
Abstract
NLR inflammasomes, caspase 1 activation platforms critical for processing key pro-inflammatory cytokines, have been implicated in the development of nonalcoholic fatty liver disease (NAFLD). As the direct role of the NLRP3 inflammasome remains unclear, we tested effects of persistent NLRP3 activation as a contributor to NAFLD development and, in particular, as a modulator of progression from benign hepatic steatosis to steatohepatitis during diet-induced NAFLD. Gain of function tamoxifen-inducible Nlrp3 knock-in mice allowing for in vivo temporal control of NLRP3 activation and loss of function Nlrp3 knockout mice were placed on short-term choline-deficient amino acid-defined (CDAA) diet, to induce isolated hepatic steatosis or long-term CDAA exposure, to induce severe steatohepatitis and fibrosis, respectively. Expression of NLRP3 associated proteins was assessed in liver biopsies of a well-characterized group of patients with the full spectrum of NAFLD. Nlrp3 −/− mice were protected from long-term feeding CDAA-induced hepatomegaly, liver injury, and infiltration of activated macrophages. More importantly, Nlrp3 −/− mice showed marked protection from CDAA-induced liver fibrosis. After 4 weeks on CDAA diet, wild-type (WT) animals showed isolated hepatic steatosis while Nlrp3 knock-in mice showed severe liver inflammation, with increased infiltration of activated macrophages and early signs of liver fibrosis. In the liver samples of patients with NAFLD, inflammasome components were significantly increased in those patients with nonalcoholic steatohepatitis (NASH) when compared to those with non-NASH NAFLD with mRNA levels of pro-IL1 beta correlated to levels of COL1A1. Our study uncovers a crucial role for the NLRP3 inflammasome in the development of NAFLD. These findings may lead to novel therapeutic strategies aimed at halting the progression of hepatic steatosis to the more severe forms of this disease.
Key message
-
Mice with NLRP3 inflammasome loss of function are protected from diet-induced steatohepatitis.
-
NLRP3 inflammasome gain of function leads to early and severe onset of diet-induced steatohepatitis in mice.
-
Patients with severe NAFLD exhibit increased levels of NLRP3 inflammasome components and levels of pro-IL1β mRNA correlate with the expression of COL1A1.
Similar content being viewed by others


Introduction
Nonalcoholic fatty liver disease (NAFLD) is the hepatic manifestation of the metabolic syndrome and most abundant chronic liver disease in the Western world [1, 2]. It is predicted that nonalcoholic steatohepatitis (NASH), the most severe form of NAFLD will be the leading cause of liver transplantation in the USA by the year 2020 [3]. The prevalence of NAFLD ranges from 20 to 30 % in the general population and up to 75–100 % in obese individuals. It is estimated that about 10 to 25 % of patients with NASH may progress to cirrhosis [4, 5]. The clinical pathologic picture resembles that of alcohol-induced liver injury but occurs in patients who consume little or no alcohol [6]. Despite its high prevalence, the factors driving the progression of NAFLD to NASH remain poorly understood and no treatment has been proven effective [7].
The current, and most accepted, concept outlining the pathogenesis of NAFLD involves multiple “hits.” These hits are characterized by the occurrence of parallel events that involve a complex interaction and cross talk between environmental factors and host genetics [8, 9]. These traits may promote isolated steatosis (a process of fat accumulation in the liver), innate immune activation, inflammation, cell death, and progressive liver damage [10].
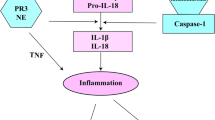
The NLRP3 inflammasome is a multi-protein complex that recognizes various pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) [11]. It is composed of the nucleotide-binding oligomerization domain (NOD) leucine-rich repeat containing receptors (NLR) containing pyrin domain 3 (NLRP3), adaptor proteins such as the apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC), and the serine protease caspase 1 (Casp1) [12, 13]. Upon sensing of the aforementioned patterns, Casp1 is cleaved and therefore activated further inducing interleukin-1β (IL-1β) maturation. Casp1 activation and IL-1 signaling have been implicated in the pathogenesis of alcoholic liver disease (ALD) and NAFLD, as well as liver fibrosis [14, 15]. Indeed, the importance of Casp1 activation and IL-1 signaling has been demonstrated in numerous diet-induced mouse models of ALD and NAFLD; these studies utilized various knockouts in the IL-1 pathway, all of which reduced the pathologic features of liver damage [16–18].
We have recently shown that constitutively global and to a lesser extent myeloid-specific NLRP3 inflammasome activation results in severe liver inflammation and activated fibrogenesis, while identifying hepatocyte pyroptotic cell death as a novel mechanism of NLRP3-mediated liver damage [19]. In the present study, we address the role of NLRP3 activation in the context of NAFLD development and hypothesize that it plays a central role in the progression to more severe stages of disease. We approached this hypothesis using loss-of-function and gain-of-function approaches using both Nlrp3 knockout mice and a novel knock-in mouse model allowing for temporal control of NLRP3 activation. To mimic the full spectrum of human disease progression from isolated hepatic steatosis to NASH and severe fibrosis in conjunction with weight gain in mice, we choose the choline-deficient amino acid-defined (CDAA) diet [20]. Time course experiments revealed that 16 weeks of this diet results in the development of NASH and fibrosis, whereas 4 weeks of diet results in early changes of isolated hepatic steatosis [20]. Moreover, translational studies on the relevance of the NLRP3 inflammasome in human NASH were conducted in a large and well-characterized cohort of patients with the full spectrum of disease.
