




Abstract
Drug resistance is one of the main obstacles to the successful treatment of cancer. The availability of agents that are highly effective against drug-resistant cancer cells is therefore essential. The present study was performed to examine the anticancer effects of evodiamine, a major constituent of the Chinese herb Evodiae fructus , in adriamycin-resistant human breast cancer NCI/ADR-RES cells. Evodiamine inhibited the proliferation of NCI/ADR-RES cells in a concentration-dependent manner with a GI 50 of 0.59 ± 0.11 μM. This agent also caused a substantial apoptosis at 1 μM. FACScan flow cytometric analysis of cell cycle progression revealed that a G 2 /M arrest was initiated after a 12-h exposure to the drug. Evodiamine increased tubulin polymerization as determined by the immunocytochemical and in vivo tubulin polymerization analyses. In a time- and concentration-dependent manner, evodiamine also promoted the phosphorylations of Raf-1 kinase and Bcl-2. The phosphorylation site of Raf-1 kinase was identified to be serine 338 . The in vivo anticancer effects of evodiamine were evaluated in Balb-c/nude mice following a tumor xenograft implantation of NCI/ADR-RES cells. The antitumor activity of evodiamine against the human multiple-drug resistant tumor xenograft was found to be superior to that of paclitaxel. Evodiamine therefore represents a highly promising chemotherapeutic agent in the treatment of human multiple-drug resistant cancer cells.
Introduction
Microtubules, which are formed by a self-assembly of α and β tubulin heterodimers, are integral components of the mitotic spindle, a structure that is critical to a variety of fundamental functions, including the regulation of cell proliferation, differentiation and apoptosis ( 1 ). Antimitotic drugs that target microtubules have potential clinical utility and are usually classified into two main groups. The first group is represented by microtubule-stabilizing agents that promote microtubule polymerization. Members of this group include paclitaxel, docetaxel and the epothilones. The second group is represented by agents that destabilize microtubules and thereby inhibit microtubule polymerization. Members of this group inlcude the vinca alkaloids, estramustine, colchicines and cryptophycins ( 2 ). Clinically, the taxenes and vinca alkaloids are among the best characterized antimitotic drugs and have therefore been used widely to treat various kinds of human cancers. The utility of these drugs, however, is limited by the development of drug resistance ( 3 , 4 ).
Microtubule-targeting agents act primarily to suppress spindle-microtubule dynamics, resulting in a mitotic slowing or blockade at the metaphase/anaphase transition and the consequent triggering of apoptotic cell death ( 5 ). Disruption of microtubule dynamics is also associated with alterations in the activities of several protein kinases, including Raf-1, JNK and p34cdc2 ( 6 , 7 ). These kinases are known to be involved in Bcl-2 phosphorylation either directly or indirectly. Bcl-2 phosphorylation has been shown to be induced by microtubule-targeting agents but not by DNA-damaging agents ( 8 ). Phosphorylation of Bcl-2 inactivates this protein resulting in apoptosis ( 9 ). Inactivation may be associated with the release of Bax from Bcl-2/Bax heterodimers ( 10 ).
The Ser/Thr kinase Raf-1, a ubiquitously expressed homologue of the v-raf oncogene, functions at the entry to the mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) cascade and in the transmission of mitogenic, developmental and survival signals in eukaryotic cells ( 11 ). However, Raf-1 kinase is stimulated during both mitogenic activation and mitosis ( 12 ). The mechanism whereby Raf-1 is activated during mitosis and the subsequent downstream effects of activation are distinct from those involved in growth factor stimulation. In mitotic cells, Ras-independent signaling causes the cytoplasmic activation of Raf-1 kinase without the activation of the MEK/ERK pathway ( 13 – 15 ). Microtubule-binding agents are reported to promote Raf-1/Bcl-2 phosphorylation and subsequent disruption of the microtubule network, leading to apoptotic cell death ( 16 , 17 ).
Multiple-drug resistance is the major clinical obstacle in cancer therapy. Drug resistance can be intrinsic (failure of the first chemotherapy) or acquired (response to the first chemotherapy but failure of the second). In either case, however, tumors become refractory to a variety of structurally diverse anticancer drugs. Failure of clinical chemotherapy may be attributable to pharmacokinetic, tumor environment, or cancer cell-specific issues. The best characterized mechanism of drug resistance to microtubule inhibitors is the overexpression of the P-glycoprotein drug efflux pump ( 18 ). In addition, structural and functional alterations in the α- and β-tubulins of tumor cells, resulting from either genetic mutations or post-translational modifications, are associated with an acquired resistance to taxenes and vinca alkaloids ( 4 ). Identification of natural and synthetic compounds with therapeutic effects on multiple-drug resistant tumors remains an attractive goal.
Evodiamine is one of the major bioactive compounds that have been isolated and purified from the traditional Chinese herbal medicine, Wu-Chu-Yu. Evodiamine has been shown to possess anti-inflammatory ( 19 ), anti-obesity ( 20 ) and anti-nociceptive effects ( 21 ). Furthermore, evodiamine is documented to inhibit tumor cell proliferation, cell migration with invasion and lung metastasis ( 22 , 23 ). In this study, evodiamine was found to possess inhibitory activity against the multiple-drug resistant human breast cancer NCI/ADR-RES cells. The mechanism whereby evodiamine produces this inhibition was also explored in these cells.
