
Abstract
The purpose of this study was to determine the maximum tolerated dose (MTD) and recommended dose (RD) of pemetrexed with folate and vitamin B12 supplementation (FA/VB12) in Japanese patients with solid tumours and to investigate the safety, efficacy, and pharmacokinetics of pemetrexed. Eligible patients had incurable solid tumours by standard treatments, a performance status 0–2, and adequate organ function. Pemetrexed from 300 to 1200 mg m−2 was administered as a 10-min infusion on day 1 of a 21-day cycle with FA/VB12. Totally, 31 patients were treated. Dose-limiting toxicities were alanine aminotransferase (ALT) elevation at 700 mg m−2, and infection and skin rash at 1200 mg m−2. The MTD/RD were determined to be 1200/1000 mg m−2, respectively. The most common grade 3/4 toxicities were neutropenia (grade (G) 3:29, G4:3%), leucopenia (G3:13, G4:3%), lympopenia (G3:13%) and ALT elevation (G3:13%). Pemetrexed pharmacokinetics in Japanese were not overtly different from those in western patients. Partial response was achieved for 5/23 evaluable patients (four with non-small cell lung cancer (NSCLC) and one with thymoma). The MTD/RD of pemetrexed were determined to be 1200/1000 mg m−2, respectively, that is, a higher RD than without FA/VB12 (500 mg m−2). Pemetrexed with FA/VB12 showed a tolerable toxicity profile and potent antitumour activity against NSCLC in this study.
Similar content being viewed by others


Main
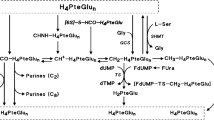
Pemetrexed (LY231514, Alimta®, Eli Lilly and Company, IN, USA) is a novel antifolate (Taylor and Patel, 1992) that is approved in the United States and a number of European Union countries , for treatment of patients with malignant pleural mesothelioma (MPM) in combination with cisplatin, and non-small cell lung cancer (NSCLC) after prior chemotherapy as a single agent. In vitro experiments show that pemetrexed inhibits three enzymes in folate metabolism: thymidylate synthase (TS), dihydrofolate reductase (DHFR), and glycinamide ribonucleotide formyltransferase (GARFT) (Shih et al, 1998). Given the schedule dependency observed preclinically, three regimens were explored in phase I studies: (1) 0.2–5.2 mg m−2 daily for 5 days every 3 weeks (McDonald et al, 1998); (2) 10–40 mg m−2 weekly for 4 weeks repeated every 6 weeks (Rinaldi et al, 1995); and (3) 50–700 mg m−2 every 3 weeks (Rinaldi et al, 1999).
The third regimen (one dose every 3 weeks) was chosen for subsequent phase II studies because of its convenient administration, ability to give repeated doses, and occurrence of objective responses. The original maximum tolerated dose (MTD) and the recommended dose (RD) was 600 mg m−2, but was decreased to 500 mg m−2 owing to toxicities experienced early in phase II studies. The initial phase I and II studies showed that myelosuppression was the principle drug-related toxicity, with a frequency of grade 3/4 neutropenia of 50% and grade 3/4 thrombocytopenia of 15% (Hanauske et al, 2001). Less than 10% of patients experienced gastrointestinal toxicities such as diarrhoea or mucositis. Although the prevalence of gastrointestinal toxicities and severe hematologic toxicities was low, these toxicities were associated with a high risk of mortality.
Infrequent severe myelosuppression with gastrointestinal toxicity has been observed not only for pemetrexed, but for the class of antifolates, including the DHFR inhibitor methotrexate (Morgan et al, 1990), the TS inhibitor raltitrexed (Maughan et al, 1999), and the GARFT inhibitor lometrexol (Alati et al, 1996; Mendelsohn et al, 1996). Clinical experience and nonclinical studies with methotrexate and lometrexol indicated that severe toxicity may be associated with nutritional folate status (Morgan et al, 1990; Alati et al, 1996; Mendelsohn et al, 1996). In fact, in the study of lometrexol, a significant effect of folate supplementation on toxicity was observed (Laohavinij et al, 1996). Based on these experiences, Niyikiza et al (2002a) investigated relationships between toxicity and baseline patient characteristics for early pemetrexed studies. They found total plasma homocysteine and methylmalonic acid levels to predict severe neutropenia and thrombocytopenia, with or without grade 3/4 diarrhoea, mucositis, or infection. Homocysteine and methylmalonic acid are known as indicators of folate and vitamin B12 deficiencies (Rosenberg and Fenton, 1989; Savage et al, 1994). Thus, it was hypothesized that a patient's risk for severe toxicity could be reduced by decreasing the levels of homocysteine and methylmalonic acid with folate and vitamin B12 supplementation (FA/VB12) (Niyikiza et al, 2002a).