Materials and methods
Mouse strains
The following mouse strains were used in this study: Nlrp3 −/−(provided by J. Bertin, E. Grant, A. Coyle, and Millennium Pharmaceuticals) [21]. Nlrp3 knock-in mice were generated as previously described, with an alanine 350 to valine (A350V) substitution and the presence of an intronic floxed neomycin resistance cassette, in which expression of the mutation does not occur unless the Nlrp3 mutants are first bred with mice expressing Cre recombinase [22]. Nlrp3 knock-in mice were bred to B6.Cg-Tg(Cre/Esr1)5Amc/J mice (obtained from Jackson Labs) to allow for mutant Nlrp3 expression in adult models after administration of tamoxifen [23]. University of California at San Diego Institutional Animal Care and Use Committee approved all protocols.
Mutant NLRP3 protein expression induction
Nlrp3 mutant and wild-type (WT) mice were injected i.p. with 50 mg/kg tamoxifen-free base (MP Biomedicals, Solon, OH) in 90 % sunflower seed oil from Helianthus annus (Sigma, St. Louis, MO) and 10 % ethanol daily for 4 days, followed by twice weekly injections as previously described [24].
Dietary isolated hepatic steatosis and NASH fibrosis induction
Mice were fed with CDAA diet or choline-supplemented amino acid-defined (CSAA) diet as control [25]. Long-term feeding of this diet (16 weeks) results in a phenotype that closely resembles human NASH: Mice become obese, dyslipidemic, and insulin-resistant and exhibit steatohepatitis and perisinusoidal/pericellular fibrosis, whereas short-term feeding (4 weeks) is associated with early disease characterized mainly by isolated hepatic steatosis [20]. Nlpr3 knockouts mice or WT mice were placed on the CDAA or CSAA control diet for 16 weeks, starting at 7 weeks of age. Tamoxifen-inducible Nlrp3 mutants were placed on CDAA diet or regular chow for 4 weeks and compared to WT mice fed with the same diets starting at 7 weeks of age.
NAFLD/NASH study population
To assess the role of the NLRP3 inflammasome in the development of NAFLD and NASH in humans, we analyzed messenger RNA (mRNA) levels of proteins related to the NLRP3 inflammasome in liver samples of 77 patients with a diagnosis of NASH or non-NASH NAFLD. These patients are part of a cohort of morbidly obese patients who underwent bariatric surgery at a German center for bariatric surgery. Liver samples were obtained while patients underwent surgery. This cohort covers the full spectrum of disease from isolated hepatic steatosis to NASH to advanced fibrosis and cirrhosis [26, 27]. Table 1 describes the characteristics of the patient population.
This study was approved by the ethics committee (Institutional Review Board) of the University Hospital Essen. Patients volunteering were informed about intraoperative risks and benefits of wedge liver biopsy and provided informed written consent.
Liver sample preparation
At the selected time interval, mice were anesthetized (ketamine 60 mg/kg plus xylazine 10 mg/kg intraperitoneally), the peritoneal and thoracic cavities opened, and blood samples were taken via cardiac puncture. Next, using phosphate-buffered saline (PBS), blood was flushed out of the liver via a catheter in the left main cardiac chamber. The liver was harvested, and the tissue was divided: (i) A representative section was fixed in 10 % formalin for 24 h and embedded in paraffin or directly frozen in embedding medium for frozen tissue sections; (ii) samples of 50 μg were placed in 500 μl of RNAlater® solution (Life Technologies, Carlsbad, CA, USA); and (iii) the remaining liver tissue was quickly frozen in liquid nitrogen and stored in −80 °C.
Histology, immunohistochemistry, and Sirius Red staining
Steatosis, inflammation, and ballooning were graded on the basis of the NAFLD activity score by an experienced pathologist (Dr. Bettina Papouchado) in routinely stained tissue sections [28]. Liver fibrosis was assessed using Sirius Red, whereby liver sections were incubated for 2 h at room temperature with an aqueous solution of saturated picric acid containing 0.1 % Fast Green FCF and 0.1 % Direct Red. Red stained collagen fibers were quantitated by digital image analysis (ImageJ, NIH, US). Immunohistochemistry staining for myeloperoxidase (Myeloperoxidase Ab-1, Thermo Scientific, Waltham, MA, USA) and F4/80 (a global marker for murine macrophages) (F4/80, AbD Serotec, Hercules, CA, USA) were performed in formalin-fixed, paraffin-embedded liver sections according to the manufacturer’s instruction. Immunohistochemistry for lymphocyte antigen 6 complex, locus C1 (Ly6c) (Abcam, Cambrigde, MA, USA) was performed on frozen liver sections. Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay was performed per manufacturer’s instructions (ApopTag® peroxidase in situ Apoptosis Detection kit, Millipore, Billerica, MA, USA).
Real-time PCR
Total RNA was isolated from liver tissue and analyzed as previously described [19]. The sequences of the primers used for quantitative PCR are given in Supplemental Table 1.
Immunoblot analysis
Immunoblot analysis were performed as previously described [19]. Anti-Casp1 p10, anti-IL-18, anti-desmin (all three from Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-IL-1β (Abcam, Cambridge, MA, USA), and anti-alpha smooth muscle actin (α-SMA; GeneTex, Irvine, CA, USA) antibodies were used in combination with appropriate peroxidase-conjugated secondary antibodies. Protein load was verified with an α-tubulin antibody (dilution 1:10,000) (Hybridomabank, University of Iowa) (kindly provided by M. Kaulich). Bands were visualized with the enhanced chemiluminescence reagent and digitized using a CCD camera (ChemiDoc®, Bio-Rad, Hercules, CA, USA). Expression intensity was quantified by ImageLab (Bio-Rad).