Materials and methods
Materials
Evodiamine was obtained from Matsuura Yakugyo Co., Ltd. (Nagoya, Japan). Paclitaxel and vincristine were purchased from Sigma Chemical Co. (St Louis, MO). The compounds were dissolved in dimethyl sulfoxide and an equal volume of vehicle was added in the control experiments. RPMI-1640 medium, fetal bovine serum (FBS) and the other cell culture reagents were obtained from Gibco BRL Life Technologies (Grand Island, NY). Propidium iodide, anti-β-tubulin monoclonal antibody and secondary antibodies (mouse, rabbit or goat IgG horseradish peroxidase conjugate and FITC-conjugated mouse antihuman antibody) were bought from Sigma (St Louis, MO). The fluorescein-conjugated paclitaxel (Oregon green 488 paclitaxel) was purchased from Molecular Probes Inc. (Eugene, OR). The sources of the primary antibodies were listed: the caspase-8, caspase-9, phospho-p44/42 MAP kinase (Thr202/Tyr204) and phospho-SAPK/JNK (Thr183/Tyr185) antibodies from Cell Signaling Technologies (Boston, MA), the anti-human caspase-6 and caspase-7 antibodies from BD Biosciences PharMingen (SanDiego, CA), the actin, cell division cycle 25c (cdc25c), cyclin B1, cdc2 kinase, Bcl-2 and phospho-Bcl-2 (Ser70) antibodies from Santa Cruz Biotechnology (Santa Cruz, CA) and the phospho-Raf-1 (Ser338) antibody from Upstate Biotechnology (Lake Placid, NY).
Cell lines and cell culture
NCI/ADR-RES cell line, formerly known as MCF-7/adr, was obtained from the DTP Human Tumor Cell Line Screen (Developmental Therapeutics Program, NCI). Human breast carcinoma cells, MCF-7 was purchased from American Type Culture Collection (Rockville, MD). Both breast cancer cell lines were propagated in RPMI-1640 supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin and were cultured at 37°C in a humidified atmosphere of 95% air–5% CO 2 .
Sulforhodamine B (SRB) assay
For the proliferation assay of tumor cells, NCI/ADR-RES cells and MCF-7 cells are fixed in situ with 10% trichloroacetic acid (TCA) after cell inoculation, to represent a measurement of the cell population at the time of drug addition. The plates were incubated for an additional 48 h following drug addition and the assay was terminated by 10% TCA. SRB dye at 0.4% (w/v) in 1% acetic acid was added to stain the cells. Unbound dye was removed by repeated washing with 1% acetic acid and the plates were air dried. Bound stain was subsequently solubilized with 10 mM trizma base, and the absorbance is read on a microplate reader at a wavelength of 515 nm. Growth inhibition of 50% (GI 50 ) is calculated as the drug concentration, which caused a 50% reduction in the net protein increase in control cells during the drug incubation.
Apoptosis assay
For a quantitative assessment of oligonucleosomal DNA fragmentation, 10 4 cells were incubated with medium containing the agents tested. Apoptosis was detected by Cell Death ELISA PLUS kit (Roche Molecular Biochemicals, Mannheim, Germany).
Colony-formation assay
Cells were suspended in 0.36% bactoagar (Difco, Detroit, MI) and seeded into 24-well plates over a 0.6% agar base layer in RPMI with 10% FBS. Every 3 days, ∼250 μl of media with treatments was added to each plate. After 2 weeks, colonies were stained overnight with 0.5 mg/ml 3-(4,5-dimethyldiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT, Sigma) in phosphate-buffered saline (PBS) and counted. Each experiment was performed in triplicate and repeated at least three times.
Flow cytometric cell cycle analysis
Following drug treatment, the cells were harvested by trypsinization, washed with PBS, then pellets were resuspended and fixed in ethanol (70%, v/v) and incubated at −20°C for 6 h. Cells were stained with propidium iodide staining buffer containing Triton X-100 (0.1%, v/v), RNase A (100 μg/ml) and propidium iodide (80 μg/ml) for 30 min. Cell cycle distribution was performed using a FACScan flow cytometry with CellQuest software (Becton Dickinson, San Jose, CA).
Immunocytochemistry and confocal microscopy
Cells were grown and treated on eight-chamber slides. After treatment, the cells were fixed with methanol, blocked with 2% bovine serum albunin (BSA) stained with anti-β-tubulin monoclonal antibody (1:100 dilution, Sigma, UK) and then the FITC-conjugated mouse antihuman antibody (1:100 dilution, Sigma). Nuclear staining was performed with 1 μg/ml 4,6-diamidino-2-phenylindole. Cells were imaged with Leica TCS SP2 Spectral Confocal System.
In vivo tubulin polymerization assay
To quantitate the degree of in vivo tubulin polymerization in response to tubulin-binding agents, the in vivo tubulin polymerization assay was performed as described previously ( 24 ). Briefly, treated cells are lysed with 200 μl of hypotonic buffer containing 1 mM MgCl 2 , 2 mM EGTA, 0.5% NP-40, 2 mM phenylmethylsulfonyl fluoride (PMSF), 200 U/ml aprotinin, 100 μg/ml soybean trypsin inhibitor, 5.0 mM ε-amino caproic acid, 1 mM benzamidine and 20 mM Tris–HCl, pH 6.8. The cell lysate in which the cytosolic and cytoskeletal fractions containing soluble (s) and polymerized (p) tubulin, respectively, was separated by centrifugation, resolved by electrophoresis through SDS–PAGE, and immunoblotted with an antibody against β-tubulin.