FA/VB12 is now required for all patients participating in pemetrexed studies. Using this strategy, the pivotal phase III studies for MPM and NSCLC were successfully conducted with amelioration of severe drug-related toxicity (Niyikiza et al, 2002b; Vogelzang et al, 2003; Hanna et al, 2004).
One may expect that pemetrexed administration with supplementation would be more tolerable for patients and permit significant dose escalation above the current RD of 500 mg m−2. Therefore, we conducted a phase I study to determine the MTD of pemetrexed with FA/VB12 for Japanese patients with solid tumours and to identify the RD for subsequent Japanese phase II studies. Our secondary objectives were to investigate the safety, antitumour effect, and pharmacokinetics of pemetrexed with supplementation in Japanese patients. A similar phase I study has been conducted outside Japan, but only preliminary data are available at this time (Hammond et al, 2003).
Patients and methods
Patient selection
Eligible patients had histologic or cytologic diagnosis of solid cancer that was incurable by standard treatments. Patients also must have been between 20 and 75 years of age, have an Eastern Cooperative Oncology Group (ECOG) performance status of 0–2, and have an estimated life expectancy of at least 3 months. Adequate organ function was required, which included bone marrow reserve (white blood cell count 4.0–12.0 × 103 mm−3, platelets ⩾100 × 103 mm−3, haemoglobin ⩾9.0 g dl−1, and absolute granulocyte count ⩾2.0 × 103 mm−3), hepatic function (bilirubin ⩽1.5 × upper limit of normal, aspartate/alanine transaminase (AST/ALT) ⩽2.5 × upper limit of normal, and serum albumin ⩾2.5 g dl−1), renal function (serum creatinine ⩽upper limit of normal and Cockcroft and Gault creatinine clearance ⩾60 ml min−1), and lung function (PaO2 ⩾60 torr).
Prior chemotherapy or hormone therapy was allowed if it was carried out ⩾14 days before study entry (⩾35 days for nitrosourea or mitomycin-C). Previous radiotherapy was also allowed, but only if ⩽25% of marrow was irradiated and if it was completed ⩾21 days before study entry. Pretreated patients must have recovered from all toxicities before study entry. Prior surgery was allowed if patients recovered from the effect of the operation. Patients were excluded from this study for active infection, symptomatic brain metastasis, interstitial pneumonitis, or pulmonary fibrosis diagnosed by chest X-ray, serious concomitant systemic disorders incompatible with the study, clinically significant effusions, or the inability to discontinue aspirin and other nonsteroidal anti-inflammatory agents during the study.
This study was conducted in compliance with the guidelines of good clinical practice and the Declaration of Helsinki Principles, and it was approved by the local institutional review boards. All patients gave written informed consent before study entry.
Treatment
Pemetrexed was administered as a 10-min infusion on day 1 of a 21-day cycle. Patients remained on study unless they were discontinued because of disease progression, unacceptable adverse events, inadvertent enrollment, use of excluded concomitant therapy, cycle delay >42 days, or patient refusal.
Patients were instructed to take a daily 1 g multivitamin with 500 μg of folate beginning 1 week before day 1 of cycle 1 until study discontinuation. Vitamin B12 (1000 μg) was intramuscularly injected, starting 1 week before day 1 of cycle 1 and repeated every 9 weeks until study discontinuation.
Patients enrolled in pemetrexed clinical studies have received dexamethasone prophylactically to avoid pemetrexed-induced rash. As this was the first study of pemetrexed in Japanese patients and the incidence of the drug-induced rash in Japanese patients was unknown, the steroid was not to be administered prophylactically.