Liver function tests
Serum values of alanine aminotransferase (ALT) were measured in blood samples at the end of the feeding cycles according to the manufacturer’s instruction (Infinity™ ALT, Thermo Scientific, Waltham, MA, USA).
Unless stated otherwise, all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Statistical analyses
Analyses were performed with GraphPad (version 5.03, GraphPad Software Inc., CA, USA) and R (version 3.0.2; www.r-project.org). The significance level was set at α = 5 % for all comparisons. If data or log-transformation of data had approximately normal distributions, the interaction or main effects of genotype and diet were tested by ANOVA. Least-squared means were used for group comparisons. If data deviated from the normal distribution, nonparametric Mann–Whitney tests were used for group comparisons, or stratified Mann–Whitney tests were used for testing the effect of a single factor. For group comparisons, we limit this to three comparisons that are of particular interest. In the knockout experiment, we compared WT CDAA versus WT CSAA, Nlrp3 −/− CDAA versus Nlrp3 −/− CSAA, and Nlrp3 −/− CDAA versus WT CDAA. Similar comparisons were done for the knock-in experiment. Holm’s method was used for multiple group comparison adjustment for each measurement. Correlations between two variables were described by Spearman’s rank correlation coefficient (rho). Unless otherwise stated, data are expressed as mean ± SEM or as absolute number or percentage for categorical variables.
Results
Nlrp3 knockout mice are protected from CDAA-induced hepatomegaly and liver injury
Nlrp3 −/− and WT mice placed on CDAA diet or control diet for 16 weeks showed significant weight gain throughout the investigational period (Fig. 1). There was no significant interaction effect between the type of diet and genetic background on body weight after 16 weeks on diet. However, while CDAA diet significantly increased liver weights in WT mice compared with the WT mice fed with CSAA (p = 0.006), Nlrp3 −/− mice did not show a significant increase and were thus protected from CDAA-induced hepatomegaly. Moreover, serum ALT levels were significantly elevated in WT mice fed with CDAA in comparison to WT mice on CSAA control diet (p = 0.002). Nlrp3 −/− mice were protected from this CDAA-induced increase, as their ALT levels were not significantly different between diet types (Fig. 1). Liver steatosis was present in all groups. Quantification of liver inflammation showed an increase in the grade of inflammation in WT mice fed with CDAA compared to WT mice on CSAA diet (Fisher’s exact test p = 0.06). Nlrp3 −/− mice on CDAA showed a trend toward lower, albeit not significantly lower, grades of inflammation when compared to WT mice on CDAA diet. TUNEL staining was significantly increased in WT mice fed with CDAA diet when compared to WT mice on CSAA diet or Nlrp3 knockout mice on either diet (Fig. 1).

Nlrp3 −/− are protected from CDAA-induced hepatomegaly and liver damage. Nlrp3 −/− and WT mice weaned after 21 days were placed on normal chow (NC) diet for 4 weeks and thereafter placed either on CDAA or CSAA diet for an additional 16 weeks (a); all groups gained weight throughout the investigational period (b). Notably, Nlrp3 −/− mice were protected from CDAA-induced hepatomegaly (b) and liver damage—assessed by ALT serum levels (c). Analysis of liver histology revealed marked liver steatosis in all groups, with mice fed with CDAA showing larger droplets and higher variability in droplet size (d, e). WT mice fed with CDAA diet showed higher grades of inflammation when compared to WT on CSAA diet, and Nlrp3 −/− mice on CDAA diet showed a trend toward lower grades when compared to diet-matched WT mice (e). TUNEL analysis showed a significant increase in WT mice fed with CDAA diet when compared to WT mice on CSAA diet; this increase was not found in Nlrp3 knockout mice (d, f)
CDAA-induced inflammation is ameliorated in mice with Nlrp3 loss of function
Inflammation and neutrophilic infiltration are known elements in the development of NASH. We therefore assessed infiltration of myeloid cells and their regulatory chemokines in liver samples. Quantification of immunohistochemical staining for myeloperoxidase (MPO) revealed that CDAA diet significantly increased MPO-positive cells compared with CSAA diet in both Nlrp3 −/− and WT mice (p < 0.01 and p < 1e − 5, respectively) (Fig. 2). mRNA levels for chemokine (C-X-C motif) ligand 2 (CXCL2) were increased in WT mice on CDAA diet when compared to WT mice on CSAA diet and were significantly reduced in Nlrp3 −/− when compared to WT mice (p < 0.01 for both markers, Mann–Whitney test stratified by diet) (Fig. 2). Expression of F4/80, a pan marker of murine macrophages, was increased in WT mice when compared to Nlrp3 −/− regardless of diet fed (p < 0.001, stratified Mann–Whitney test). However, mRNA levels of lymphocyte antigen 6 complex (Ly6c), monocyte chemotactic protein 1 (MCP1), inducible form of nitric oxide synthase (iNOS), and TNF-α (markers for inflammatory or M1 polarized macrophages) were significantly increased in WT mice fed with CDDA when compared to WT mice on CSAA diet, while Nlrp3 −/− mice fed with CDAA had significantly lower levels of MCP1, iNOs, and TNF-α than WT mice on CDAA diet (Fig. 2). Protein analysis of the NLRP3 inflammasome related proteins, Casp1, and IL-1β showed significantly elevated levels of their precursor proteins, pro-Casp1 and pro-IL-1β, in WT and Nlrp3 −/− mice on CDAA compared to CSAA diet, respectively (pro-Casp1 p < 1e − 04 for both types; pro-IL-1β p < 0.05 for both types). However, Casp1 fragments (P20 and P10) and mature IL-1β were only significantly increased in WT mice on CDAA diet when compared to CSAA diet, while Nlrp3 −/− exhibited no increase in Casp1 fragments or mature IL-1β on either diet (Fig. 2).

