to albumin or IgG-containing moieties have prolonged circulatory half-lives due to engagement with the recycling neonatal Fc receptor and reduced kidney filtration (Robbie et al., 2013; Larsen et al., 2018). Other siRNA features currently under investigation that could be exploited for protein-binding modulation include linker chemistry and conjugation to lipophilic molecules, peptides, monoclonal antibodies, or siRNA oligomers (Smith and Nikonowicz, 2000; Khan et al., 2016; Gandioso et al., 2017; Tushir-Singh, 2017).
Developing a standardized assessment of what makes a specific siRNA-protein interaction “meaningful” and characterizing protein-siRNA interactions in terms of specificity and affinity remains an active area of research. While we demonstrated that surfaced-based binding assays have utility in qualitatively identifying siRNA-binding partners (Fig. 5), we also observed significant orientation effects that are likely steric-driven (Supplemental Figs. 4 and 5). In the future, to better rank-order molecules by binding affinity, solution-based equilibrium measurements such as fluorescence polarization [recently applied to ASOs (Gaus et al., 2018)] or kinetic exclusion assays are recommended. To identify novel binders, rather than use a bottom-up approach such as the one performed here using purified proteins of interest, siRNA-protein pull-down combined with mass spectrometry proteomics will provide a nonbiased, comprehensive assessment of the siRNA-protein-binding landscape. To delineate complex relationships between siRNA structure, protein binding, and pharmacological effect, additional studies addressing variation in sequence, chemical modification, modification pattern, and conjugation ligands are needed.
siRNA-Protein Interactions: Changing the Binding Paradigm in a Therapeutic Context.
siRNA-protein interactions depend upon numerous factors, including protein structure, siRNA sequence and chemical modifications, kinetics, and concentration. In biologic matrices, additional considerations apply, including competition with other proteins for siRNA binding and competition with other oligonucleotides for protein binding. Affinities between chemically modified therapeutic oligonucleotides and specific proteins range from low nM to >500 μM (Gaus et al., 2018). Due to these complexities, siRNA-protein interactions are not well understood, and they cannot currently be anticipated a priori. To advance siRNA therapeutics, a paradigm shift in experimental design and interpretation is needed.
Rather than conforming to small-molecule-like “lock and key” or “induced fit” principles (Koshland, 1995) or protein-protein interactions, where a certain threshold of specificity and stability is required to achieve meaningful binding (Vishwanath et al., 2017), siRNA-protein interactions are governed by multiple weak complementary forces. These forces are effectively enhanced by the high surface area and high surface area-to-density ratio of siRNA relative to other therapeutic modalities. They include electrostatic and hydrophobic interactions, hydrogen bonding, and base stacking (Luscombe et al., 2001; Jayaram and Jain, 2004; Tolstorukov et al., 2004; Koh et al., 2011). The consequences are complex binding events arising from a convolution of association and dissociation rates, reflecting a distribution of local affinities driven by chemical modification pattern, GalNAc or other conjugate, 5′ phosphorylation state, or 3′ base identity, blurring boundaries of how we think about interaction specificity (Jankowsky and Harris, 2015). Thus, to advance understanding of siRNA-protein interactions in a therapeutic setting, establishment of a new metric of what constitutes a “relevant” binding event in the context of PK-PD analysis is required.
A central rationale guiding us in this work has been to address the following question: Does PPB matter for therapeutic siRNA? In establishing an fu assay to measure siRNA PPB and liver protein binding, and in developing an siRNA-protein interaction screening platform, we have established a bioanalytical toolkit to build knowledge in this understudied domain. In the future, the in vitro techniques described here can aid in vivo PK-PD data interpretation for this emerging modality and guide design of the next generation of siRNA therapeutics.
Acknowledgments
Thank you to Christina Shen, Ben Jiang, Yun Ling, Zhican Wang, Justin Murray, Babak Basiri, and Fang Xie for their work supporting aspects of this project.
Authorship Contributions
Participated in research design: Humphreys, Rock, Lade, Thayer, Smith.
Conducted experiments: Humphreys, Lade, Basiri, Hao, Huang.
Contributed new reagents or analytic tools: Wu, Sham, Thayer, Basiri, Hao, Huang.
Performed data analysis: Humphreys, Lade.
Wrote or contributed to the writing of the manuscript: Humphreys, Thayer, Lade, Rock, Smith.
Footnotes
- Received February 22, 2019.
- Accepted May 6, 2019.
All authors are employees and stock holders of Amgen, Inc.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ASGPR
- asialoglycoprotein receptor
- ASO
- antisense oligonucleotide
- BLI
- biolayer interferometry
- bn-siRNA
- biotinylated siRNA
- CHAPS
- 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate
- dsRNA
- double-stranded RNA
- 2′-F
- 2′deoxy 2′-fluoro
- FDA
- Food and Drug Administration
- GalNAc
- N-acetylgalactosamine
- MW
- molecular weight
- MWCO
- molecular-weightcutoff
- 2′-OMe
- 2′O-methyl
- pAb
- polyclonal antibody
- PBST
- PBS with Tween-20
- PK-PD
- pharmacokinetic-pharmacodynamic
- PPB
- plasma protein binding
- PS
- phosphorothioate
- RISC
- RNA-induced silencing complex
- siRNA
- small interfering RNA
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}