
Abstract
Purpose
Axitinib, a potent and selective inhibitor of vascular endothelial growth factor receptors 1, 2, 3, is metabolized by cytochrome P450 3A4 and glucuronidation. This study evaluated the effect of rifampin, a potent inducer of drug-metabolizing enzymes, on axitinib plasma pharmacokinetics. Equal numbers of Japanese and Caucasian subjects were enrolled to assess the potential differences in axitinib pharmacokinetics between the two ethnicities.
Methods
Forty healthy volunteers were randomized to receive 5 mg axitinib alone and with 600 mg rifampin.
Results
Rifampin expectedly decreased AUCinf and C max of axitinib (geometric mean reduced by 79 and 71%, respectively). However, differences in axitinib pharmacokinetics were not observed between Japanese and Caucasian subjects (geometric mean ratios for axitinib treatment alone for AUCinf and C max were 103 and 96%).
Conclusions
The results support a common axitinib starting dose in both populations. Potent inducers of drug-metabolizing enzymes reduce axitinib exposure and dose adjustments may be needed for optimal efficacy.
Similar content being viewed by others

Introduction
Axitinib (AG-013736) is an oral, potent, and selective inhibitor of vascular endothelial growth factor receptors 1, 2, 3 [1]. Early clinical trial experience in over 500 cancer patients has demonstrated that it has activity and is well-tolerated as a single agent [2–4] and in combination with various chemotherapeutic regimens [5, 6]. Axitinib is currently in phase III development for advanced renal cell carcinoma and in phase II development for various other tumor types.
Clinically, axitinib is administered 5 mg twice daily as an oral starting dose and its pharmacokinetics are characterized by rapid oral absorption (with peak plasma concentrations occurring 1–2 h after dosing in the fasted state) and a terminal plasma half-life of 3–5 h. Axitinib has an oral absolute bioavailability of approximately 58% (Pfizer Inc., Data on file 2008) with generally linear pharmacokinetics [7].
Axitinib is primarily metabolized by cytochrome P450 (CYP) 3A4 and to a lesser extent by CYP1A2, CYP2C19 and uridine diphosphate glucuronosyltransferase (UGT) 1A1 [6]. Therefore, axitinib plasma exposure has the potential to be affected by the co-administration of drugs that inhibit or induce CYP3A. This has been previously observed with the co-administration of CYP3A inhibitor ketoconazole, which increased the area under the plasma concentration–time curve from time 0 to 24 h (AUC(0–24)) and maximal plasma concentration (C max) of axitinib approximately 2- and 1.5-fold, respectively [8]. Rifampin is a known potent CYP3A4 inducer along with other drug-metabolizing enzymes including CYP1A2 and UGT1A1, and is hence likely to affect the pharmacokinetics of axitinib [9–11]. In addition, subjects who are homozygous for UGT1A1*28 variant gene have the potential for reduced glucuronidation of axitinib. Due to differences in the frequency of UGT1A1*28 genotype between Caucasians (12%, range 9–16%) and Japanese (4%, range 1–6%) [12–22], this may also contribute to differences in the pharmacokinetics between these ethnicities.
The primary objective of this phase I study was to investigate potential pharmacokinetic interaction in healthy volunteers when axitinib is administered in combination with rifampin. Secondary objectives included: (1) assessing potential inter-ethnic differences between Caucasian and Japanese subjects, (2) evaluating the effect of UGT1A1 genotype on the pharmacokinetics of axitinib, (3) evaluating the safety and tolerability of axitinib alone and when co-administered with rifampin.
Methods
Subjects
This study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki (Revised Edinburgh, 2000) and in accordance with all International Conference on Harmonization and Good Clinical Practice guidelines. The final protocol and informed consent documentation were reviewed and approved by the Institutional Review Board at the study center (Radiant Covance, Hawaii, USA).