Dose escalation
In this study, 10 dose levels of pemetrexed, 300, 500, 600, 700, 800, 900, 1000, 1200, 1450, and 1750 mg m−2, were to be examined with a starting dose of 300 mg m−2. At dose levels from 300 to 1000 mg m−2, three patients were to be treated initially. If no dose-limiting toxicities (DLTs) occurred during cycle 1, escalation proceeded to the next dose level. If 1 DLT occurred, three patients were added. If no additional DLTs were observed, escalation proceeded to the next dose level. At dose levels from 1200 to 1750 mg m−2, six patients were to be treated at once. If two or more patients had DLTs at any dose level, dose escalation stopped, and this dose level was considered the MTD. The RD was then established by discussion with principal investigators, and the Efficacy and Safety Evaluation Committee.
A DLT was defined as the occurrence of one of the following toxicities during cycle 1: any grade 3/4 nonhematologic toxicity (except grade 3 nausea/vomiting and AST, ALT, or alkaline phosphatase elevation <10 × upper limit of normal that returns to grade 0–1 by the beginning of cycle 2), grade 3/4 febrile neutropenia (<1000 mm−3 with ⩾38.0°C), grade 4 leucopenia (<1000 mm−3) or neutropenia (<500 mm−3) lasting ⩾4 days, thrombocytopenia (<20 000 mm−3), or thrombocytopenia (⩾20 000 mm−3) requiring platelet transfusion. A failure to start the second cycle by day 42 owing to toxicity was also considered a DLT. All toxicities were assessed according to National Cancer Institute-Common Toxicity Criteria (NCI-CTC) version 2.
Treatment assessments
Tumour response was assessed by the Response Evaluation Criteria in Solid Tumors (RECIST) criteria. Evaluable patients were subjected to CT or MRI measurement to determine the size of tumours at anytime at the discretion of investigators.
Pharmacokinetic analysis
Blood and urine were collected from each patient over a period of 72 h following administration in cycle 1. Blood samples were taken just before administration, at the end of infusion, and approximately 5, 15, 30 min and 1, 2, 4, 6, 8, 24, 48 and 72 h after the start of infusion. Urine was collected over the following time intervals: 0–4, 4–8, 8–12, 12–24, 24–36, 36–48, 48–60, and 60–72 h. Plasma and urine samples were analysed for pemetrexed at Taylor Technology Inc., Princeton, NJ, USA. Plasma samples were analysed using a validated liquid chromatography/electrospray ionisation-tandem mass spectrometry method that generated a linear response over the concentration ranges of 10–2000 ng/ml and 1000–200 000 ng/ml (Latz et al, 2006). Urine samples were analysed using a similar analytical technique (Chaudhary et al, 1999).
Pharmacokinetics were evaluated using noncompartmental methods (WinNonlin Professional Version 3.1; Pharsight Corporation, Cary NC, USA). Pharmacokinetic parameters determined based on plasma concentration vs time data were maximum plasma concentration (Cmax), elimination half-life (t1/2), area under the plasma concentration vs time curve (AUC) from time 0 to infinity (AUC0-∞), volume of distribution at steady-state (Vss) and plasma clearance (CLp) (Rowland and Tozer, 1995). The fraction of drug excreted unchanged in urine (Fe) was calculated by dividing the cumulative amount of pemetrexed excreted unchanged in urine within 72 h (Ae0–72) by the administered dose (Rowland and Tozer, 1995).
Results
Patient disposition and characteristics
From October 2001 to September 2004, a total of 35 Japanese patients were enrolled and 31 were treated at four centres in Japan. Four patients were not treated owing to protocol criteria not met (n=3) and investigator decision (n=1). The majority of patients were male (65%), had an ECOG performance status of 1 (84%), were diagnosed with NSCLC (61%), and received prior chemotherapy (94%) (Table 1).
Dose escalation and dose-limiting toxicities
Three or six patients were enrolled at each dose level from 300 to 1200 mg m−2, except the 900 mg m−2 dose level (Table 2). At this dose level, one additional patient was enrolled because a patient was excluded from the DLT analysis. Before the dose initiation, this patient had grade 3 fasting hyperglycemia that was aggravated after the start of dosing. Therefore, this patient was rated as inappropriate for evaluation.
The first DLT was observed at the 700 mg m−2 dose level. This 66-year-old woman with NSCLC experienced grade 3 ALT elevation. After an additional three patients were enrolled, no other DLTs were observed.
