


Abstract
Cytochrome P4503A4 (CYP3A4) is the principal drug-metabolizing enzyme in human liver. Drug-drug interactions (DDIs) caused by induction of CYP3A4 can result in decreased exposure to coadministered drugs, with potential loss of efficacy. Immortalized hepatocytes (Fa2N-4 cells) have been proposed as a tool to identify CYP3A4 inducers. The purpose of the current studies was to characterize the effect of known inducers on CYP3A4 in Fa2N-4 cells, and to determine whether these in vitro data could reliably project the magnitude of DDIs caused by induction. Twenty-four compounds were chosen for these studies, based on previously published data using primary human hepatocytes. Eighteen compounds had been shown to be positive for induction, and six compounds had been shown to be negative for induction. In Fa2N-4 cells, all 18 positive controls produced greater than 2-fold maximal CYP3A4 induction, and all 6 negative controls produced less than 1.5-fold maximal CYP3A4 induction. Subsequent studies were conducted to determine the relationship between in vitro induction data and in vivo induction response. The approach was to relate in vitro induction data (Emax and EC50 values) with efficacious free plasma concentrations to calculate a relative induction score. This score was then correlated with decreases in area under the plasma concentration versus time curve values for coadministered CYP3A4 object drugs (midazolam or ethinylestradiol) from previously published clinical DDI studies. Excellent correlations (r2 values >0.92) were obtained, suggesting that Fa2N-4 cells can be used for identification of inducers as well as prediction of the magnitude of clinical DDIs.
Cytochrome P4503A4 (CYP3A4) is the major drug-metabolizing enzyme in human liver and is responsible for the clearance of many commonly used drugs, including benzodiazepines, statins, calcium channel blockers, and HIV protease inhibitors. Certain drugs can modulate the level of CYP3A4 activity, thereby causing changes in clearance of coadministered drugs that are CYP3A4 substrates. Levels of CYP3A4 activity can be decreased by inhibition of enzyme activity, or increased by induction of new protein synthesis. Changes in CYP3A4 activity, either through inhibition or induction, can result in potentially serious drug-drug interactions (DDIs). Whereas assays for evaluating inhibition of CYP3A4 are routine and the relationship between in vitro data and in vivo effects relatively well understood (Bjornsson et al., 2003), assays for evaluating induction are less well characterized and the relationships between in vitro and in vivo data less clear.
Induction of CYP3A4 is thought to occur primarily through transcriptional activation of the gene. The 5′-regulatory region of the CYP3A4 gene contains elements that bind various transcription factors that can up- or down-regulate transcription. One such transcription factor is the pregnane X receptor (PXR). PXR is a ligand-activated transcription factor that is activated by a variety of drugs and endogenous compounds to increase transcription of CYP3A4 as well as other drug-metabolizing enzymes and transporters (Kliewer et al., 2002). Most drugs that induce levels of CYP3A4 are thought to do so primarily via PXR activation, e.g., rifampicin, phenytoin, hyperforin, and clotrimazole (Lehmann et al., 1998).
There are several in vitro tools for assessment of CYP3A4 induction. To this point, primary human hepatocyte cultures have been considered the gold standard for in vitro induction assessment (Madan et al., 2003). Primary hepatocytes are typically treated with test compound for 2 or 3 days; then, CYP3A4 levels are compared with those in vehicle-treated cells. CYP3A4 expression can be evaluated by measuring enzymatic activity with a selective probe substrate or by measuring CYP3A4 mRNA. Primary hepatocytes are considered a reliable way to assess induction; however, ability to attain data can be limited by quality and availability of human hepatocytes. Also, there is considerable interindividual variability in induction responses in primary hepatocytes, with some preparations showing very little response to inducers, and others showing a high degree of response (Madan et al., 2003). A weak inducer could be missed if hepatocytes from “low responders” are used. Also, this system is not amenable to higher-throughput applications and, as such, does not meet the needs of early drug discovery. In contrast, PXR-based reporter assays have provided a means for high-throughput induction assessment. These assays typically involve cultured cell lines that have been transfected with both an expression vector for PXR and a reporter gene construct containing a responsive element for PXR (Goodwin et al., 1999). Induction potential is then measured by the ability of a test compound to activate transcription of the reporter gene. These systems generally have a wide dynamic range and are good indicators of a test compound's ability to activate PXR. However, there are several drawbacks to the use of PXR-based reporter assays. They are typically run in tumorigenic cell lines, which might or might not have all the coregulatory factors that are required for full transcriptional activation. Also, reporter gene assays do not measure native gene expression or native enzyme activity.