Western blot analysis
The breast cancer cells were cultured and treated in 60-mm tissue culture Petri dishes. To harvest the cells, they were scraped from culture dishes with lysis buffer containing 1 mM EGTA, 1 mM EDTA, 150 mM NaCl, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM PMSF, 1 mM NaVO 4 and 1 μg/ml leupeptin and 1 μg/ml aprotinin in 20 mM Tris–HCl buffer, pH 7.5. Cell lysates were centrifuged at 13 000 g for 30 min. Total protein was determined and equal amounts of protein were separated by SDS–PAGE and immunoblotted with specific antibodies. Proteins were visualized by enhanced chemiluminescence (Amersham, Buckinghamshire, UK). For detection of the Raf-1 protein expression, the membrane was probed with phospho-ser338-Raf-1 in advance. After visualization, it was then stripped and re-probed with Raf-1 antibody.
In vivo antitumor effect
Female BALB/c athymic ( nu+/nu+ ) mice, 4–6 weeks of age, were purchased from National Science Council (Taipei, Taiwan). Mice were acclimatized at the Animal Facility of National Taiwan University before being injected with cancer cells. Animals were provided with sterilized food and water. Mice were injected subcutaneously with 10 7 NCI/ADR-RES cells in 0.1 ml. After 2 weeks, when established tumors of ∼100 mm 3 were detected, 6 mice per group were treated. Evodiamine (10 mg/kg) suspended in 0.5% carboxymethyl cellulose (CMC) was taken orally twice a day. The stock of paclitaxel injection was 6 mg/ml soluble in cremophor EL/ethanol and was diluted to 2 mg/ml with PBS before injection. Paclitaxel (20 mg/kg) was given as an intraperitoneal injection twice a week and the injection volume was 0.2 ml. A separate control group for paclitaxel-treated mice received cremophor EL/ethanol/PBS via an intraperitoneal injection as well. Tumor volume was determined by caliper measurements (mm) and using the formula for an ellipsoid sphere: 0.5 × L × W 2 = mm 3 . All animal studies were conducted in accordance with the guidelines of the National Institutes of Health ‘Guide for the Care and Use of Animals’.
Statistical analysis
Results are expressed as means ± SEM of the indicated number of independent experiments. Student's t -test was calculated to compare the mean of each group with that of the control group and P -values <0.05 were considered significant.
Results
Evodiamine suppressed growth and survival of NCI/ADR-RES cells
The antiproliferative effect of evodiamine was evaluated in human multiple-drug resistant breast cancer NCI/ADR-RES cells. Evodiamine inhibited NCI/ADR-RES cell proliferation in a concentration-dependent manner with a GI 50 of 0.59 ± 0.11 μM ( Figure 1A ). In contrast, NCI/ADR-RES cells exhibited a much lower susceptibility to paclitaxel and vincristine, chemotherapeutic agents that are used widely in the treatment of breast cancer ( Table I ). Additionally, the long-term viability of NCI/ADR-RES cells, as measured by the colony-formation assay, was found to be significantly decreased after exposure of the cells to evodiamine ( Figure 1B ).
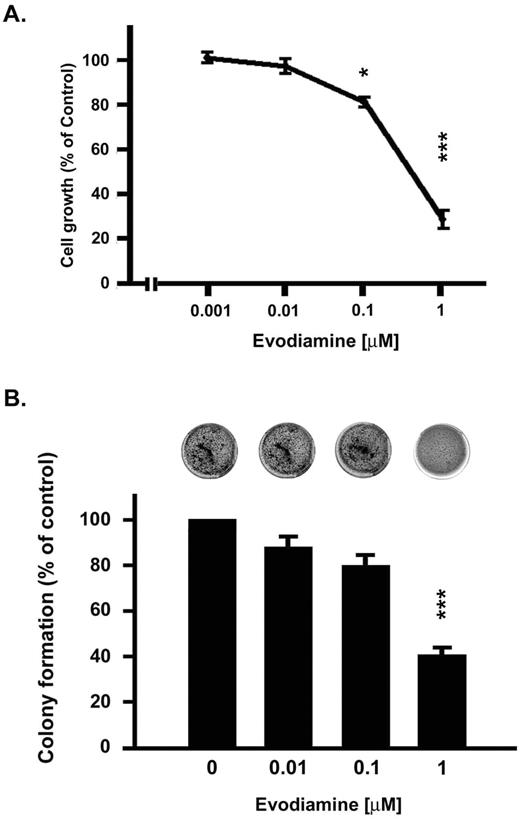
Evodiamine inhibited the growth of NCI/ADR-RES cells. ( A ) Effects on proliferation. Cells were incubated with evodiamine for 48 h, and incubations were terminated by the addition of TCA. SRB dye (0.4% w/v in 1% acetic acid) was added to stain the cells. Bound dye was subsequently solubilized with 10 mM Trizma base, and absorbance of the solution at 515 nm was determined. ( B ) Effects on colony formation. Cells were suspended in 0.36% bactoagar and seeded into 24-well plates over a 0.6% agar base layer in RPMI medium containing 10% FBS. After 2 weeks, colonies were stained overnight with 0.5 mg/ml MTT in PBS and counted. Results were expressed as mean ± SEM of four independent experiments. *P < 0.05, ***P < 0.001 compared with control group.