The next DLTs were observed at the 1200 mg m−2 dose level, which enrolled six patients at once. One patient, a 72-year-old woman with MPM, had grade 3 infection at day 6 of cycle 1. Neutropenia was not simultaneously observed in this cycle. After 12 days, the event was resolved with antibiotics. This patient continued in study with dose reduction to 1000 mg m−2. The other patient, a 68-year-old man with NSCLC, had grade 2 rash at day 5 of cycle 1. The severity of the event reached grade 3 at day 7. After 9 days from the occurrence, rash was resolved with dexamethasone and H1-antihistamine. This patient continued in study without dose reduction. As two DLTs were observed, the 1200 mg m−2 dose level was considered as the MTD. The RD for subsequent phase II studies was then evaluated to be pemetrexed 1000 mg m−2. Both events were considered as drug-related events by investigators.
Safety
The safety evaluation was completed from data obtained from cycle 1–6 for all dose levels except 1200 mg m−2 (cycle 1–3). These data were collected and analysed to evaluate safety when the MTD and RD were determined. The major toxicities observed in >50% of patients during all cycles evaluated for this report included rash, nausea, anorexia, fatigue, ALT elevation, AST elevation, lactate dehydrogenase elevation, leucopenia, neutropenia, lymphopenia, hematocrit decreased, haemoglobin decreased and erythropenia (Table 3). The most commonly reported grade 3/4 toxicity was neutropenia: nine patients (29%) had grade 3 neutropenia, and one patient (3%) had grade 4 neutropenia. Other grade 3/4 hematologic toxicities were grade 3 leucopenia in four patients (13%), grade 4 leucopenia in one patient (3%), grade 3 lymphopenia in four patients (13%), and grade 3 haemoglobin decreased in two patients (6%). The most commonly reported grade 3 nonhematologic toxicity was ALT elevation (four patients (13%)). Other grade 3 toxicities included AST elevation in one patient (3%), anorexia in one patient (3%), infection in one patient (3%), malaise in one patient (3%), and rash in one patient (3%) were observed. No grade 4 nonhematologic toxicities were reported.
The only serious adverse event was observed at the 900 mg m−2 level. This 71-year-old man with NSCLC experienced grade 1 pyrexia at day 18 of cycle 3 and was hospitalized; however, the event was resolved the next day. The investigator did not consider it as a drug-related event. One patient at 900 mg m−2 level discontinued treatment owing to adverse events (neutropenia, anorexia, and pyrexia). No deaths were observed during the study period or for 31 days after the last dose.
At the 900 mg m−2 and higher dose levels, all patients had either grade 1/2 or grade 3/4 rash. At cycle 1, 25 patients experienced rash. Of these, 20 patients received corticosteroid. At or after cycle 2, corticosteroid treatment was given only for nine rash events, whereas rash events were observed in 20 cycles in cumulative total among patients. In addition, the severity of rash quickly improved or disappeared after administration of corticosteroid. Although the protocol allowed corticosteroid use for prevention of rash from cycle 2, only seven patients actually received the preventive treatment. Among those who did not receive the prophylactic corticosteroid, the incidence of a rash observed at, or after, cycle 2 was about one-third of the incidence observed in cycle 1.
Pharmacokinetic analysis
Mean dose-normalised pemetrexed plasma concentration vs time profiles following single doses of 300–1200 mg m−2 pemetrexed are provided in Figure 1. This body surface area (BSA)-normalized dose range represents absolute doses of 414–2018 mg in Japanese patients with a mean BSA of 1.64 m2 (range, 1.36–1.97 m2).

Mean dose-normalised pemetrexed plasma concentration–time profiles following single-dose administration in Japanese patients.
Pharmacokinetic parameters for each dose group are summarised in Table 4. Lack of a monotonic trend in CLp and Vss between cohorts indicated that pemetrexed pharmacokinetics are consistent across dose groups. Consistency of pemetrexed pharmacokinetics across dose groups is also illustrated by the lack of systematic pattern across dose groups in the dose-normalised plasma concentration vs time profiles (Figure 1). The overall mean t1/2 is approximately 2.74 h and was essentially similar across all dose groups (range, 2.28–3.62 h).