Recently, Mills et al. (2004) described the initial characterization of a nontumorigenic immortalized human hepatocyte line, Fa2N-4, and demonstrated the inducibility of CYP3A4 in response to prototypical inducers. The Fa2N-4 cell line is derived from immortalization of primary human hepatocytes and maintains expression and inducibility of various drug-metabolizing enzymes and transporters, as well as morphological characteristics of primary hepatocytes (Xenotech, 2003). However, in contrast to primary hepatocytes, the immortalized hepatocytes are easily maintained and propagated in culture, are amenable to higher-throughput applications, and provide a system with less intrinsic variability. In addition, Fa2N-4 cells do not require transfection of PXR or reporter genes, and induction assessment can be done using either mRNA levels or native enzyme activity. In addition to CYP3A4, other PXR-regulated genes (CYP2C9, MDR1) are induced in response to prototypical inducers (Mills et al., 2004).
The purpose of these studies is to further characterize CYP3A4 induction in Fa2N-4 cells, using a series of positive and negative controls. In addition, a method is proposed whereby potency and efficacy of induction in Fa2N-4 cells can be used to predict the magnitude of induction observed in clinical studies.
Materials and Methods
Chemicals. β-Naphthoflavone, carbamazepine, dexamethasone, methotrexate, methylprednisolone, mifepristone, nifedipine, phenobarbital, phenytoin, pioglitazone, probenecid, reserpine, rifampicin, roxithromycin, and sulfinpyrazone were purchased from Sigma-Aldrich (St. Louis, MO). Clarithromycin, clotrimazole, efavirenz, rifabutin, rifapentine, ritonavir, and rosiglitazone were purchased from Sequoia Research Products (Oxford, UK). Troglitazone was purchased from TRC Research Chemicals (Toronto, ON, Canada). Avasimibe and compound A [6-methoxy-1-methyl-1-trifluoromethyl-isochroman-7-ylmethyl)-(2-phenyl-piperidin-3-yl)-amine] were produced at Pfizer Global Research and Development (Groton, CT).
Cell Culture. The Fa2N-4 cell line of immortalized hepatocytes was generated in collaboration with MultiCell Technologies (Warwick, RI), as described previously (Mills et al., 2004). Cells were grown in Biocoat Type I collagen-coated flasks (BD Biosciences, Bedford, MA) in a humidified 37°C incubator (5% CO2) in serum-free Multi-Function Enhancing (MFE) medium (Xenotech LLC, Lenexa, KS).
Induction Experiment. Cells were maintained in collagen-coated flasks until 2 to 3 days postconfluence. Medium was removed, and cells were trypsinized and resuspended in MFE medium containing 10% fetal bovine serum (Invitrogen, Carlsbad, CA), at a concentration of approximately 750,000 cells per ml. One hundred microliters of cell solution was added to each well of 96-well collagen-coated plates (75,000 cells/well). After cell attachment (approximately 3 h), serum-containing medium was replaced with serum-free MFE medium. Cells were incubated for 48 h before treating with test compounds. This adaptation period was required for the cells to exhibit robust induction response. Stock solutions of test compounds (1000×) were prepared in DMSO and diluted 1000-fold with serum-free medium for cell treatment (0.1% DMSO final). After 24 h of treatment with test compounds or vehicle control, medium was removed and fresh medium containing test compounds was added, and cells were incubated for an additional 24 h. At the end of the incubation period, medium was removed and cells were washed twice with saline. Cells were lysed and mRNA was harvested as described below.
Rifampicin (1–50 μM) was tested with each plate of compounds as a positive control. The experiment was considered acceptable if the maximal induction response with rifampicin was greater than 8-fold. This degree of response is needed to ensure adequate dynamic range to detect weak inducers. Test compounds were initially assayed at 1, 10, 30, and 50 μM. If no evidence of induction was observed at these concentrations, the compound was considered negative for induction. Some compounds showed a positive response with at least one of these initial concentrations, but a full dose-response curve was not achieved. In these cases, the range was extended to either higher or lower concentrations until either a maximal fold-induction was achieved, or further extension was limited by cell toxicity.
Cell Toxicity Assessment. Parallel sets of plated cells were treated and cell viability was measured using a WST1 Cell Proliferation Assay (Roche Diagnostics, Indianapolis, IN), which measures mitochondrial dehydrogenase activity as a marker for metabolically active cells. In brief, after the 48-h treatment period, WST1 reagent was added to cell culture medium (1 part reagent to 10 parts cell culture medium), mixed gently, and incubated at 37°C for 20 min. Plates were shaken vigorously on an orbital shaker for 1 min, and absorbance was measured at 440 nm. Wells containing culture medium, but no cells, were used as background (measured at 660 nm). Treatments were deemed toxic if there was a significant decrease in absorbance compared with vehicle treatment.