Key inclusion criteria included: female of non-childbearing potential or male, age 18–55 years; Caucasian or first-generation Japanese (both parents of Japanese descent, born in Japan and resident outside of Asia for ≤5 years); and body mass index 18–28 kg/m2 with total body weight >45 kg (99 lb). Key exclusion criteria included: clinically significant medical or surgical conditions with the potential to interfere with the absorption, distribution, metabolism or excretion of the study drugs; history of alcohol consumption >14 drinks/week for men or 7 drinks/week for women within 6 months of screening; smoker; electrocardiogram (ECG) abnormality; blood pressure >140/90 mmHg on two evaluations at least 10 min apart; and treatment with an investigational drug or any known CYP450 enzyme-inducing/-inhibiting agents or herbal supplements within 30 days prior to first dose of study medication. Subjects were to abstain from using prescription and non-prescription drugs (other than acetaminophen as deemed necessary), vitamins and dietary supplements within 7 days prior to the first dose of study medication and throughout the study.
Treatments
This single-center, open-label study involved subjects being assigned to two separate treatment sequences according to a two-way crossover design. The two treatments involved subjects receiving single 5 mg axitinib as an oral dose on day 1 (treatment A), and subjects receiving rifampin (Rifadin®, Aventis Pharmaceuticals Inc., Bridgewater, NJ, USA) 600 mg/day orally for 9 days with a single 5 mg oral dose of axitinib co-administered on day 8 (treatment B). In both treatment arms, axitinib was administered in the morning after an overnight fast of at least 10 h. To standardize conditions, subjects were not allowed to eat or drink (except water) for at least 4 h following dosing. To ensure full compliance, all doses of study medication were administered under medical supervision at the clinic.
Subjects received treatments A and B according to a crossover design. Volunteers initially receiving treatment A adhered to a 7-day washout period before commencing treatment B; those initially receiving treatment B adhered to a 21-day washout before treatment A began. Washout was required to allow elimination of axitinib from plasma and for recovery from rifampin-mediated enzyme induction.
Study assessments
Sample collection
Blood samples (5 mL) for determination of axitinib plasma concentration were collected pre-dose and at 0.5, 1, 1.5, 2, 4, 6, 8, 12, 16, 24 and 32 h following axitinib administration on both treatment arms.
Bioanalytical methods
Plasma concentrations of axitinib were measured using a validated high-performance liquid chromatography with tandem mass spectrometric detection method (Charles River Discovery and Development Services; Worcester, MA, USA) [7]. The limit of quantification for the axitinib assay in plasma was 0.1 ng/mL, the between-day precision was <15.7% and bias <12.8% for quality control samples evaluated in the concentration range of interest. Pharmacodynamic evaluations consisted of the ratio of urinary 6, β-hydroxycortisol/cortisol concentrations as a marker of CYP3A4 induction [23]. The limit of quantification for the cortisol assay in urine was 2.5 ng/mL, the between-day precison was <12.5% and the bias was <15%. For 6, β-hydroxycortisol in urine, the limit of quantification was 12.5 ng/mL, the between-day precision was <8.7% and bias was <16%. The urinary 6, β-hydroxycortisol/cortisol ratio was calculated for all subjects. During treatment A, urine samples were collected pre-dose (baseline). During treatment B, urine samples were collected pre-dose (baseline) and in the morning on days 8 and 9 following combination treatment with axitinib and rifampin. Urine concentrations of cortisol and 6, β-hydroxycortisol were determined using a validated high-performance liquid chromatography method with tandem mass spectrometric detection (ABC Laboratories; Columbia, MO, USA).
Genotyping
To assess possible differences in metabolism, subjects were genotyped for metabolizing enzymes including UGT1A1. DNA was extracted from whole blood using Qiagen’s QIAamp kit. The polymerase chain reaction was used to amplify DNA samples. The Sequenom MassArray was used to perform the genotyping of the UGT1A1*28 (Sequenom Discovery) genotyping assay. The genotypes were determined with MassARRAY RT™ software (SpectroTYPER™ Version 3.4, Sequenom). The UGT1A1*28 genotype was investigated as an exploratory variable to determine the potential correlation with pharmacokinetic variability. Specifically, the variants of UGT1A1 to be assessed included UGT1A1*60, UGT1A1-3156 (−3156 G→A nucleotide change), UGT1A1 promoter TA repeat (*28, *36, *37), UGT1A1*6 and UGT1A1*27.