Nlrp3 loss of function alleviates CDAA-diet-induced inflammation. Myeloperoxidase (MPO) expressing cells are significantly increased in mice fed with CDAA diet compared to mice on CSAA diet in both WT mice and Nlrp3 −/− mice (a, b). Similarly, mRNA levels of CXCL2 and MCP1 were increased in WT mice on CDAA diet when compared to WT mice on CSAA diet and were significantly reduced in Nlrp3 −/− when compared to WT mice (e, f).Expression of F4/80 was increased in WT mice when compared to Nlrp3 −/−regardless of diet fed (a, c). mRNA levels of Ly6c (a, d), iNOS (g), and TNF-α (h) (markers for inflammatory or M1-polarized macrophages) were significantly increased in WT mice fed with CDAA when compared to WT mice fed with CSAA, while iNOS and TNF-α expression were significantly lower in Nlrp3 −/− mice fed CDAA than WT mice fed with CDAA. Protein levels of pro-Casp1 were increased in Nlrp3 −/− and WT mice fed with CDAA, while only WT mice showed the corresponding increase in Casp1 subunits P10 and P20 (d). The protein levels of pro-IL-1β were elevated in mice fed with CDAA diet compared to mice fed with CSAA diet and Nlrp3 −/− mice on CDAA exhibited significantly lower IL-1β protein levels when compared to WT mice on CDAA (d)
Nlrp3 deletion results in protection from CDAA-induced liver fibrosis
As long-term feeding with CDAA diet has been shown to induce fibrosis in mice, we next explored changes in liver fibrogenesis and fibrosis in Nlrp3 knockout mice. Therefore, liver sections were stained with Sirius Red, and representative sections were quantitated by digital image analysis (Fig. 3). As expected, WT mice showed a significant increase in collagen deposition when comparing CDAA with CSAA diet (p < 1e − 13). Notably, Nlrp3 knockout mice were almost completely protected from diet-induced increases in collagen deposition (Fig. 3). Conclusively, while WT mice fed on CDAA showed significant increases in mRNA levels of α-SMA (p < 0.01), collagen type 1A1(COL1A1) (p < 0.05) and matrix metalloproteinase-2 (MMP-2) (p < 0.01) over WT mice fed on CSAA, Nlrp3 −/− mice on CDAA had significantly lower values of these markers than WT mice on CDAA (Mann–Whitney test with Holm adjustment) (Fig. 3). Tissue inhibitor of matrix metalloproteinase 1 (TIMP1), a marker for hepatic stellate cell (HSC) activation, was significantly elevated in mice fed with CDAA compared to CSAA in both WT and Nlrp3 −/− mice (p < 1e − 07 for both genotypes), while its expression was significantly reduced in Nlrp3 −/− mice on CDAA diet when compared to WT mice on CDAA diet (p < 0.05) (Fig. 3). In agreement with expression data, protein levels of α-SMA were significantly lower in Nlrp3 −/− than in WT mice on CDAA (p < 1e − 09).

Nlrp3 −/− mice are protected from CDAA-induced liver fibrosis. Quantitative image analysis of liver sections stained with Sirius Red (a) reveals that CDAA diet significantly increases collagen deposition in WT mice when compared to WT mice fed with CSAA diet (b). Notably, Nlrp3 −/− mice on CDAA diet showed a significantly lower percentage of Sirius Red stained liver area when compared to diet-matched WT mice (a, b). In line with this histological finding, mRNA levels of α-SMA, COL1A1, and MMP2 were significantly higher in liver samples of WT mice fed with CDAA diet when compared to WT mice fed with CSAA diet. Moreover, aforementioned mRNA levels were significantly reduced in Nlrp3 −/− mice on CDAA diet when compared to diet-matched WT mice (c). Expression levels of TIMP1 were significantly increased in Nlrp3 −/− and WT mice on CDAA when compared to mice fed with CSAA, while its expression was reduced in Nlrp3 −/− mice when compared to WT mice on CDAA diet (c). Protein levels of α-SMA were increased in WT mice fed with CDAA diet when compared to WT mice fed with CSAA diet, and those levels were significantly reduced in Nlrp3 −/− mice fed with CDAA when compared to WT mice on CDAA diet for 16 weeks (d)
Persistent NLRP3 activation results in chemokine production and liver inflammatory changes
We have recently shown that constitutive NLRP3 activation leads to severe liver inflammation with infiltration of myeloid cells in mouse pups alongside severe growth retardation and significantly impaired survival [19]. In order to test the direct effects of persistent NLRP3 activation as a contributor to NAFLD development and, in particular, as a modulator of progression from benign hepatic steatosis to steatohepatitis during diet-induced NAFLD, we generated a gain of function tamoxifen-inducible knock-in adult mouse model for temporal control over NLRP3 inflammasome activation (Fig. 4) [24]. WT and inducible mutants (Nlrp3 A350V/+ CreT) were weaned as usual at 21 days of age and placed on normal chow (NC) for 4 weeks. Thereafter, mice were injected with tamoxifen to allow for nuclear translocation of Cre and, therefore, expression of mutant Nlrp3 in knock-in mice for an additional 4 weeks. Assessment of liver histology showed that Nlrp3 mutant mice developed extensive liver inflammation with increased mRNA levels of pro-IL-1β and pro-Casp1 (Fig. 4). We found a significant increase of myeloperoxidase expressing cells in inflammatory foci, and these changes were associated with significant chemokine production of intercellular adhesion molecule 1 (ICAM1) and CXCL2 (Fig. 4). Analysis of liver macrophages revealed not only an increase in the overall number, when assessed by immunohistochemistry and mRNA expression of F4/80, but also a significant increase of inflammatory Ly6c-positive macrophages (Fig. 4). Macrophages exhibited a profile of M1 polarization with increases in levels of iNOS and unchanged expression of arginase 1 (Arg1) in Nlrp3 mutant mice when compared to WT mice (Fig. 4).