Quantification of CYP3A4 mRNA. Total RNA was extracted from cells using the RNeasy 96 kit according to instructions provided by the manufacturer (QIAGEN, Valencia, CA). RNA (100 ng) was analyzed using the Invader RNA assay reagent kits according to instructions provided by the manufacturer (Third Wave Technologies, Madison, WI), and as described previously (Mills et al., 2004).
Statistics and Curve-Fitting. Experiments were carried out in triplicate and each compound was tested on at least 2 different days. All statistical analyses were carried out using Microsoft Excel (Microsoft, Redmond, WA). All curve-fitting was carried out using SigmaPlot 8.0 (Systat Software, Inc., Chicago, IL). To determine EC50 and Emax values, concentration-response data were fit to a three-parameter sigmoid (Hill) function, according to the following equation: y = (Emax · xγ)/(EC50γ + xγ).
Results
Characterization of Fa2N-4 Cells Using Positive and Negative Controls. To assess the utility of the Fa2N-4 cell line to correctly identify inducers of CYP3A4, cells were treated with a variety of compounds that had previously been identified as positive or negative for CYP3A4 induction. The criterion for inclusion was that data from primary human hepatocyte experiments conclusively identified a compound as either positive or negative for induction of CYP3A4 using either enzyme activity or mRNA as an endpoint. Compounds used as negative controls were β-naphthoflavone, methotrexate, methylprednisolone, clarithromycin, roxithromycin, and probenecid (Pichard et al., 1992; Ledirac et al., 2000; Luo et al., 2002; Madan et al., 2003). Compounds used as positive controls were mifepristone, troglitazone, carbamazepine, dexamethasone, efavirenz, rosiglitazone, clotrimazole, avasimibe, ritonavir, phenytoin, pioglitazone, phenobarbital, reserpine, rifabutin, sulfinpyrazone, nifedipine, rifapentine, and rifampicin (Kocarek et al., 1995; Li et al., 1997; Drocourt et al., 2001; Luo