In this study, pemetrexed was primarily excreted unchanged in urine, which is consistent with its known elimination pathway (i.e., renal excretion). The Fe averaged 0.752 (range, 0.645–0.827). Mean Fe values were consistent across dosing cohorts.
Tumour response
In this study, 23 of the 31 patients were evaluable for response by RECIST criteria (Table 5). Partial responses (PRs) were observed in four patients with NSCLC (one patient each at 500, 700, 800, and 1200 mg m−2) and one patient with thymoma at 500 mg m−2. In addition, one patient with NSCLC at 500 mg m−2 had a PR by the World Health Organization criteria, but was not evaluable via RECIST.
Discussion
This is the first phase I study of pemetrexed in Japanese patients. The MTD for pemetrexed administered with FA/VB12 was 1200 mg m−2 and determined the RD for subsequent phase II studies was 1000 mg m−2.
In contrast with the previously determined MTD (600 mg m−2) without vitamin supplementation (Rinaldi et al, 1999), our MTD increased by a factor of 2 whereas maintaining a tolerable safety profile. Niyikiza et al (2002a, 2002b) conducted a multivariate analysis on 246 patients in phase II pemetrexed studies without vitamin supplementation, and the incidence of grade 4 neutropenia was 32% and grade 4 thrombocytopenia was 8% . Also 6% of patients had grade 3/4 diarrhoea, 5% had grade 3/4 mucositis, and a 5% incidence of drug-related death occurred. In contrast, our study had grade 4 neutropenia of only 3% (one patient) and no grade 4 thrombocytopenia. In addition, no grade 3/4 diarrhoea or mucositis, and no drug-related deaths were observed.
In the pivotal phase III study of NSCLC patients, those who received pemetrexed (500 mg m−2) plus vitamin supplementation had a lower incidence of severe toxicities compared to those who received docetaxel (75 mg m−2), including grade 3/4 neutropenia (5.3 vs 40.2%) and grade 3/4 diarrhoea (0.4 vs 2.5%) (Hanna et al, 2004).
Dose-dependency for toxicity of pemetrexed plus supplementation was further investigated to understand the effect of supplementation on safety. The patients in this study were divided into three groups by doses: low dose (300–600 mg m−2 (n=9)), middle dose (700–900 mg m−2 (n=13)), and high dose (1000 and 1200 mg m−2 (n=9)). Grade 1/2 toxicity such as erythropenia, lymphopenia, hematocrit decreased, ALT and AST elevation, and anorexia increased dose dependently from approximately 20–50% to approximately 75%. However, there was no obvious correlation between grade 3/4 toxicity and dose group. Therefore, high dose levels of pemetrexed with FA/VB12 is expected to be tolerable enough for patients.
In this study, severe rash was rarely observed even without the prophylactic corticosteroid. Although this result suggests that the steroid premedication for prevention of severe rash is no longer necessary for patients with pemetrexed treatment if the FA/VB12 is concomitantly conducted, it would be too early to conclude it as the data of patients untreated with the premedication are limited at this moment.
The pharmacokinetic results in our study were consistent with a phase I study of pemetrexed without vitamin supplementation in western patients by Rinaldi et al (1999) In that study, pemetrexed t1/2 was 3.1 h; and CL was 85 ml/min (Rinaldi et al, 1999 and unpublished results). In our study, the t1/2 of pemetrexed was about 2.7 h; and CL was 81.9 ml/min. Additionally, the Fe of pemetrexed was similar for Japanese patients (75% in our study) and western patients (78% in the Rinaldi study (Rinaldi et al, 1999)). These results indicate that pharmacokinetics of pemetrexed in Japanese patients are similar to those in western patients.
Although our study is the first phase I study to evaluate pemetrexed with FA/VB12 in Japanese patients, a similar phase I study has been conducted in western patients. In the preliminary results of that study, heavily pretreated patients had a MTD of 925 mg m−2, and lightly pretreated patients had a MTD of 1050 mg m−2 (Hammond et al, 2003). The comparison of these two studies suggests that the improved tolerability experienced by Japanese patients when pemetrexed is administered with FA/VB12 is not attributable to ethnic differences; rather, it is attributable to the vitamin supplementation.