The antiproliferative effects of evodiamine and other clinical chemotherapeutic compounds, paclitaxel and vincristine
| GI 50 (μM) | Verapamil 30 μM | NCI/ADR-RES cells | MCF-7 cells |
|---|---|---|---|
| Evodiamine | − | 0.5933 ± 0.1102 | 0.6598 ± 0.0517 |
| + | 0.1333 ± 0.0447 * | 0.3388 ± 0.1262 | |
| Paclitaxel | − | 4.9150 ± 1.2218 | 0.0029 ± 0.0017 |
| + | 0.0048 ± 0.0012 ** | 0.0002 ± 0.0001 | |
| Vincristine | − | 5.0989 ± 1.2639 | 0.0952 ± 0.0418 |
| + | 0.0071 ± 0.0010 ** | 0.0049 ± 0.0021 |
| GI 50 (μM) | Verapamil 30 μM | NCI/ADR-RES cells | MCF-7 cells |
|---|---|---|---|
| Evodiamine | − | 0.5933 ± 0.1102 | 0.6598 ± 0.0517 |
| + | 0.1333 ± 0.0447 * | 0.3388 ± 0.1262 | |
| Paclitaxel | − | 4.9150 ± 1.2218 | 0.0029 ± 0.0017 |
| + | 0.0048 ± 0.0012 ** | 0.0002 ± 0.0001 | |
| Vincristine | − | 5.0989 ± 1.2639 | 0.0952 ± 0.0418 |
| + | 0.0071 ± 0.0010 ** | 0.0049 ± 0.0021 |
The proliferation of human breast cancer cells, NCI/ADR-RES, was evaluated by SRB assay as previously described in Material and methods. Results were expressed as mean ± SEM of four independent experiments.
P < 0.05,
P < 0.01 compared with untreated verapamil group.
The antiproliferative effects of evodiamine and other clinical chemotherapeutic compounds, paclitaxel and vincristine
| GI 50 (μM) | Verapamil 30 μM | NCI/ADR-RES cells | MCF-7 cells |
|---|---|---|---|
| Evodiamine | − | 0.5933 ± 0.1102 | 0.6598 ± 0.0517 |
| + | 0.1333 ± 0.0447 * | 0.3388 ± 0.1262 | |
| Paclitaxel | − | 4.9150 ± 1.2218 | 0.0029 ± 0.0017 |
| + | 0.0048 ± 0.0012 ** | 0.0002 ± 0.0001 | |
| Vincristine | − | 5.0989 ± 1.2639 | 0.0952 ± 0.0418 |
| + | 0.0071 ± 0.0010 ** | 0.0049 ± 0.0021 |
| GI 50 (μM) | Verapamil 30 μM | NCI/ADR-RES cells | MCF-7 cells |
|---|---|---|---|
| Evodiamine | − | 0.5933 ± 0.1102 | 0.6598 ± 0.0517 |
| + | 0.1333 ± 0.0447 * | 0.3388 ± 0.1262 | |
| Paclitaxel | − | 4.9150 ± 1.2218 | 0.0029 ± 0.0017 |
| + | 0.0048 ± 0.0012 ** | 0.0002 ± 0.0001 | |
| Vincristine | − | 5.0989 ± 1.2639 | 0.0952 ± 0.0418 |
| + | 0.0071 ± 0.0010 ** | 0.0049 ± 0.0021 |
The proliferation of human breast cancer cells, NCI/ADR-RES, was evaluated by SRB assay as previously described in Material and methods. Results were expressed as mean ± SEM of four independent experiments.
P < 0.05,
P < 0.01 compared with untreated verapamil group.
Quantitative assessment of oligonucleosomal DNA fragmentation was used to measure apoptotic cell death. Massive apoptosis of NCI/ADR-RES cell was detected ( Figure 2A ) after 24 h of treatment with evodiamine. Traditionally two pathways, termed the mitochondrial ‘intrinsic’ and the transmembrane ‘extrinsic’ pathways, by which caspase is triggered, are held responsible for the induction of apoptotic cell death ( 25 , 26 ). The activation states of caspase-3, -6, -7 -8 and -9 were therefore examined in the NCI/ADR-RES cells after 24 h of treatment with evodiamine. Although no changes in procaspase-3, -6 or -8 were observed, caspase-9 and caspase-7 were found to be activated during evodiamine-induced apoptosis ( Figure 2B ).
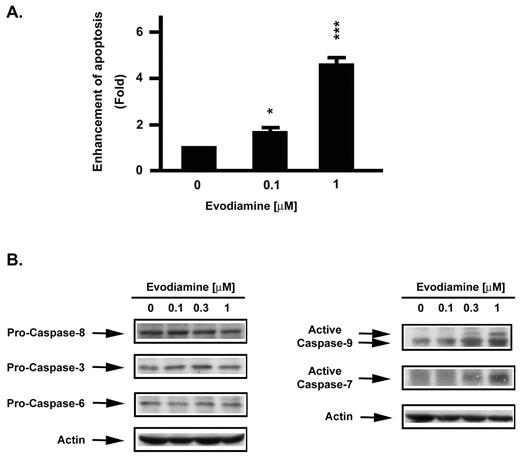
Evodiamine induced apoptosis and promoted activation of caspases in NCI/ADR-RES cells. ( A ) Measurements of apoptosis. For quantitative assessment of oligonucleosomal DNA fragmentation, cells were treated with the agents indicated. Apoptosis was detected with the Cell Death ELISA PLUS kit. Enhancement of apoptosis was determined in relation to control cells receiving vehicle alone. Results were expressed as mean ± SEM of three independent experiments. *P < 0.05, ***P < 0.001 compared with control group. ( B ) Caspase activations. After 24 h treatments with vehicle or evodiamine, cells were harvested and subjected to lysis. Protein measurements were performed, and equal amounts of protein were subjected to SDS–PAGE. Caspase-3, -6, -7, -8 and -9 were detected by immunoblotting and the proteins were visualized by enhanced chemiluminescence.
P-glycoprotein is known to be overexpressed in NCI/ADR-RES cells ( 27 ). However, efflux of the fluorescent dyes calcein and rhodamine 123, which serves as a measure of P-glycoprotein activity, was not affected by evodiamine (data not shown). Furthermore, the GI 50 value of evodiamine did not change prominently during incubations of the drug in combination with verapamil, a traditional P-glycoprotein inhibitor. In view of these findings, evodiamine is unlikely to serve as a substrate for P-glycoprotein in tumor cells.