Safety
Adverse event (AE) monitoring, vital signs (heart rate and blood pressure), 12-lead ECG recordings, and clinical laboratory tests were carried out. AE severity was based on the National Cancer Institute Common Terminology Criteria for Adverse Events criteria; relationship to treatment was determined by the investigator.
Pharmacokinetic and statistical evaluations
The pharmacokinetic evaluation of axitinib was performed with WinNonlin (Version 4.01, Pharsight, Mountain View, CA) using a non-compartmental approach. Pharmacokinetic parameters estimated for orally administered axitinib included AUC from time zero extrapolated to infinity (AUCinf) and from time zero to the time for the last quantifiable concentration (AUClast), C max, time to C max (T max), terminal half-life, oral clearance and apparent volume of distribution. Pharmacokinetic parameters were summarized by treatment, ethnicity and genotype.
For the parameters AUCinf, AUClast and C max, a statistical analysis to determine the estimated ratio of adjusted geometric means and the associated 90% confidence interval (CI) was performed. A sample size of 32 subjects was determined to have sufficient power to detect treatment differences in AUCinf and C max. An additional eight subjects were enrolled to account for possible dropouts.
Results
Subjects
A total of 40 male subjects (Caucasian, n = 20; Japanese, n = 20) with a mean (±standard deviation) age of 29 (±7.6) years (range 21–52 years) and a mean weight of 72.3 (±9.5) kg (range 54.0–96.7 kg) were enrolled. During the enrollment period of 6 months for this study, no women of non-childbearing potential who otherwise met study entry criteria were found; hence, only male subjects eventually were enrolled in this study. One subject failed to complete both treatment periods for reasons unrelated to treatment (failed drug test) but was included in the evaluable population. Hence, all subjects (n = 40) received at least one axitinib 5 mg oral dose and 39 subjects received axitinib and rifampin combination therapy.
CYP3A4 induction
The mean urinary 6, β-hydroxycortisol/cortisol ratio was 4.2 ± 3.5 at baseline and increased to 18.6 ± 15.8 after 8 days of rifampin dosing. After cessation of rifampin treatment, the mean 6, β-hydroxycortisol/cortisol ratio returned to near baseline values (4.6 ± 3.4). Similar results were observed in Caucasian and Japanese subjects. In Caucasian subjects, mean 6, β-hydroxycortisol/cortisol ratio was 4.5 ± 2.6 at baseline and increased to 15.3 ± 9.1 after 8 days of rifampin dosing and in Japanese subjects the mean ratio was 4.0 ± 4.3 at baseline and 21.7 ± 20.0 after 8 days of rifampin dosing.
Pharmacokinetics
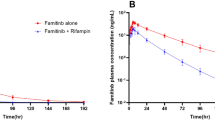
The plasma concentration–time profiles of axitinib in the absence and presence of rifampin are shown in Fig. 1a. Axitinib plasma concentrations were reduced when co-administered with rifampin. Axitinib pharmacokinetic parameters for the entire evaluable subject population are reported in Table 1 and Fig. 1b, c. Geometric means of AUCinf and AUClast for axitinib were decreased by 79 [geometric mean ratio 21% (90% CI 18–24)] and 80% [geometric mean ratio 20% (90% CI 17–23)] respectively, when co-administered with rifampin. The geometric mean for axitinib C max was reduced by 71% when co-administered with rifampin [geometric mean ratio 29% (90% CI 24–35)]. The axitinib plasma half-life appeared to be decreased in the presence of rifampin (Table 1); however this should be interpreted with caution since this is likely due to axitinib plasma concentrations falling below the limit of quantification at later time-points in the presence of rifampin which resulted in poorly estimated terminal half-life in many subjects.