In our phase I study, four NSCLC patients and one thymoma patient had PRs. Except for one, all of the patients with PR had ⩾3 prior chemotherapy regimens. The NSCLC patients with PRs received doses of pemetrexed higher than 500 mg m−2, which is the approved dose for NSCLC treatment in a number of countries. Therefore, subsequent phase II studies using our RD of 1000 mg m−2 with vitamin supplementation could show more prominent antitumour activity for cancer patients. To examine this hypothesis, a Japanese phase II study is being conducted, examining pemetrexed 500 or 1000 mg m−2 every 3 weeks with full supplementation for patients with locally advanced or metastatic NSCLC. Clinical trials for other tumours, including MPM, are also ongoing. For the prophylactic corticosteroid, as severe rash was not frequently observed in this study, the steroid is not to be administered prophylactically in both currently on-going studies.
In conclusion, pemetrexed with FA/VB12 resulted in a tolerable toxicity profile. The MTD was 1200 mg m−2. The RD was 1000 mg m−2.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Alati T, Worzalla JF, Shih C, Bewley JR, Lewis S, Moran RG, Grindey GB (1996) Augmentation of the therapeutic activity of lometrexol -(6-R)5,10-dideazatetrahydrofolate- by oral folic acid. Cancer Res 56: 2331–2335
Chaudhary AK, Schannen V, Knadler MP (1999) Analysis of LY231514 in plasma and urine using perchloric acid with LC/MS/MS. In Presented at the Proceedings of 47th ASMS Conference on Mass Spectrometry and Allied Topics. Dallas, TX, June 13–17 (abstract)
Hammond LA, Forero L, Beeram M, Forouzesh B, De Bono J, Tolcher A, Patnaik A, Monroe P, Clark R, Rowinsky EK (2003) Phase 1 study of pemetrexed (LY231514) with vitamin supplementation in patients with locally advanced or metastatic cancer. Proc Am Soc Clin Oncol 22: 133 (abstract 532)
Hanauske A, Chen V, Paoletti P, Niyikiza C (2001) Pemetrexed disodium: a novel antifolate clinically active against multiple solid tumors. Oncologist 6: 363–373
Hanna N, Shepherd FA, Fossella FV, Pereira JR, De Marinis F, von Pawel J, Gatzemeier U, Tsao TC, Pless M, Muller T, Lim HL, Desch C, Szondy K, Gervais R, Shaharyar, Manegold C, Paul S, Paoletti P, Einhorn L, Bunn Jr PA (2004) Randomized phase III trial of pemetrexed versus docetaxel in patients with non-small-cell lung cancer previously treated with chemotherapy. J Clin Oncol 22: 1589–1597
Laohavinij S, Wedge SR, Lind MJ, Bailey N, Humphreys A, Proctor M, Chapman F, Simmons D, Oakley A, Robson L, Gumbrell L, Taylor GA, Thomas HD, Boddy AV, Newell DR, Calvert AH (1996) A phase I clinical study of the antipurine antifolate lometrexol (DDATHF) given with oral folic acid. Invest New Drugs 14: 325–335
Latz JE, Chaudhary A, Ghosh A, Johnson RD (2006) Population pharmacokinetic analysis of ten phase II clinical trials of pemetrexed in cancer patients. Cancer Chemother Pharmacol 57: 401–411
Maughan TS, James RD, Kerr D, McArdle C, Ledermann JA, Seymour M, Johnston C, Stephens RJ (1999) Preliminary results of a multicentre randomised trial comparing 3 chemotherapy regimens (de Gramont, Lokich and Raltitrexed) in metastatic colorectal cancer. Proc Am Soc Clin Oncol 18: 262a (abstract 1007)
McDonald AC, Vasey PA, Adams L, Walling J, Woodworth JR, Abrahams T, McCarthy S, Bailey NP, Siddiqui N, Lind MJ, Calvert AH, Twelves CJ, Cassidy J, Kaye SB (1998) A phase I and pharmacokinetic study of LY231514, the multitargeted antifolate. Clin Cancer Res 4: 605–610
Mendelsohn LG, Gates SB, Habeck LL, Shackelford KA, Worzalla J, Shih C, Grindey GB (1996) The role of dietary folate in modulation of folate receptor expression, folylpolyglutamate synthetase activity and the efficacy and toxicity of lometrexol. Adv Enzyme Regul 36: 365–381