Evodiamine induced cell cycle progression arrest at the G 2 /M phase in NCI/ADR-RES cells
The effect of evodiamine on cell cycle progression was examined by FACScan flow cytometric analysis. NCI/ADR-RES cells were exposed for 24 h to varying concentrations of evodiamine. A concentration-dependent block at the G 2 /M phase was observed in association with a loss of cells in the G 0 /G 1 phase. The number of cells in the sub-G 1 phase, which represents an apoptotic state, was also clearly increased ( Figure 3 ). The cell cycle progression arrest at the G 2 /M phase was imposed after 12 h of exposure to evodiamine, whereas arrest at the sub-G 1 phase required 24 h of treatment (data not shown).
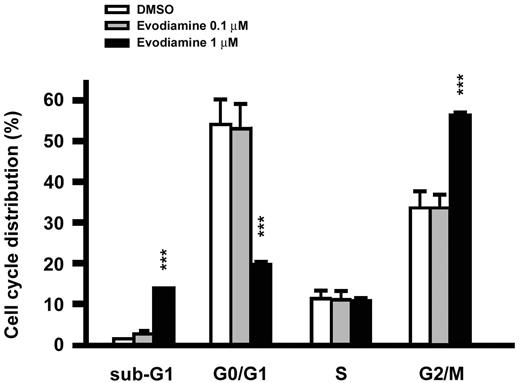
Evodiamine induced cell cycle progression arrest at the G 2 /M phase in NCI/ADR-RES cells. Following drug treatments, the cells were harvested by trypsinization, collected by centrifugation, resuspended and fixed in ethanol, and incubated at −20°C. Cells were then stained with propidium iodide for 30 min, and cell cycle distribution was determined using a FACScan flow cytometry with CellQuest software. Results were expressed as mean ± SEM. of four independent experiments. ***P < 0.001 compared with control group.
Effects of evodiamine on microtubule organization in NCI/ADR-RES cells
Anti-microtubule agents characteristically disrupt the cell cycle. The organization of the microtubule network in NCI/ADR-RES cells was therefore examined by immunocytochemical and in vivo tubulin polymerization measurements. The microtubule network in control cells exhibited normal organization and arrangement. Treatment with evodiamine (1 μM) enhanced microtubule polymerization as visualized by an increased density of cellular microtubules and formation of microtubule bundles ( Figure 4A ). The distribution of tubulin present in the cytosolic (soluble) and cytoskeletal (polymerized) forms was separated by centrifugation and determined. In lysates of evodiamine-treated cultures, a shift in tubulin from soluble to polymerized forms was observed ( Figure 4B ), indicating a promotion of tubulin polymerization. However, a high concentration of paclitaxel (10 μM) was required to promote the microtubule polymerization under similar incubation conditions. These high concentrations may have been required to compensate for the paclitaxel efflux through the P-glycoprotein pump.
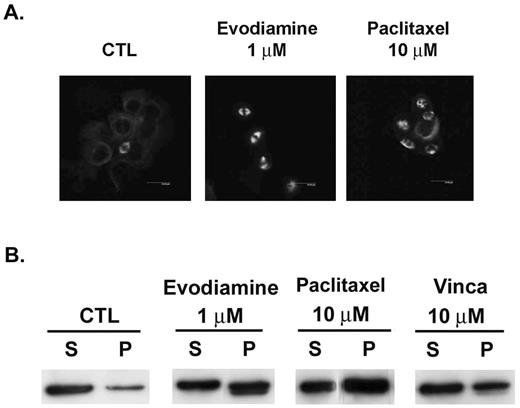
Evodiamine promoted microtubule polymerization in NCI/ADR-RES cells. ( A ) Organization and arrangement of the microtubule network. After 24 h treatments with vehicle or drug, cells were fixed with methanol and blocked with 2% BSA. Cells were then stained with β-tubulin antibody at 37°C and FITC-conjugated secondary antibody. Imaging was performed with the Leica TCS SP2 Spectral Confocal System. ( B ) After 24-h treatments, cells were washed with PBS and lysed with hypotonic buffer. Cytosolic (soluble, S) and cytoskeletal (polymerized tubulin, P) tubulin fractions were separated by centrifugation. Fractions were resolved by SDS–PAGE, followed by immunoblotting with antibody against β-tubulin.
Fluorescein-conjugated paclitaxel was utilized to ascertain whether evodiamine and paclitaxel bound to the same site on microtubules. The fluorescence intensity of bound drug was quantitated by flow cytometry for paclitaxel-treated cultures incubated with varying concentrations of evodiamine. At concentrations up to 30 μM, evodiamine did not reduce the fluorescence intensity provided by paclitaxel alone (data not shown), indicating separate binding sites for these two agents.
Modulation of G 2 /M cell cycle regulatory proteins in NCI/ADR-RES cells by evodiamine
On account of microtubule damage, the spindle-assembly checkpoint was activated during evodiamine treatment. To explore the mechanism by which evodiamine induces mitotic arrest, various cell cycle regulatory proteins were examined in NCI/ADR-RES cells as a function of evodiamine treatment. The phosphorylation state of the protein phosphatase known as cdc25c, a key activator of cyclin B1/cdc2 kinase (also known as cyclin-dependent kinase 1, Cdk1), which controls M-phase entry in eukaryotic cells, was of particular interest. After a 24 h treatment with vehicle or evodiamine at varying concentrations, slower migrating forms of cdc25c were observed for evodiamine-treated cultures, indicating a change in the phosphorylation status of this protein ( Figure 5 ). The observed changes in the migration of this protein were evodiamine concentration-dependent. Bcl-2 was also found to be phosphorylated in cells exposed to 1 μM evodiamine, but no alterations in cyclin B1 or cdc2 kinase were observed ( Figure 5 ). The phosphorylation site of Bcl-2 protein was further identified as Ser 70 by a specific phosphor-Bcl-2-antibody ( Figure 5 ).