NLRP3 activation leads to severe liver inflammation. WT and Nlrp3 mutant mice were weaned after 21 days and placed on a normal chow for 4 weeks prior to induction of the knock-in mutation (in Nlrp3 mutants) by subcutaneous injections of tamoxifen. Analysis of liver histology revealed that Nlrp3 mutant mice fed with normal chow develop significantly more liver inflammation when compared to WT mice (a, b). Nlrp3 mutant mice also showed a significant increase in myeloid cells (assessed via immunohistochemistry for MPO) when compared to WT mice (a, d). In line with this finding, mRNA levels of Casp1, pro-IL-1β (c), CXCL2, and ICAM1 (e) were significantly elevated (p < 0.05) in Nlrp3 mutant mice. Total macrophages (a, f) (assessed via immunohistochemistry and mRNA levels for F4/80) and Ly6c-positive macrophages (a, h) were increased in Nlrp3 mutant mice when compared to WT mice. Also, macrophages from Nlrp3 mutant mice showed an inflammatory M1 profile as evidenced by increased iNOS (g) and unchanged Arg1 (i) mRNA expression when compared to WT mice
Nlrp3 mutant mice on short-term CDAA feeding show HSC activation and a marked increase in liver fibrogenesis
In order to test whether persistent NLRP3 activation is a factor that contributes to disease progression in NAFLD, Nlrp3 mutant and WT mice were placed on CDAA diet and simultaneously injected with tamoxifen for 4 weeks as described (Fig. 5). This time point on the CDAA diet was selected to mimic the initial isolated hepatic steatosis stage of disease progression. Nlrp3 knock-ins showed significantly increased Sirius Red staining when compared to WT animals (p < 1e − 05) (ANOVA). Analysis of mRNA levels showed significant increases in the expression of key pro-fibrogenic genes (α-SMA and COL1A1) and markers of HSC activation (TIMP1 and connective tissue growth factor (CTGF)) in Nlrp3 mutants when compared to WT mice fed with CDAA diet. Comparing Nlrp3 mutants fed with CDAA with those fed with NC revealed similar increases (Fig. 5). Protein levels of desmin (marker for HSCs) and α-SMA were significantly elevated in mice fed with CDAA diet when compared to mice fed with NC (p < 0.001) (ANOVA). Furthermore, Nrp3 mutants on CDAA diet had significantly elevated levels of the aforementioned proteins when compared to WT mice on CDAA (desmin p < 1e − 05 and α-SMA p < 1e − 03).

Short-term CDAA feeding induces liver fibrogenesis in Nlrp3 mutant mice. After receiving normal chow for 4 weeks, Nlrp3 mutant and WT mice were placed on either CDAA or NC diet and injected with tamoxifen for 4 weeks (a). Quantitative analysis of Sirius Red staining revealed significantly increased areas in Nlrp3 mutant mice fed with CDAA diet (b). Expression analysis of pro-fibrogenic genes (α-SMA and COL1A1) and genes associated with hepatic stellate cell activation (TIMP1 and CTGF) showed significant increases in Nlrp3 mutants fed with CDAA diet when compared to Nlrp3 mutants fed with NC diet (with the exception of TIMP1) and also WT mice fed with CDAA (c). Protein analysis showed significantly increased levels of desmin and α-SMA in mice fed with CDAA diet when compared to mice on NC with protein levels being further elevated in Nlrp3 mutants on CDAA diet (d)
Expression of NLRP3, pro-IL-1β, and pro-IL-18 are increased in the livers of patients with NASH and correlate to liver fibrosis
To test the potential clinical relevance of data generated from the various mouse models, we next investigated mRNA levels of NLRP3 inflammasome related proteins in the liver samples of an extensively characterized cohort of 77 patients with the full spectrum of NAFLD, consistent with patients undergoing bariatric surgery at the center for bariatric surgery at the University of Essen, Germany. These patients displayed the entire range of NAFLD and exhibited a rather high percentage of NASH (43 %). Thirty three samples from patients with NASH and 44 samples from patients diagnosed with non-NASH NAFLD were analyzed (Table 1). Liver biopsies were analyzed for mRNA levels of NLRP3, pro-IL-1β, pro-IL-18, and COL1A1. Notably, we found that in patients diagnosed with NASH, expression of NLRP3 inflammasome associated proteins (NLRP3, pro-IL-1β, and pro-IL-18) were significantly higher when compared to patients with non-NASH (Fig. 6). Moreover, mRNA levels of COL1A1 and pro-IL-1β had a moderate but significant Spearman rank correlation (r = 0.48, p = 0.01) (Fig. 6). No multiplicity adjustment was done for these exploratory analyses.