Morgan SL, Baggott JE, Vaughn WH, Young PK, Austin JV, Krumdieck CL, Alarcon GS (1990) The effect of folic acid supplementation on the toxicity of low-dose methotrexate in patients with rheumatoid arthritis. Arthritis Rheum 33: 9–18
Niyikiza C, Baker SD, Seitz DE, Walling JM, Nelson K, Rusthoven JJ, Stabler SP, Paoletti P, Calvert AH, Allen RH (2002a) Homocysteine and methylmalonic acid: markers to predict and avoid toxicity from pemetrexed therapy. Mol Cancer Ther 1: 545–552
Niyikiza C, Hanauske A-R, Rusthoven JJ, Calvert AH, Allen R, Paoletti P, Bunn Jr PA (2002b) Pemetrexed safety and dosing strategy. Semin Oncol 29: 24–29
Rinaldi DA, Burris HA, Dorr FA, Woodworth JR, Kuhn JG, Eckardt JR, Rodriguez G, Corso SW, Fields SM, Langley C (1995) Initial phase I evaluation of the novel thymidylate synthase inhibitor, LY231514, using the modified continual reassessment method for dose escalation. J Clin Oncol 13: 2842–2850
Rinaldi DA, Kuhn JG, Burris HA, Dorr FA, Rodriguez G, Eckhardt SG, Jones S, Woodworth JR, Baker S, Langley C, Mascorro D, Abrahams T, Von Hoff DD (1999) A phase I evaluation of multitargeted antifolate (MTA, LY231514), administered every 21 days, utilizing the modified continual reassessment method for dose escalation. Cancer Chemother Pharmacol 44: 372–380
Rosenberg LE, Fenton WA (1989) Disorders of propionate and methylmalonate metabolism. In The Metabolic Basis of Inherited Disease Scriver CR, Sly WL, (eds). 6th edition, pp 821–844. McGraw-Hill: New York
Rowland M, Tozer TN (1995) Clinical Pharmacokinetics: Concepts and Applications (ed 3). Lippincott Williams & Wilkins: Baltimore
Savage DG, Lindenbaum J, Stabler SP, Allen RH (1994) Sensitivity of serum methylmalonic acid and total homocysteine determinations for diagnosing cobalamin and folate deficiencies. Am J Med 96: 239–246
Shih C, Habeck LL, Mendelsohn LG, Chen VJ, Schultz RM (1998) Multiple folate enzyme inhibition: mechanism of a novel pyrrolopyrimidine-based antifolate LY231514 (MTA). Adv Enzyme Regul 38: 135–152
Taylor EC, Patel HH (1992) Synthesis of pyrazolo[3,4-d]pyrimidine analogues of the potent antitumor agent N-{4-[2-(2-amino-4(3H)-oxo-7H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl}-L-glutamic acid (LY231514). Tetrahedron 48: 8089–8100
Vogelzang NJ, Rusthoven JJ, Symanowski J, Denham C, Kaukel E, Ruffie P, Gatzemeier U, Boyer M, Emri S, Manegold C, Niyikiza C, Paoletti P (2003) Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol 21: 2636–2644
Acknowledgements
This study has been supported and funded by Eli Lilly Japan KK, Kobe, Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Nakagawa, K., Kudoh, S., Matsui, K. et al. A phase I study of pemetrexed (LY231514) supplemented with folate and vitamin B12 in Japanese patients with solid tumours. Br J Cancer 95, 677–682 (2006). https://doi.org/10.1038/sj.bjc.6603321
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6603321
Keywords
This article is cited by
-
Effect of renal function on pemetrexed-induced haematotoxicity
Cancer Chemotherapy and Pharmacology (2016)
-
A patient with malignant pleural mesothelioma complicated with Dubin–Johnson syndrome treated with cisplatin/pemetrexed combination therapy: the need to consider mutation to the ATP-binding cassette-C2 gene and the pharmacokinetics of pemetrexed
International Cancer Conference Journal (2015)
-
Third-space fluid distribution of pemetrexed in non-small cell lung cancer patients
Cancer Chemotherapy and Pharmacology (2014)
-
Feasibility study of two schedules of sunitinib in combination with pemetrexed in patients with advanced solid tumors
Investigational New Drugs (2012)
-
Safety and pharmacokinetics of intrathecal administration of pemetrexed in rats
Cancer Chemotherapy and Pharmacology (2011)