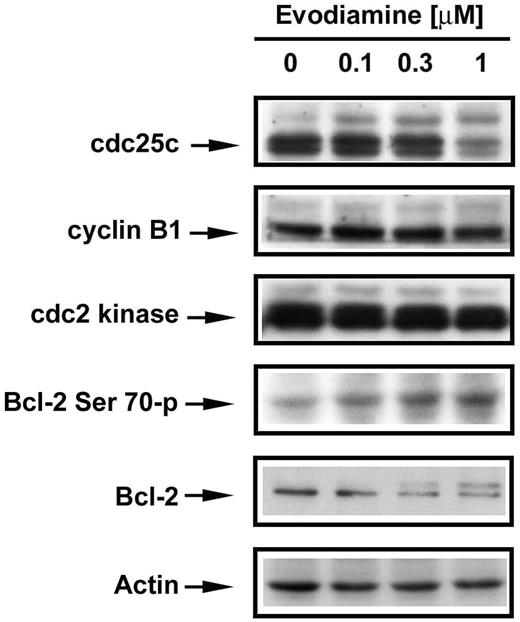
Effects of evodiamine on G 2 /M cell cycle regulatory proteins. After the 24-h treatments with vehicle or evodiamine, NCI/ADR-RES cells were harvested and lysed. Protein measurements were performed, and equal amounts of protein were subjected to SDS–PAGE. Cdc25c, cyclin B1, cdc2 kinase and Bcl-2 were detected by immunoblotting and visualized by enhanced chemiluminescence.
Effects of evodiamine on Raf-1 kinase and Bcl-2 phosphorylation in NCI/ADR-RES cells
Several protein kinases are thought to phosphorylate Bcl-2 in vivo , including Raf-1, JNK and cyclin B1/cdc2 kinases ( 6 ). Neither upregulation of cyclin B1 nor activation of JNK kinases was detected in the NCI/ADR-RES cells exposed to evodiamine ( Figures 5 and 6 ). However, the electrophoretic mobility of Raf-1 kinase was found to be retarded in evodiamine-treated cells, indicating a modification by phosphorylation. The phosphorylation of Raf-1 kinase was detectable at 12 h and prominent at 24 h of treatment. In contrast, no significant change in ERK activation was detected ( Figure 6 ). The phosphorylation site of Raf-1 kinase was identified as Ser 338 through the use of a Raf-1 kinase phospho-specific antibody. Phosphorylation was time-dependent and was associated with the slower migrating forms of Raf-1 kinase. Phosphorylation of Bcl-2 in evodiamine-treated cells was also found to be time-dependent ( Figure 6 ).
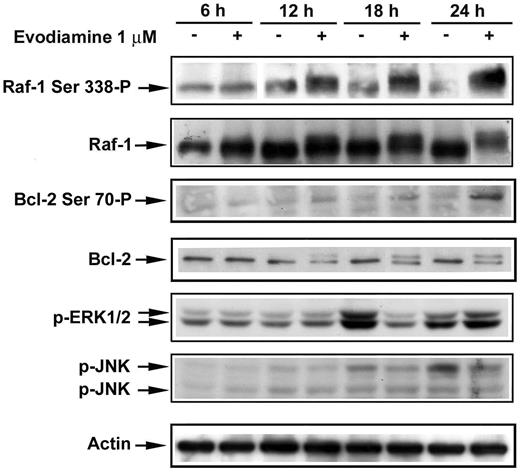
Effects of evodiamine on Raf-1 and Bcl-2 phosphorylation. After treatments with vehicle or evodiamine for the indicated times, NCI/ADR-RES cells were harvested and subjected to lysis. Equal amounts of lysate protein were subjected to SDS-PAGE. Phospho-JNK, phospho-Ser 338 -Raf-1, phospho-ERK1/2 and Bcl-2 were detected by immunoblotting and visualized by enhanced chemiluminescence. For detection of Raf-1 protein expression, the membrane probed with phospho-Ser 338 -Raf-1 antibody was stripped and re-probed with Raf-1 antibody.
Effects of evodiamine and paclitaxel on NCI/ADR-RES xenografts in nude mice
The in vivo effects of evodiamine were evaluated in Balb-c/nude mice subjected to tumor xenograft implantation of NCI/ADR-RES cells. The antitumor activity of evodiamine was compared with that for paclitaxel in these mice. Evodiamine (10 mg/kg) was administrated orally twice daily, whereas paclitaxel (20 mg/kg) was administered by intraperitoneal injection twice weekly. Each respective control group was administered via the same route. During the treatment, the tumor volume in control groups gradually increased and the two control groups via different administered routes exhibited similar progression (data not shown). Initially, evodiamine and paclitaxel were found to slow the tumor growth to comparable degrees ( Figure 7A ). At later times, however, the tumor growth rate was faster in paclitaxel-treated, compared with evodiamine-treated, animals. At the end of the drug treatment period, evodiamine was found to be significantly more effective than paclitaxel in inhibiting tumor growth. Moreover, death due to evodiamine toxicity was not observed in the xenograft model and body weight was not significantly affected ( Figure 7B ). The antitumor activity of evodiamine against human multiple-drug resistant tumor xenograft was superior to that of paclitaxel.