et al., 2002; Madan et al., 2003; Sahi et al., 2003a,b; Hariparsad et al., 2004). Fa2N-4 cells were treated for 2 days with varying concentrations of test compounds and CYP3A4 mRNA was measured. CYP3A4 mRNA levels were normalized to the vehicle control, and expressed as fold-induction. Maximal fold-induction values for the 24 test compounds and the vehicle control are shown in Fig. 1. Note that the concentrations required for maximal fold-induction varied from 0.1 μM for mifepristone to 1000 μM for phenobarbital. Maximal fold-induction produced with compounds identified as negative controls was less than 1.5-fold, and the maximal fold-induction produced with positive controls was greater than 2-fold. No false positives or false negatives were observed.
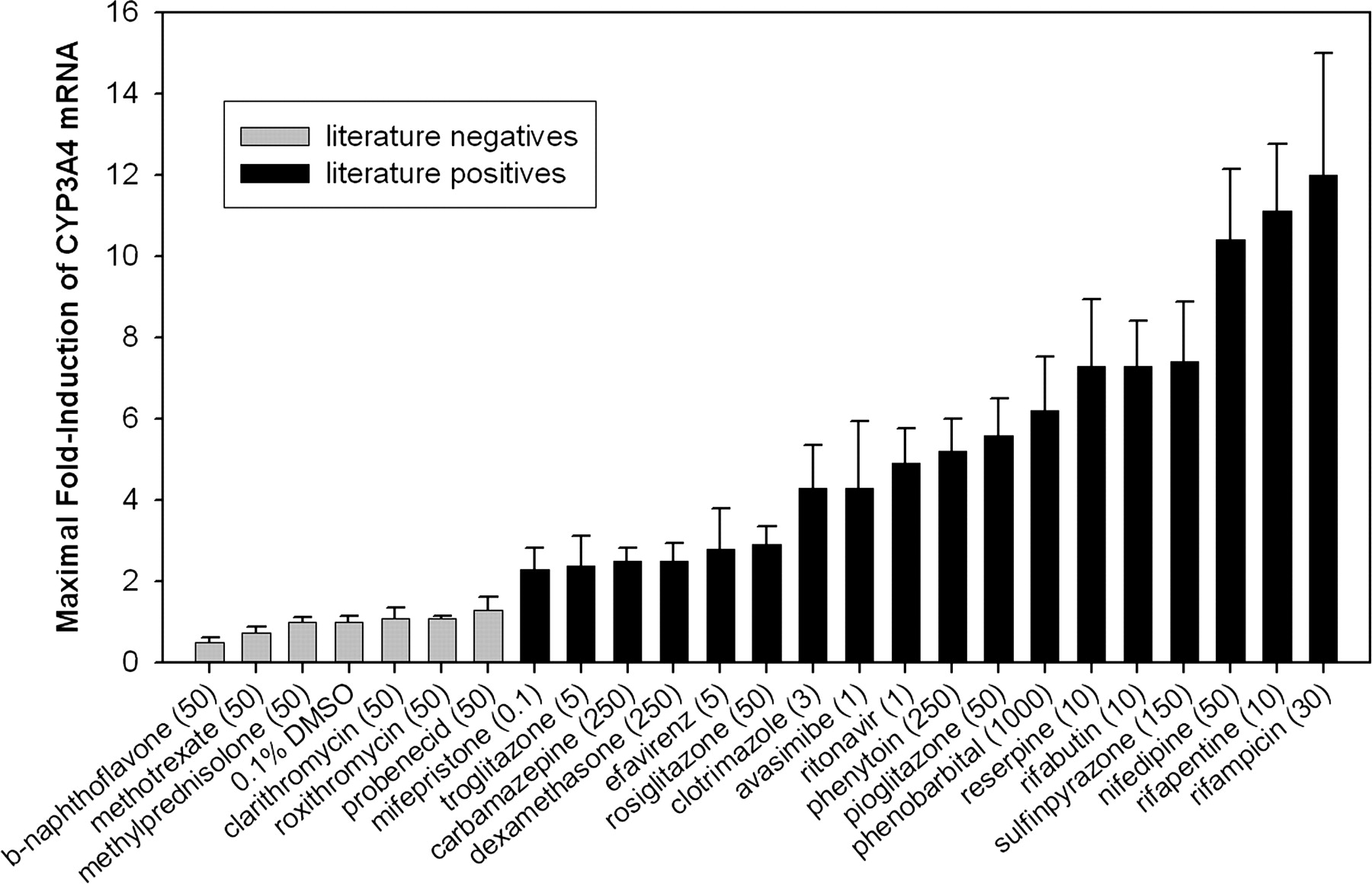
Maximal fold-induction of CYP3A4 in Fa2N-4 cells treated with various inducers and noninducers. Gray bars indicate compounds previously identified as negative for CYP3A4 induction in primary hepatocytes, and black bars indicate compounds previously identified as positive for induction of CYP3A4 in primary hepatocytes. Cells were treated for 2 days with compounds in 0.1% DMSO, followed by cell harvest, RNA isolation, and measurement of CYP3A4 mRNA. Data were normalized to vehicle control (0.1% DMSO). Concentrations shown (μM) are those that produced maximal fold-induction of CYP3A4 for a given compound.
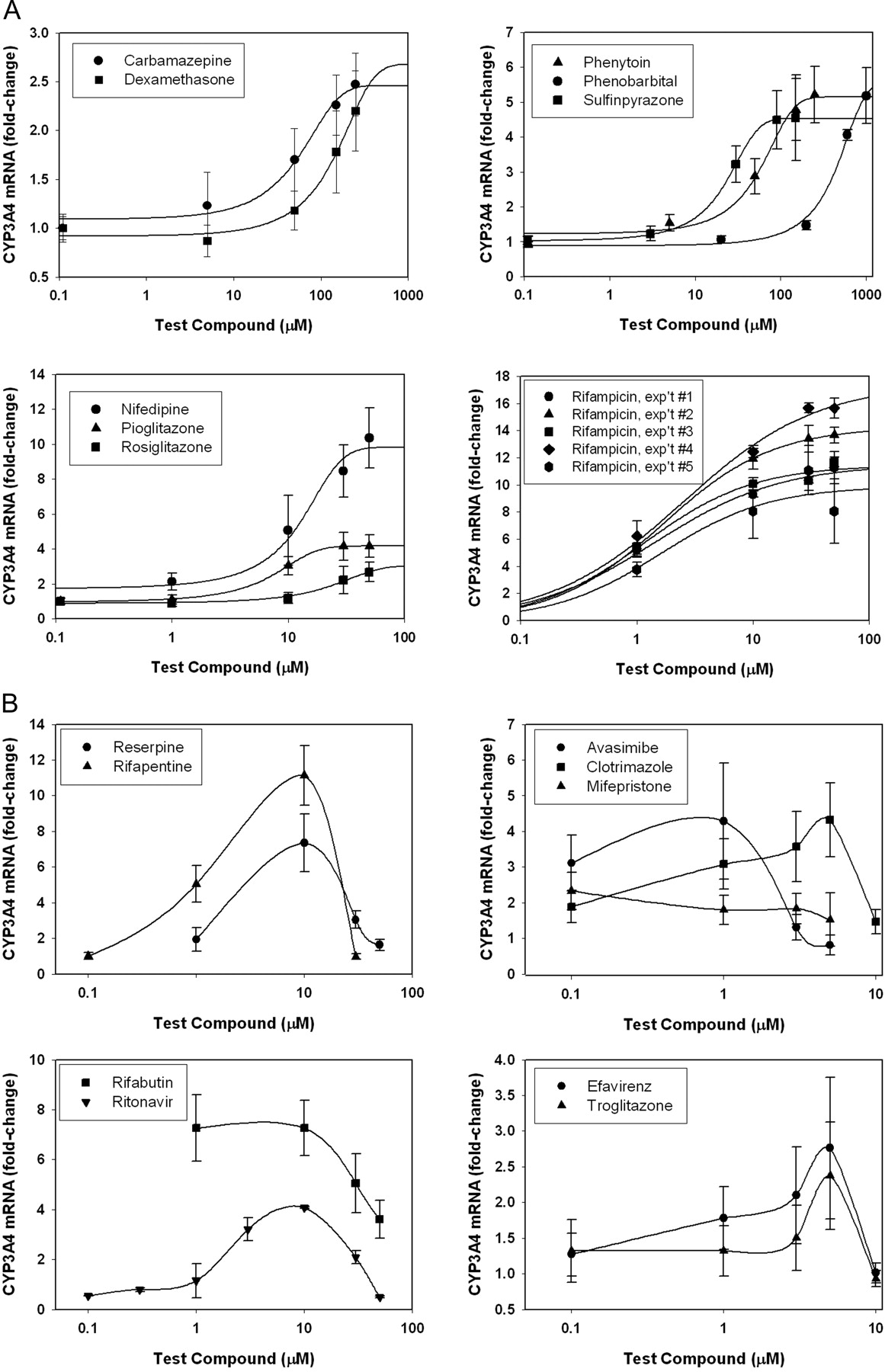
Concentration-Response for Inducers in Fa2N-4 Cells. Since induction of CYP3A4 is a result of receptor-mediated processes, it should be concentration-dependent and saturable. Concentration-response relationships for the current set of compounds fell into one of three categories. Nine compounds, carbamazepine, dexamethasone, nifedipine, phenobarbital, phenytoin, pioglitazone, rifampicin, rosiglitazone, and sulfinpyrazone, showed standard dose-response curves with fold-induction increasing with dose to a