a Mean (±standard deviation) axitinib plasma concentration profile in subjects administered axitinib alone (open symbols) (n = 40), and co-administered axitinib and rifampin (closed symbols) (n = 39) (Inset same plot with y-axis on log scale). b Comparative box plot of axitinib area under the plasma concentration–time curve from 0 h to infinity (AUCinf; ng h/mL) in all subjects in the absence (n = 40) and presence (n = 39) of rifampin. c Comparative box plot of axitinib maximal plasma concentration (C max; ng/mL) in all subjects in the absence (n = 40) and presence (n = 39) of rifampin. Box plot represents 25th and 75th percentiles, whiskers extend to 5th and 95th percentiles. Median is indicated by line within box. Circles represent values for individual subjects
Effect of ethnicity on axitinib pharmacokinetics
Axitinib pharmacokinetics were comparable between Caucasian and Japanese subjects in the presence and absence of rifampin (Table 2; Figs. 2, 3). The geometric mean ratio (Japanese versus Caucasian) for axitinib AUCinf and C max was 103 (90% CI 71–151) and 96% (90% CI 64–144) respectively. When axitinib was administered alone, median peak plasma concentrations of axitinib were achieved in 1.5 h (range 1.5–2 h) after dosing in Caucasian volunteers and 1.75 h (range 0.5–2 h) in Japanese subjects as shown in Fig. 2a. In subjects administered axitinib and rifampin in combination, axitinib mean peak plasma concentrations were observed at 1.5 h after dosing in both ethnic groups (Fig. 2b). Figure 3a, b compares the AUCinf and C max, respectively, of axitinib in Caucasian and Japanese volunteers in the presence and absence of rifampin and demonstrate no differences. Inter-subject variability for axitinib AUCinf measured by coefficient of variation was similar in Japanese and Caucasian subjects at 65 and 54%, respectively.

a Mean (±standard deviation) axitinib plasma concentration profile in Japanese (closed symbols) (n = 20) and Caucasian (open symbols) (n = 20) subjects after administration of axtitinib, 5 mg single dose, alone (Inset same plot with y-axis log scale) b Mean (±standard deviation) axitinib plasma concentrations observed over time in Japanese (closed symbols) (n = 20) and Caucasian (open symbols) (n = 19) subjects in the presence of rifampin (Inset log scale)

Comparative box plot of axitinib a area under the plasma concentration–time curve from 0 h to infinity (AUCinf; ng h/mL) and b maximal plasma concentration (C max; ng/mL) in Japanese (n = 20) and Caucasian (n = 20) subjects in the absence and presence of rifampin. Box plot represents 25th and 75th percentiles, whiskers extend to 5th and 95th percentiles. Median is indicated by line within box. Circles represent values for each subject
Effect of UGT1A1 genotype on axitinib pharmacokinetics
During the 6-month enrollment period of the study, recruitment was kept open in an effort to enroll at least three Caucasian and three Japanese subjects who were variant for the UGT1A1*28 allele. Of the 40 subjects eventually recruited over the enrollment period, three Caucasian and one Japanese subject were homozygous variant for UGT1A1*28. In both Caucasian and Japanese subjects, pharmacokinetic parameters (AUCinf and C max) were similar for subjects with all UGT1A1*28 genotypes as shown in Fig. 4a and b respectively.

Comparative box plot of axitinib a area under the plasma concentration–time curve from 0 h to infinity (AUCinf, ng h/mL) b maximal plasma concentration (C max, ng/mL) in UGT1A1*28 wild type (6/6, n = 23), heterozygous (6/7, n = 13), and variant (7/7, n = 4) subjects. Data provided in this figure are following administration of axitinib alone (i.e., in the absence of rifampin). Box plot represents 25th and 75th percentiles, whiskers extend to 5th and 95th percentiles. Median is indicated by line within box. Circles represent values for each subject
Safety
None of the AEs were treatment-related or resulted in treatment discontinuation, dose reduction, or death during the study. No clinically relevant mean changes in hematology, vital sign or ECG findings were noted in the overall safety population or in either ethnic groups.