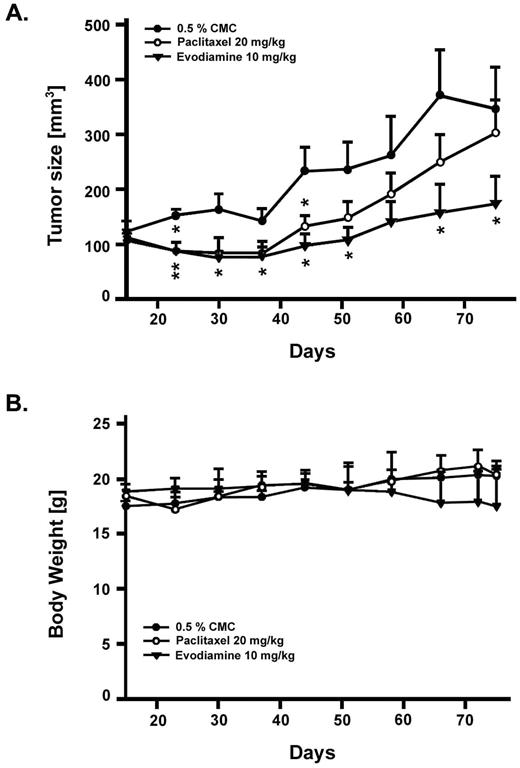
Antitumor effects of evodiamine and paclitaxel in nude mice bearing human mammary carcinoma NCI/ADR-RES xenografts. Female BALB/c athymic ( nu+/nu+ ) mice, 4–6 weeks of age, were provided with sterilized food and water. Mice were injected s.c. with NCI/ADR-RES cells. After 2 weeks, established tumors of ∼100 mm 3 were detected. Six mice per group were treated with the agent of interest or vehicle. Evodiamine suspended in 0.5% CMC was administered orally twice daily. Paclitaxel was given by intraperitoneal injection twice weekly. Tumor volume was determined by caliper measurements (mm) and by using the formula for an ellipsoid sphere: L × W × W/2 = mm 3 .
Discussion
In the present study, evodiamine, a primary constituent of Evodiae fructus , was found to exhibit significant antitumor actions against adriamycin-resistant human breast cancer NCI/ADR-RES cells. Cell proliferation and long-term viability were significantly suppressed and massive cell apoptosis was induced ( Figures 1 and 2 ). Two pathways exist whereby caspases are activated to signal apoptotic cell death. The extrinsic apoptotic pathway is activated by death receptor engagement, which in turn, signals caspase-8 activation. The intrinsic apoptotic pathway, triggered by various extracellular and intracellular stresses, promotes mitochondrial formation of apoptosomes, large protein complexes containing cytochrome c, apoptotic protease activation factor 1 (Apaf-1) and caspase-9. Initiator caspases such as caspase-8 and -9 promote the activation of the procaspase cascade by processing and activating the effector caspases, caspase-3, -6 and -7 ( 25 , 26 ). Treatment with evodiamine resulted in a prominent caspase-9 but not caspase-8 activation, indicating that intrinsic mitochondrial damage was involved in this apoptotic signaling. Downstream activation of the executioner caspases was also examined in evodiamine-treated preparations. Evodiamine treatment resulted in a significant induction of caspase-7 activation but failed to promote the cleavage of caspase-3 and -6 in the NCI/ADR-RES cells ( Figure 2B ). Caspase-3 and -6 probably play a minor role in apoptosis in the NCI/ADR-RES cells since they are known to express low concentrations of these caspases ( 28 , 29 ).
Cell cycle progression is subject to tight regulation by several different Cdk regulatory mechanisms ( 30 ). Prior to mitosis, the cyclin B1/cdc2 kinase complex is inactive. When cyclin B1/cdc2 kinase is activated by the phosphatase cdc25c, the complex drives cell cycle progression from G 2 phase into M phase. Mitosis and cytokinesis both occur during the M phase. During mitosis two sister chromatids align and are subsequently segregated into two daughter cells. In this report as well as in previous reports ( 31 , 32 ), evodiamine was observed to promote significant cell cycle arrest at the G 2 /M transition ( Figure 3 ). This finding implicates cyclin B1/cdc2 kinase in evodiamine-induced mitotic arrest. Although the cyclin B1 expression was not upregulated during evodiamine treatment ( Figure 5 ), phosphatase cdc25c was observed to be hyperphosphorylated. This observation is consistent with the proposal that mitotic arrest in evodiamine-treated cells results from an increased cyclin B1/cdc2 kinase activity.
Bcl-2 was also found to be phosphorylated during evodiamine treatment. An alternative possibility is that increased Bcl-2 phosphorylation, which is considered a marker of microtubule-binding agent-induced cell cycle arrest ( 8 ), was primarily responsible for the drug-induced mitotic arrest. Phosphorylation of Bcl-2 protein can be mediated by cyclin B1/cdc2, JNK, or Raf-1 kinases in cancer cells exposed to microtubule-binding agents ( 6 , 7 ). JNK kinase was unlikely to be involved in evodiamine-induced cell cycle arrest for two reasons. First, JNK kinase activation was not observed in treated preparations ( Figure 6 ). Second, the GI 50 value for evodiamine as well as for Bcl-2 phosphorylation did not change significantly when cells were incubated with evodiamine in combination with sp600125, a known JNK inhibitor (data not shown). Raf-1, which is reported to be activated by mitogens or by mitosis ( 13 , 14 ), may participate in distinct signal transduction pathways depending on its subcellular localization. The hyperphosphorylation of Raf-1 kinase induced by evodiamine in NCI/ADR-RES cells occurred coordinately with cell cycle progression arrest at the G 2 /M transition. In addition, no significant ERK activation downstream of Raf-1 kinase phosphorylation was observed ( Figure 6 ). When the cells were treated with evodiamine in combination with PD98059, a selective MEK inhibitor, phosphorylation of Bcl-2 was not abrogated. Raf-1/Bcl-2 phosphorylation is therefore proposed to play a critical role in evodiamine-induced apoptosis in the NCI/ADR-RES cells. This proposal is in full agreement with the published findings on evodiamine in other systems ( 13 , 15 ).