point of saturation. These concentration-response data were fitted to a sigmoidal curve, and EC50 and Emax parameters were determined (Fig. 2A; see Table 2). Nine compounds showed atypical dose-response curves, in that fold-induction increased with dose to a point, then decreased at higher concentrations (Fig. 2B). In five of these cases (avasimibe, clotrimazole, efavirenz, mifepristone, and troglitazone), the bell-shaped dose-response could be attributed to toxicity (Fig. 2B, right graphs). However, in the other four cases, the atypical dose-response was not associated with toxicity, as assessed by the mitochondrial dehydrogenase assay (reserpine, rifabutin, rifapentine, and ritonavir) (Fig. 2B, left graphs). The reason for this bell-shaped dose-response is not known, but has been observed in other studies of induction (Reinach et al., 1999; Raucy, 2003; Xenotech, 2003).
List of compounds with corresponding in vitro induction data and published clinical data used to generate RIS
Assay Reproducibility. To assess the interassay variability in CYP3A4 induction in Fa2N-4 cells, concentration-response experiments with rifampicin were included each time the assay was run. Intra-assay variability (variability among replicate wells) was generally less than 25% (Table 1). Interassay variability in maximal fold-induction and EC50 values was also assessed (Table 1). Using mRNA as an endpoint, maximal fold-induction values ranged from 9.7- to 19-fold with an average of 13-fold and standard deviation of 3.7-fold (coefficient of variation of 28%). EC50 values ranged from 1.3 μM to 3.1 μM, with an average of 1.9 μM and standard deviation of 0.70 (coefficient of variation of 37%). Concentration-response curves for the additional 23 test compounds were carried out at least twice. Intra-assay variability was generally less than 25%. Interassay variability ranged from 3% to 53%, with a mean (±S.D.) interassay variability of 30 ± 16% (data not shown).