Discussion
The results of this study demonstrate three important findings. First, as expected, axitinib exposure is decreased when concomitantly administered with rifampin in healthy volunteers. Second, there is no difference in the plasma pharmacokinetics of axitinib in healthy Caucasian and Japanese subjects. Third, UGT1A1*28 polymorphism does not appear to affect the pharmacokinetics of axitinib.
The ratio of urinary 6, β-hydroxycortisol/cortisol has been used previously as a measure of CYP3A4 induction [23]. In this study, the ratio of urinary 6, β-hydroxycortisol/cortisol concentrations in subjects was consistent with maximal induction of CYP3A4 and returned to normal following washout after the end of rifampin dosing. The maximally observed effect was similar between Japanese and Caucasian subjects after 8 days of rifampin dosing. In addition, there was no difference in the 6, β-hydroxycortisol/cortisol ratios between Caucasian and Japanese subjects. These results are consistent with effective CYP3A induction with rifampin in both Caucasian and Japanese subjects.
Results from this study indicate that geometric mean AUCinf and C max for axitinib were decreased by 79 and 71%, respectively, when co-administered with rifampin. Induction of CYP3A4 with rifampin 600 mg/day has been demonstrated in previous studies using the ratio of 6, β-hydroxycortisol/cortisol in urine [23]. In addition, rifampin has also been shown to induce UGT1A1 and other drug-metabolizing enzymes in clinical studies [11, 24]. Induction of axitinib metabolism as mediated by CYP3A4 in healthy, non-smoking volunteers resulted in a substantial decrease in axitinib plasma concentration. This study therefore provides the lower bound for (minimum) plasma concentrations expected following an oral dose of 5 mg axitinib, i.e., in the presence of a potent metabolic inducer.
First-generation Japanese subjects recruited in this study were required to have been born in Japan, not lived outside of Asia for more than 5 years, and should have had both parents of Japanese descent. Ethnic differences in axitinib pharmacokinetics between Japanese and Caucasian subjects were minimal; the geometric mean ratios for axitinib AUCinf and C max were not statistically different between Caucasian and Japanese subjects and pharmacokinetic parameters were similar between the subjects, indicating that these ethnicities do not contribute significantly to the axitinib pharmacokinetic variability observed in this study. In addition, safety data were similar for Caucasian and Japanese subjects.
Previous in vitro studies with human liver microsomes have indicated the involvement of UGT1A1 in the glucuronidation of axitinib, and so the effect of the UGT1A1*28 genotype on axitinib pharmacokinetics was also explored in Caucasian and Japanese subjects. Subjects who are variant for UGT1A1*28 have the potential for reducing glucuronidation of axitinib. The frequency of variant UGT1A1*28 is 12% (range 9–16%) for Caucasians and 4% (range 1–6%) for Japanese populations [12–22]. Although this study was not statistically powered to detect genotype-mediated changes, axitinib pharmacokinetic parameters were similar across UGT1A1*28 genotypes and ethnicity, implying that this genotype does not likely contribute to the axitinib pharmacokinetic variability observed in this study.
Single-dose axitinib, alone or in combination with rifampin, was well tolerated by healthy volunteers in this study. Combination treatment with rifampin for 8 days did not produce any drug-related AEs. All observed AEs were deemed not related to study drug by the investigator. No serious or clinically significant AEs were reported.
Conclusions
A reduction in axitinib exposure is observed when co-administered with repeated doses of rifampin. Concomitant treatment with axitinib and potent inducers of CYP3A4 would require axitinib dose adjustment for optimal clinical efficacy. The similarity of axitinib pharmacokinetics in Caucasian and Japanese subjects supports a common starting dose and regimen in these populations.