Raf-1 kinase is phosphorylated exclusively on serine residues during mitosis ( 33 ), and a variety of serine/threonine kinases have been implicated in this phosphorylation. The activation of Raf-1 by p21-activated kinases (Paks) is mediated by the phosphorylation of Raf-1 at Ser 338 ( 34 , 35 ). In the NCI/ADR-RES cells, Raf-1 phosphorylation, as measured by decreases in the electrophoretic mobility of the kinase, was significant and time-dependent. Furthermore, the phosphorylation was shown to occur at Ser 338 . These findings are consistent with the proposal that Raf-1 phosphorylation in evodiamine-treated NCI/ADR-RES cells is attributable to Paks.
Through the use of immunocytochemical and in vivo tubulin polymerization assays, evodiamine treatment was found to enhance microtubule polymerization and the formation of microtubule bundles in NCI/ADR-RES cells. Similar enhancements were observed during paclitaxel treatment. However, the concentration of paclitaxel (10 μM) required for these enhancements in the NCI/ADR-RES cells was much higher than in non-resistant MCF-7 breast cancer cells (0.1 μM). As paclitaxel serves as a substrate for P-glycoprotein, addition of higher doses of the drug are probably required to achieve adequate intracellular concentrations in the NCI/ADR-RES cells. Similar to its effects on microtubules, evodiamine was found to potently inhibit the proliferation of MCF-7 cells, with a GI 50 of 0.66 ± 0.052 μM observed ( Table I ).
This study is the first to demonstrate that evodiamine possesses anti-proliferative and apoptotic activity against multiple-drug resistant breast cancer cells both in vitro and in vivo . Evodiamine is therefore predicted to hold great promise for the chemotherapy of drug-resistant cancers in human patients and clearly warrants further investigation.
This study was supported by a research grant from National Science Council of the Republic of China (NSC 93-2320-B-002-115). We thank Prof. C.-L.Chien (National Taiwan University, Taipei, Taiwan) for guidance on confocal microscopy.
References
Wittmann,T., Hyman,A. and Desai,A. (
Jordan,M.A. and Wilson,L. (
Drukman,S. and Kavallaris,M. (
Orr,G.A., Verdier-Pinard,P., McDaid,H. and Horwitz,S.B. (
Bhalla,K.N. (
Wang,L.G., Liu,X.M., Kreis,W. and Budman,D.R. (
Srivastava,R.K., Mi,Q., Hardwick,J.M. and Longo,D.L. (
Haldar,S., Basu,A. and Croce,C.M. (
Srivastavaa,R.K., Sasakia,C.Y., Hardwickb,J.M. and Longoa,D.L. (
Haldar,S., Chintapalli,J. and Croce,C.M. (
Dhillon,A.S. and Kolch,W. (
Laird,A.D., Taylor,S.J., Oberst,M. and Shalloway,D. (
Ziogas,A., Lorenz,I.C., Moelling,K. and Radziwill,G. (
Laird,A.D., Morrison,D.K. and Shalloway,D. (
Blagosklonny,M.V., Chuman,Y., Bergan,R.C. and Fojo,T. (
Blagosklonny,M.V., Schulte,T., Nguyen,P., Trepel,J. and Neckers,L.M. (
Blagosklonny,M.V., Giannakakou,P., el-Deiry,W.S., Kingston,D.G., Higgs,P.I., Neckers,L. and Fojo,T. (
Gottesman,M.M., Fojo,T. and Bates,S.E. (
Chiou,W.F., Sung,Y.J., Liao,J.F., Shum,A.Y. and Chen,C.F. (
Kobayashi,Y., Nakano,Y., Kizaki,M., Hoshikuma,K., Yokoo,Y. and Kamiya,T. (
Kobayashi,Y. (
Ogasawara,M., Matsubara,T. and Suzuki,H. (
Ogasawara,M. and Suzuki,H. (
Liao,C.H., Pan,S.L., Guh,J.H. and Teng,C.M. (
Strasser,A., O’Connor,L. and Dixit,VM. (
Okada,H and Mak,T.W. (
Wu,L., Smythe,A.M., Stinson,S.F., Mullendore,L.A., Monks,A., Scudiero,D.A., Paull,K.D., Koutsoukos,A.D., Rubinstein,L.V. and Boyd,M.R. (
Nieves-Neira,W. and Pommier,Y. (
Chadderton,A., Villeneuve,D.J., Gluck,S., Kirwan-Rhude,A.F., Gannon,B.R., Blais,D.E. and Parissenti,A.M. (
Stewart,Z.A., Westfall,M.D. and Pietenpol,J.A. (
Huang,Y.C., Guh,J.H. and Teng,C.M. (
Kan,S.F., Huang,W.J., Lin,L.C. and Wang,P.S. (
Lovric,J. and Moelling,K. (
Zang,M., Waelde,C.A., Xiang,X., Rana,A., Wen,R. and Luo,Z. (
Author notes
1Pharmacological Institute and 2School of Pharmacy, College of Medicine, National Taiwan University, Taipei 100, Taiwan

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}