Intraday (boldface) and interday (italicized) assay variability from five rifampicin concentration-response experiments conducted in triplicate
The stability of induction response with passage number has been shown in previous publications, and results indicated that fold-induction is stable at least up to passage number 47 (Xenotech, 2003; Mills et al., 2004). In the current studies, the passage number of the cells used was between passages 22 and 35.
Modeling the Relationship between Induction of CYP3A4 in Fa2N-4 Cells and Clinical Induction. To establish a correlation between data generated in vitro and data observed in clinical studies, the potency and efficacy of inducers in Fa2N-4 cells were related to exposures achieved in vivo. Inducers were scored based on the relationship of the EC50 and Emax values in Fa2N-4 cells and the efficacious concentration observed in vivo, according to the following equation:  The value Ceff, free is the published plasma concentration of inducer achieved after a standard therapeutic dose, corrected for protein binding. This concentration was chosen to approximate the concentration of drug available for binding to PXR and effecting induction of CYP3A4. The RIS values for various inducers using the above equation are shown in Table 2. The published clinical changes in either midazolam or ethinylestradiol AUC upon coadministration with inducers are also shown. Note that the relationship between RIS and clinical DDI was evaluated only for inducers. Compounds that are capable of simultaneous inhibition and induction of CYP3A4 at clinically relevant concentrations (e.g., ritonavir and efavirenz) were excluded from this analysis.
The value Ceff, free is the published plasma concentration of inducer achieved after a standard therapeutic dose, corrected for protein binding. This concentration was chosen to approximate the concentration of drug available for binding to PXR and effecting induction of CYP3A4. The RIS values for various inducers using the above equation are shown in Table 2. The published clinical changes in either midazolam or ethinylestradiol AUC upon coadministration with inducers are also shown. Note that the relationship between RIS and clinical DDI was evaluated only for inducers. Compounds that are capable of simultaneous inhibition and induction of CYP3A4 at clinically relevant concentrations (e.g., ritonavir and efavirenz) were excluded from this analysis.
The correlation between RIS and clinical DDI was assessed by plotting the RIS versus the percentage decrease in AUC of a coadministered CYP3A substrate, data obtained from published clinical DDI trials. Figure 3 shows the correlation plots for the widely used CYP3A probe substrate, midazolam (Fig. 3A) and ethinylestradiol (Fig. 3B). The relationship between RIS and percentage decrease in midazolam AUC fits a Hill function with an Emax of 97%. A positive correlation is also apparent between RIS and percentage decrease in ethinylestradiol AUC, and achieves an Emax of 66%. Excellent correlations between the RIS and clinical DDI were obtained using either midazolam or ethinylestradiol clinical data (r2 ≥ 0.92).
Discussion
Since induction of CYP3A4 can result in significant DDIs, it is important to assess a new drug candidate's ability to cause CYP3A4 induction. In the current work, we demonstrate that the Fa2N-4 line of immortalized hepatocytes not only can be used to identify inducers, but is a robust and reproducible enough system to be used to predict extent of clinical induction.
Induction of CYP3A4, CYP2C9, and MDR1 had previously been demonstrated in Fa2N-4 cells in response to rifampicin, phenobarbital, or dexamethasone (Mills et al., 2004). The current work indicates that these immortalized hepatocytes can appropriately identify a larger diversity of inducers, including those compounds that are relatively weak inducers in vitro. Since the 18 positive controls tested had maximal fold-induction values of greater than 2, and the 6 negative controls had maximal fold-induction values of less than 1.5, it seems appropriate when screening compounds with unknown induction potential to consider a compound in vitro positive if it produces a maximal fold-induction of greater than or equal to 2. Using this criterion, even the weak in vitro inducers such as carbamazepine and dexamethasone would be correctly identified.

Concentration-response curves for compounds producing a positive response in Fa2N-4 cells. Carbamazepine, dexamethasone, phenytoin, phenobarbital, sulfinpyrazone, nifedipine, pioglitazone, rosiglitazone, and rifampicin exhibited typical concentration-response relationships that could be fit using a sigmoidal curve-fitting program [symbols denote observed data (±S.D.), whereas lines denote curve fit] (A). Reserpine, rifabutin, rifapentine, and ritonavir exhibited bell-shaped concentration-response relationships (B, left graphs). Attainment of full concentration-response relationships for avasimibe, clotrimazole, efavirenz, mifepristone, and troglitazone was limited by cell toxicity (B, right graphs).