NLRP3 inflammasome-associated proteins are increased in livers of patients with NASH and correlate with collagen deposition. The mRNA levels of NLRP3, pro-IL-1β, and pro-IL-18 were analyzed in the liver samples of a cohort with NAFLD and NASH. Table 1 describes the characteristics of the patient population. The mRNA levels of NLRP3 (a), pro-IL-18 (b), and pro-IL-1β (c) were significantly increased in patients with NASH when compared to patients with non-NASH NAFLD. Moreover, mRNA levels of pro-IL-1β were significantly correlated with levels of COL1A1 (d) (mRNA values are graphed at log10 scale)
Discussion
The key findings of the present study relate to the identification of the NLRP3 inflammasome as an important contributor to liver damage and fibrosis during NASH development [17, 29, 30]. Our results demonstrate that Nlrp3 −/− mice are almost completely protected from CDAA-diet-induced fibrosis. Temporally controlled activation of the Nlrp3 inflammasome using a novel tamoxifen-inducible mouse model resulted in spontaneous liver inflammation. Moreover, inducible persistent NLRP3 activation can induce HSC activation and marked fibrogenesis after short-term feeding with CDAA diet, which typically results in only isolated benign hepatic steatosis in WT mice. Notably, expression of NLRP3 was significantly increased in livers of patients with NASH, and levels of pro-IL1 beta mRNA correlate with expression levels of COL1A1.
Emerging evidence supports a central role of NLRP3 activation, Casp1 activation and IL-1 signaling in the pathogenesis of many liver diseases including: ischemia/reperfusion injury and drug-, pathogen-, or endotoxin-mediated pathology, as well as NAFLD and liver fibrosis [14, 18, 29, 31]. Moreover, HSCs, the principal hepatic non-parenchymal cells responsible for liver fibrosis, have been shown to express all components of the NLRP3 inflammasome. Direct activation of NLRP3 by monosodium urate crystals was also demonstrated in primary HSC and LX-2 cells. This was associated with upregulation of TGF-β and collagen 1, suggesting a direct role for NLRP3 in HSC activation and the development of liver fibrosis [32]. The contribution of NLRP3 inflammasome activation in the development of NAFLD and the regulation of inflammatory liver changes and fibrosis characteristic of NASH has remained unclear. While Nlrp3 knockouts fed a high-fat diet for 9 months have been shown to demonstrate significantly less pathologic liver changes [33, 34], another study using a 4-week model of early isolated hepatic steatosis demonstrated that knockouts of NLRP3 inflammasome components and associated proteins show a modest increase in steatosis and inflammation in the liver [35]. Interestingly, this phenotype was transmissible to WT mice co-housed with knockouts, suggesting that NLRP3 inflammasome deficiency may result in an altered communicable gut microbiota that, in turn, may drive the early changes associated with NAFLD [35]. Recently, the role of IL-1α and IL-1β in the development of NASH was addressed by Kamari and coworkers. They found that selective deficiency in IL-1β in liver parenchymal cells, but not in bone-marrow-derived cells, protected mice from diet-induced steatohepatitis and fibrosis [16]. These observations suggest that inflammasome activation by different cell types may contribute to different aspects of steatohepatitis. Moreover, we found an increase in TUNEL-positive hepatocytes in WT mice fed with CDAA diet when compared to mice on CSAA diet indicating that those hepatocytes undergo a programmed cell death (apoptosis or pyroptosis). However, NLRP3 knockout mice fed with CDAA were protected from this increase in TUNEL-positive cells, suggesting an attenuation of CDAA-induced pyroptosis. Our data extend current results by further demonstrating in a dietary model characterized by the development of severe NASH and fibrosis in the presence of weight gain, which NLRP3 deficiency results in protection from liver injury, in particular, the development of liver fibrosis.
So far, important clues on the role of the NLRP3 inflammasome in NAFLD have been provided by studies, which used different inflammasome-associated knockout mouse models [17, 29, 30]. These models have important drawbacks including the potential for confounding effects as a result of deletion of NLRP3 or other inflammasome components in the developmental process or tissue/cellular adaptations that may occur to compensate for the deleted genes [36, 37]. To address these issues and to be able to directly test the effects of persistent NLRP3 activation in NAFLD, we used a gain-of-function approach. Our results showed that prolonged activation of NLRP3 for a period of 4 weeks resulted in marked upregulation of chemokines, such as ICAM1 and CXCL2, and inflammatory changes in the liver demonstrated by increased grade of inflammation, number of infiltrating myeloid cells, and mRNA levels of pro-Casp1 and pro-IL-1β. Combined stress of NLRP3 activation and diet-induced isolated hepatic steatosis further increased HSC activation with a marked fibrogenic response in the livers of these mice. These results strongly suggest that sustained NLRP3 activation acts as an important “hit” favoring the development of a more severe liver phenotype in the presence of an obesogenic stress that acts as a “first hit” during NAFLD development.