References
Hu-Lowe DD, Zou HY, Grazzini ML, Hallin ME, Wickman GR, Amundson K, Chen JH, Rewolinski DA, Yamazaki S, Wu EY, McTigue MA, Murray BW, Kania RS, O’Connor P, Shalinsky DR, Bender SL (2008) Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin Cancer Res 14:7272–7283. doi:10.1158/1078-0432.CCR-08-0652
Rixe O, Bukowski RM, Michaelson MD, Wilding G, Hudes GR, Bolte O, Motzer RJ, Bycott P, Liau KF, Freddo J, Trask PC, Kim S, Rini BI (2007) Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: a phase II study. Lancet Oncol 8:975–984. doi:10.1016/S1470-2045(07)70285-1
Cohen E, Rosen LS, Vokes EE, Kies MS, Forastiere A, Worden F, Kane MA, Sherman E, Kim S, Bycott P, Tortorici M, Shalinsky DR, Liau KF, Cohen RB (2008) Axitinib is an active treatment for all histological subtypes of advanced thyroid cancer: results from a phase II study. J Clin Oncol 26:4708–4713. doi:10.1200/JCO.2007.15.9566
Schiller JH, Larson T, Ou SI, Limentani A, Sandler AB, Vokes EE, Kim S, Liau KF, Bycott PW (2007) Efficacy and safety of axitinib (AG-013736) in patients (pts) with advanced non-small cell lung cancer (NSCLC): a phase II trial. J Clin Oncol 25:386s
Spano JP, Chodkiewicz C, Maurel J, Wong R, Wasan H, Barone C, Letourneau R, Bajetta E, Pithavala Y, Bycott P, Trask P, Liau K, Ricart AD, Kim S, Rixe O (2008) Efficacy of gemcitabine plus axitinib compared with gemcitabine alone in patients with advanced pancreatic cancer: an open-label randomised phase II study. Lancet 371:2101–2108. doi:10.1016/S0140-6736(08)60661-3
Rugo HS, Stopeck A, Joy AA, Chan S, Verma S, Lluch A, Liau KF, Kim S, Boycott P, Soulieres D (2007) A randomized, double-blind phase II study of the oral tyrosine kinase inhibitor (TKI) axitinib (AG-013736) in combination with docetaxel (DOC) compared to DOC plus placebo (PL) in metastatic breast cancer (MBC). J Clin Oncol 25:32s
Rugo HS, Herbst RS, Liu G, Park JW, Kies MS, Steinfeldt HM, Pithavala YK, Reich SD, Freddo JL, Wilding G (2005) Phase I trial of the oral antiangiogenesis agent AG-013736 in patients with advanced solid tumors: pharmacokinetic and clinical results. J Clin Oncol 23:5474–5483. doi:10.1200/JCO.2005.04.192
Pithavala Y, Klamerus KJ, Mount J, Garrett M, Hee B, Selaru P, Sarapa N (2008) Effect of ketoconazole on the pharmacokinetics of axitinib (AG-013736) in healthy volunteers. American Association for Cancer Research (AACR) Annual meeting, April 12–16, 2008, San Diego Convention Center, San Diego, Abstract 232
Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivistö KT (2003) Pharmacokinetic interactions with rifampicin: clinical relevance. Clin Pharmacokinet 42:819–850
Rae JM, Johnson MD, Lippman ME, Flockhart DA (2001) Rifampin is a selective, pleiotropic inducer of drug metabolism genes in human hepatocytes: studies with cDNA and oligonucleotide expression arrays. J Pharmacol Exp Ther 299:849–857
Nishimura M, Koeda A, Shimizu T, Nakayama M, Satoh T, Narimatsu S, Naito S (2008) Comparison of inducibility of sulfotransferase and UDP-glucuronosyltransferase mRNAs by prototypical microsomal enzyme inducers in primary cultures of human and cynomolgus monkey hepatocytes. Drug Metab Pharmacokinet 23:45–53. doi:10.2133/dmpk.23.45
Monaghan G, Ryan M, Seddon R, Hume R, Burchell B (1996) Genetic variation in bilirubin UPD-glucuronosyltransferase gene promoter and Gilbert’s syndrome. Lancet 347:578–581. doi:10.1016/S0140-6736(96)91273-8