To assess the utility of the Fa2N-4 immortalized hepatocytes for developing an in vitro-in vivo correlation for induction, we first examined the robustness and reproducibility of the data. The interassay variability observed with Fa2N-4 cells (21–37%, depending on the parameter) was considerably less than that reported for primary human hepatocyte cultures. For example, Madan et al. (2003) assessed induction response across primary hepatocytes from 62 individuals using enzyme activity as an endpoint. Fold-induction for 50 μM rifampicin ranged from 1.5- to 145-fold, with an average of 9.7-fold and standard deviation of 19-fold (coefficient of variation of 200%) (Madan et al., 2003; Xenotech, 2003). The relatively smaller variability in response in Fa2N-4 cells suggested that this system could provide a means to develop in vitro-in vivo correlations, and clinical predictions.
El-Sankary et al. (2001) demonstrated the utility of using in vitro potency and efficacy to rank order inducers. These investigators generated Emax and EC50 values for several inducers, using HepG2 cells transfected with glucocorticoid receptor, PXR, and a CYP3A4 reporter gene. Ranking of compounds was achieved by dividing maximal fold-induction by EC50. We extended this approach and demonstrated that by incorporating free plasma concentrations into a simple Emax model to generate the RIS, one could attain an excellent in vitro-in vivo correlation for CYP3A4 induction. Ideally, the RIS equation would incorporate the intracellular free concentration of drug in hepatocytes, which should reflect the amount of drug available for binding to PXR. However, concentration of drug within hepatocytes is not readily measurable, and as a surrogate, the published free plasma concentration at Cmax was used. Using free Cmax, the in vitro-in vivo correlation, with either midazolam or ethinylestradiol as probe substrate, was excellent (r2 ≥ 0.92). The in vitro-in vivo correlation was also attempted using total plasma concentration, but the correlation coefficient was low (r2 < 0.55; not shown). Note that treatment of Fa2N-4 cells with test compounds was done under serum-free conditions, and therefore, it is not surprising that free plasma concentrations produced a better correlation. The incorporation of free plasma concentrations is critical to appropriately identifying induction risk. For example, nifedipine and pioglitazone are relatively potent and efficacious inducers in vitro, and might be predicted to be strong inducers in vivo. However, by factoring in the low plasma concentrations and free fractions of these two drugs, the actual RIS is very low and, indeed, these compounds show very little to no clinical induction (Horsmans et al., 1991; Sachse et al., 1998). Conversely, carbamazepine is a very weak inducer in vitro, with a high EC50 and low Emax. However, when the high plasma concentration and free fractions of carbamazepine are considered, the RIS is at least an order of magnitude higher than those of pioglitazone and nifedipine, and this correlates with the clinical DDIs observed with this compound (Crawford et al., 1990; Backman et al., 1996b).
It is interesting to note that the EC50 values from the in vitro experiments were in many cases higher than the published plasma concentrations for a given drug, yet in several cases, the drug still produced clinically measurable induction. This finding suggests that to appropriately assess clinical induction risk, the ratio of the plasma concentration to EC50 is not enough. This is in contrast to the risk assessment for inhibition, which typically can be predicted from the ratio of the in vivo inhibitor concentrations and the in vitro Ki for inhibition. It appears that for induction, it is important to include not only the in vivo concentration and in vitro EC50, but also the maximal fold-induction achieved in vitro. The RIS equation incorporates these three parameters.

Relationship between RIS and clinical effects on coadministered midazolam (A) and ethinylestradiol (B). Compound abbreviations: CBM, carbamazepine; CP A, 6-methoxy-1-methyl-1-trifluoromethyl-isochroman-7-ylmethyl)-(2-phenylpiperidin-3-yl)-amine; NIF, nifedipine; PB, phenobarbital; PHT, phenytoin; PIO, pioglitazone; RIF, rifampicin; RSG, rosiglitazone; TRG, troglitazone. Data points were fitted to a three-parameter Hill function, y = (Emax · xγ)/(RIS50γ + xγ). For graph A, Emax = 97.0, γ = 1.24, and RIS50 = 0.031. For graph B, Emax = 65.6, γ = 0.997, and RIS50 = 0.0924.
The relationship between RIS and percentage decrease in AUC of coadministered drug can be used as a calibration curve for clinical induction, and hence it is critical when evaluating induction response for new compounds that the assay conditions are consistent with those in which this calibration curve was generated. In other words, under experimental conditions whereby rifampicin exhibits a greater fold-induction, then a test compound would also tend to have a greater fold-induction. Therefore, it is recommended that to use the in vitro-in vivo correlation shown here, the rifampicin response in the same experiment should be within the range of rifampicin responses observed in the calibration experiments; that is, Emax between 9- and 19-fold and EC50 between 1.3 and 3.1 μM. Alternatively, an abbreviated calibration curve could be generated for each experiment. For example, for each set of test compounds, rosiglitazone or nifedipine could be included as a low standard, pioglitazone or avasimibe as a middle standard, and rifampicin as a high standard, and a RIS/DDI relationship generated in each run.