In order to understand the potential implications of our experimental results on human NAFLD, we studied the expression of different inflammasome-associated proteins and liver fibrosis-linked proteins in livers of a large and well-characterized group of bariatric patients presenting with the full spectrum of disease [26, 27]. In line with our results that NLRP3, pro-IL-1β, and pro-IL-18 mRNA are increased in patients with NASH in comparison to patients with non-NASH NAFLD, a study by Czak et al. recently described increased inflammasome-associated gene expression (ASC, Casp1, and NLRP3) in patients with clinically and biopsy-proven NASH, as well as patients infected with chronic hepatitis C when compared to livers from healthy controls [38]. The authors also describe that saturated fatty acids induce expression of pro-IL-1β and NLRP3 in murine hepatocytes. Increased levels of free fatty acids in serum have been reported in several mouse models of steatohepatitis and in human NAFLD patients with either steatosis or steatohepatitis [39–42]. Moreover, serum levels and composition of free fatty acids were correlated with liver injury in the cohort which was used to measure inflammasome components in the present study [43]. One could speculate that saturated FAs in NASH may favor inflammasome activation, whereas a different composition of FFAs in simple steatosis may not trigger such events. Furthermore, bariatric surgery restored a normal fatty acid profile as early as 6 weeks after surgery and might therefore also have beneficial effects on NLRP3 inflammasome activity [43]. Future larger studies to better understand the role of the NLRP3 inflammasome in human NASH are warranted.
In summary, the present study identified the NLRP3 inflammasome as an essential contributor to liver damage and, in particular, liver fibrosis, during NASH development. These findings have important implications for understanding the pathogenesis of disease progression in NAFLD and may lead to novel therapeutic strategies aimed at halting the progression of hepatic steatosis to the more severe forms of this disease.
Abbreviations
- α-SMA:
-
Alpha smooth muscle actin
- ALT:
-
Alanine aminotransferase
- ASC:
-
Apoptosis-associated speck-like protein containing a caspase recruitment domain
- Arg1:
-
Arginase 1
- ASH:
-
Alcoholic steatohepatitis
- BMI:
-
Body mass index
- Casp1:
-
Caspase 1
- CDAA:
-
Choline-deficient amino acid-defined
- COL1A1:
-
Collagen, type I, alpha 1
- CSAA:
-
Choline-supplemented amino acid-defined
- CTGF:
-
Connective tissue growth factor
- CXCL 2:
-
Chemokine (C-X-C motif) ligand 2
- DAMPs:
-
Damage associated molecular patterns
- F4/80:
-
Murine macrophage marker
- HSC:
-
Hepatic stellate cell
- ICAM1:
-
Intercellular adhesion molecule 1
- IL:
-
Interleukin
- iNOS:
-
Inducible form of nitric oxide synthase
- Ly6c:
-
Lymphocyte antigen 6 complex
- MCP1:
-
Monocyte chemotactic protein-1
- MMP2:
-
Matrix metalloproteinase-2
- MPO:
-
Myeloperoxidase
- NAFLD:
-
Nonalcoholic fatty liver disease
- NASH:
-
Nonalcoholic steatohepatitis
- NC:
-
Normal chow
- NLRs:
-
Nucleotide-binding oligomerization domain (NOD) leucine-rich-repeat containing receptors
- PAMPs:
-
Pathogen associated molecular patterns
- PBS:
-
Phosphate-buffered saline
- TIMP1:
-
Tissue inhibitor of matrix metalloproteinase 1
- TNF-α:
-
Tumor necrosis factor alpha
- WT:
-
Wild type
References
Levene AP, Goldin RD (2012) The epidemiology, pathogenesis and histopathology of fatty liver disease. Histopathology 61:141–152
Vernon G, Baranova A, Younossi ZM (2011) Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther 34:274–285
Mahady SE, George J (2012) Management of nonalcoholic steatohepatitis: an evidence-based approach. Clin Liver Dis 16:631–645
Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P (2005) The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology 129:113–121
Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ (1999) Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology 116:1413–1419
Brunt EM (2002) Alcoholic and nonalcoholic steatohepatitis. Clin Liver Dis 6:399–420, vii
Hjelkrem MC, Torres DM, Harrison SA (2008) Nonalcoholic fatty liver disease. Minerva Med 99:583–593
Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A (2012) The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol 56:952–964
Wree A, Kahraman A, Gerken G, Canbay A (2011) Obesity affects the liver—the link between adipocytes and hepatocytes. Digestion 83:124–133
Tilg H, Moschen AR (2010) Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 52:1836–1846
Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10:417–426
Netea MG, van der Meer JW (2011) Immunodeficiency and genetic defects of pattern-recognition receptors. N Engl J Med 364:60–70
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140:805–820
Szabo G, Csak T (2012) Inflammasomes in liver diseases. J Hepatol 57:642–654
Kubes P, Mehal WZ (2012) Sterile inflammation in the liver. Gastroenterology 143:1158–1172
Kamari Y, Shaish A, Vax E, Shemesh S, Kandel-Kfir M, Arbel Y, Olteanu S, Barshack I, Dotan S, Voronov E et al (2011) Lack of interleukin-1alpha or interleukin-1beta inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol 55:1086–1094
de Roos B, Rungapamestry V, Ross K, Rucklidge G, Reid M, Duncan G, Horgan G, Toomey S, Browne J, Loscher CE et al (2009) Attenuation of inflammation and cellular stress-related pathways maintains insulin sensitivity in obese type I interleukin-1 receptor knockout mice on a high-fat diet. Proteomics 9:3244–3256