Beutler E, Gelbart T, Demina A (1998) Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism? Proc Natl Acad Sci USA 95:8170–8174
Ando Y, Chida M, Nakayama K, Saka H, Kamataki T (1998) The UGT1A1*28 allele is relatively rare in a Japanese population. Pharmacogenetics 8:357–360
Lampe JW, Bigler J, Horner NK, Potter JD (1999) UDP-glucuronosyltransferase (UGT1A1*28 and UGT1A6*2) polymorphisms in Caucasians and Asians: relationships to serum bilirubin concentrations. Pharmacogenetics 9:341–349
Iyer L, Hall D, Das S, Mortell MA, Ramirez J, Kim S, Di RA, Ratain MJ (1999) Phenotype-genotype correlation of in vitro SN-38 (active metabolite of irinotecan) and bilirubin glucuronidation in human liver tissue with UGT1A1 promoter polymorphism. Clin Pharmacol Ther 65:576–582
Hall D, Ybazeta G, stro-Bisol G, Petzl-Erler ML, Di RA (1999) Variability at the uridine diphosphate glucuronosyltransferase 1A1 promoter in human populations and primates. Pharmacogenetics 9:591–599
Ando Y, Saka H, Ando M, Sawa T, Muro K, Ueoka H, Yokoyama A, Saitoh S, Shimokata K, Hasegawa Y (2000) Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Cancer Res 60:6921–6926
Sugatani J, Yamakawa K, Yoshinari K, Machida T, Takagi H, Mori M, Kakizaki S, Sueyoshi T, Negishi M, Miwa M (2002) Identification of a defect in the UGT1A1 gene promoter and its association with hyperbilirubinemia. Biochem Biophys Res Commun 292:492–497
Fertrin KY, Gonçalves MS, Saad ST, Costa FF (2002) Frequencies of UDP-glucuronosyltransferase 1 (UGT1A1) gene promoter polymorphisms among distinct ethnic groups from Brazil. Am J Med Genet 108:117–119. doi:10.1002/ajmg.10209
Fang JL, Beland FA, Doerge DR, Wiener D, Guillemette C, Marques MM, Lazarus P (2002) Characterization of benzo(a)pyrene-trans-7, 8-dihydrodiol glucuronidation by human tissue microsomes and overexpressed UDP-glucuronosyltransferase enzymes. Cancer Res 62:1978–1986
Kitagawa C, Ando M, Ando Y, Sekido Y, Wakai K, Imaizumi K, Shimokata K, Hasegawa Y (2005) Genetic polymorphism in the phenobarbital-responsive enhancer module of the UDP-glucuronosyltransferase 1A1 gene and irinotecan toxicity. Pharmacogenet Genomics 15:35–41
Ohnhaus EE, Breckenridge AM, Park BK (1989) Urinary excretion of 6 beta-hydroxycortisol and the time course measurement of enzyme induction in man. Eur J Clin Pharmacol 36:39–46
Wenning LA, Hanley WD, Brainard DM, Petry AS, Ghosh K, Jin B, Mangin E, Marbury TC, Berg JK, Chodakewitz JA, Stone JA, Gottesdiener KM, Wagner JA, Iwamoto M (2009) Effect of Rifampin, a Potent Inducer of Drug-Metabolizing Enzymes, on the Pharmacokinetics of Raltegravir. Antimicrob Agents Chemother [Epub ahead of print]. doi:10.1128/AAC.01468-08
Acknowledgments
The authors thank Brad Rosbrook for statistical analysis and the medical writers at ACUMED® (Tytherington, UK) for assistance in drafting the manuscript, supported by Pfizer Inc. The study was sponsored by Pfizer Inc.
Conflict of interest statement
The authors were all employees of Pfizer Inc. (the study sponsor) at the time of the study and all own stock in Pfizer Inc.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Pithavala, Y.K., Tortorici, M., Toh, M. et al. Effect of rifampin on the pharmacokinetics of Axitinib (AG-013736) in Japanese and Caucasian healthy volunteers. Cancer Chemother Pharmacol 65, 563–570 (2010). https://doi.org/10.1007/s00280-009-1065-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-009-1065-y