One caveat with predicting clinical induction from in vitro data is the potential effect of metabolites. If metabolites are also inducers, and if exposure to these metabolites is significant, the RIS could under-predict clinical induction. In fact, we have encountered an example of a compound that had a major circulating metabolite that was also an inducer. If the RIS had been calculated with the parent compound, no clinical DDI would have been predicted. However, when the RIS of the metabolite was added in, the data correlated well with the actual induction observed in clinical DDI studies (unpublished observations).
Another caveat to the use of the RIS concerns compounds that are both inducers and inhibitors of CYP3A4 at clinically relevant concentrations (e.g., ritonavir and efavirenz). These compounds would not be accurately predicted by factoring in only the induction component. Note that some compounds might be competitive inhibitors of CYP3A4, but have Ki values that are significantly higher than therapeutic concentrations. In these cases, inhibition would not be expected to be clinically relevant, and the RIS could be used. Pioglitazone and troglitazone are examples of such compounds. The Ki values for CYP3A4 inhibition are approximately 12 μM and 1 μM for pioglitazone and troglitazone, respectively (Sahi et al., 2003a). These values are much higher than the unbound in vivo concentrations, and therefore, these compounds would not be expected to produce inhibition in vivo. In these cases, the RIS could be used to predict effects due to induction, without the need to incorporate an inhibition term. Work is under way to evaluate the utility of Fa2N-4 cell assay to accurately predict DDIs for compounds that are simultaneous inducers and inhibitors of CYP3A4 at clinically relevant concentrations.
It is interesting to note that the most potent clinical inducer published (rifampicin) produced a 97% decrease in midazolam AUC, but only a 67% decrease in ethinylestradiol AUC. Some substrates, such as midazolam, are cleared almost exclusively by CYP3A metabolism and, therefore, might be more susceptible to CYP3A induction than other substrates that are cleared via multiple pathways. Ethinylestradiol is cleared by a combination of CYP3A4 metabolism, sulfation, and glucuronidation (Maggs et al., 1983; Guengerich, 1990). It would also be expected that compounds that are cleared by both intestinal and hepatic CYP3A would be more susceptible to the effects of inducers than those cleared by hepatic enzymes only. An example of this is the difference in clinical DDIs observed with midazolam depending on whether it is administered orally or intravenously (Gorski et al., 2003). Since most DDI studies with midazolam are conducted using an oral route of administration, we chose to investigate the in vitro-in vivo correlation using oral midazolam data.
In conclusion, our findings indicate that Fa2N-4 immortalized hepatocytes can be used to identify a wide range of CYP3A4 inducers, including those compounds that are weak in vitro inducers. Moreover, potency and efficacy data from Fa2N-4 cells can be combined with plasma concentration data to generate a RIS that correlates with clinical DDI. The low interassay variability of the Fa2N-4 cells relative to primary hepatocytes makes this in vitro-in vivo correlation possible. Future studies will be directed at using these immortalized hepatocytes to develop clinical DDI prediction tools for additional drug-metabolizing enzymes.
Acknowledgments
We gratefully acknowledge the following Pfizer scientists for contributing data as well as scientific insights: Mike Banker, Puja Billis, Cuiping Chen, Angela Doran, John Gibbs, Jae Lee, Carol McRobie, Scott Obach, Dennis Scott, and Karthik Venkatakrishnan.
Footnotes
-
This work was presented in part at the Institute for Scientific Exchange Conferences on Drug-Drug Interactions (June 14–16, 2004, San Diego, CA; and June 15–17, 2005, Seattle, WA) and at the International Society for the Study of Xenobiotics (ISSX) National Meeting (October 23–27, 2005, Maui, HI).
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.010132.
-
ABBREVIATIONS: DDI, drug-drug interaction; AUC, area under the plasma concentration versus time curve; compound A, 6-methoxy-1-methyl-1-trifluoromethyl-isochroman-7-ylmethyl)-(2-phenyl-piperidin-3-yl)-amine; DMSO, dimethyl sulfoxide; MDR1, multidrug resistance protein 1; PXR, pregnane X receptor; RIS, relative induction score.
- Received March 10, 2006.
- Accepted June 30, 2006.
- The American Society for Pharmacology and Experimental Therapeutics
References
DMD articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|





{kind=link}
{kind=link}
{kind=link}