Dixon LJ, Berk M, Thapaliya S, Papouchado BG, Feldstein AE (2012) Caspase-1-mediated regulation of fibrogenesis in diet-induced steatohepatitis. Lab Investig J Tech Methods Pathol 92:713–723
Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, Hoffman HM, Feldstein AE (2013) NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation and fibrosis. Hepatology 59:898–910
Kodama Y, Kisseleva T, Iwaisako K, Miura K, Taura K, De Minicis S, Osterreicher CH, Schnabl B, Seki E, Brenner DA (2009) c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology 137:1467–1477, e1465
Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galan JE, Askenase PW et al (2006) Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24:317–327
Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, Putnam CD, Boyle DL, Firestein GS, Horner AA et al (2009) Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity 30:875–887
Hayashi S, McMahon AP (2002) Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 244:305–318
McGeough MD, Pena CA, Mueller JL, Pociask DA, Broderick L, Hoffman HM, Brydges SD (2012) Cutting edge: IL-6 is a marker of inflammation with no direct role in inflammasome-mediated mouse models. J Immunol 189:2707–2711
Nakae D, Yoshiji H, Mizumoto Y, Horiguchi K, Shiraiwa K, Tamura K, Denda A, Konishi Y (1992) High incidence of hepatocellular carcinomas induced by a choline deficient L-amino acid defined diet in rats. Cancer Res 52:5042–5045
Kahraman A, Sowa JP, Schlattjan M, Sydor S, Pronadl M, Wree A, Beilfuss A, Kilicarslan A, Altinbas A, Bechmann LP et al (2013) Fetuin-A mRNA expression is elevated in NASH compared with NAFL patients. Clin Sci 125:391–400
Kalsch J, Bechmann LP, Kalsch H, Schlattjan M, Erhard J, Gerken G, Canbay A (2011) Evaluation of biomarkers of NAFLD in a cohort of morbidly obese patients. J Nutr Metab 2011: 369168. doi:10.1155/2011/369168
Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A et al (2005) Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41:1313–1321
Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, Barrieau M, Min SY, Kurt-Jones EA, Szabo G (2012) IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest 122:3476–3489
Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E (2010) Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 139(323–334):e327
Dixon LJ, Flask CA, Papouchado BG, Feldstein AE, Nagy LE (2013) Caspase-1 as a central regulator of high fat diet-induced non-alcoholic steatohepatitis. PLoS One 8:e56100
Watanabe A, Sohail MA, Gomes DA, Hashmi A, Nagata J, Sutterwala FS, Mahmood S, Jhandier MN, Shi Y, Flavell RA et al (2009) Inflammasome-mediated regulation of hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol 296:G1248–G1257
Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP (2011) Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 12:408–415
Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD (2011) The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 17:179–188
Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ et al (2012) Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482:179–185
Eisener-Dorman AF, Lawrence DA, Bolivar VJ (2009) Cautionary insights on knockout mouse studies: the gene or not the gene? Brain Behav Immun 23:318–324
Davey RA, MacLean HE (2006) Current and future approaches using genetically modified mice in endocrine research. Am J Physiol Endocrinol Metab 291:E429–E438
Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G (2011) Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 54:133–144
Rizki G, Arnaboldi L, Gabrielli B, Yan J, Lee GS, Ng RK, Turner SM, Badger TM, Pitas RE, Maher JJ (2006) Mice fed a lipogenic methionine-choline-deficient diet develop hypermetabolism coincident with hepatic suppression of SCD-1. J Lipid Res 47:2280–2290
Oosterveer MH, van Dijk TH, Tietge UJ, Boer T, Havinga R, Stellaard F, Groen AK, Kuipers F, Reijngoud DJ (2009) High fat feeding induces hepatic fatty acid elongation in mice. PLoS One 4:e6066
de Almeida IT, Cortez-Pinto H, Fidalgo G, Rodrigues D, Camilo ME (2002) Plasma total and free fatty acids composition in human non-alcoholic steatohepatitis. Clin Nutr 21:219–223
Puri P, Wiest MM, Cheung O, Mirshahi F, Sargeant C, Min HK, Contos MJ, Sterling RK, Fuchs M, Zhou H et al (2009) The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 50:1827–1838
Wree A, Bechmann LP, Claudel T, Schlattjan M, Sowa JP, Baba H, Gerken G, Feldstein AE, Trauner M, Canbay A (2013) Bariatric surgery reduces adipocyte size, improves liver injury and counteracts lipotoxicity via changes in serum fatty acid composition and adiponectin levels. J Hepatol 58:S553
Acknowledgments
We thank Martin Pronadl and Rudolf Ott from the Clinic of Surgery II at Alfried Krupp Hospital Essen, Germany, for collecting tissue and serum samples during bariatric surgeries and clinical follow up of the enrolled patients. We thank Bettina Papouchado for assessing steatosis, inflammation, and ballooning in the liver samples. This work was funded by NIH (DK076852 and DK082451 to AEF and AI52430 to HMH) and the German Research Foundation (DFG-grant 173/2-1 to AW).
Conflict of interest
The authors state that they have nothing to disclose.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental Table 1
(DOCX 17 kb)
Rights and permissions
About this article

Cite this article
Wree, A., McGeough, M.D., Peña, C.A. et al. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J Mol Med 92, 1069–1082 (2014). https://doi.org/10.1007/s00109-014-1170-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-014-1170-1