





Abstract
Sulfonation is an important reaction in the metabolism of numerous xenobiotics, drugs, and endogenous compounds. A supergene family of enzymes called sulfotransferases (SULTs) catalyze this reaction. In most cases, the addition of a sulfonate moiety to a compound increases its water solubility and decreases its biological activity. However, many of these enzymes are also capable of bioactivating procarcinogens to reactive electrophiles. In humans three SULT families, SULT1, SULT2, and SULT4, have been identified that contain at least thirteen distinct members. SULTs have a wide tissue distribution and act as a major detoxification enzyme system in adult and the developing human fetus. Nine crystal structures of human cytosolic SULTs have now been determined, and together with site-directed mutagenesis experiments and molecular modeling, we are now beginning to understand the factors that govern distinct but overlapping substrate specificities. These studies have also provided insight into the enzyme kinetics and inhibition characteristics of these enzymes. The regulation of human SULTs remains as one of the least explored areas of research in the field, though there have been some recent advances on the molecular transcription mechanism controlling the individual SULT promoters. Interindividual variation in sulfonation capacity may be important in determining an individual's response to xenobiotics, and recent studies have begun to suggest roles for SULT polymorphism in disease susceptibility. This review aims to provide a summary of our present understanding of the function of human cytosolic sulfotransferases.
INTRODUCTION
Sulfonate conjugation was first reported by Baumann in 1876 (Baumann, 1876) and has since been shown to be an important pathway in the biotransformation of numerous xeno- and endobiotics such as drugs, chemical carcinogens, hormones, bile acids, neurotransmitters, peptides, and lipids. The universal sulfonate donor for these reactions is 3′-phosphoadenosine 5′-phosphosulfate (PAPS), and the transfer of sulfonate (
Two broad classes of SULTs have been identified: (1) membrane-bound SULTs that are located in the Golgi apparatus of the cell and are responsible for the sulfonation of peptides (e.g., CCK), proteins, lipids, and glycosaminoglycans, affecting both their structural and functional characteristics (Falany, 1997; Negishi et al., 2001) and (2) cytosolic SULTs that are responsible for the metabolism of xenobiotics and small endogenous substrates such as steroids, bile acids, and neurotransmitters. The focus of this review is on the human cytosolic SULTs, with particular emphasis on those isoforms that have been shown to metabolize a broad range of drug, xenobiotic, and endobiotic substrates. Their particular role in the metabolic activation of xenobiotics to mutagens and carcinogens will be addressed.
The sulfonation of xenobiotics and small endogenous substrates such as steroids and neurotransmitters is widely distributed in nature and occurs in organisms ranging from microbes to man (Blanchard et al., 2004; Nagata and Yamazoe, 2000; Rikke and Roy, 1996). The process of sulfonation involves the transfer of a sulfonyl (
SULFOTRANSFERASES
Rat SULT2A2 was the first SULT cloned and was originally identified as a senescence marker protein (Chatterjee et al., 1987). Since then at least 47 mammalian SULT isoforms, one insect isoform, and eight plant enzymes, which represent nine separate SULT families and 14 subfamilies, have been cloned and characterized (Blanchard et al., 2004). The latter paper provides a significant breakthrough in nomenclature of SULTs, which until then was even confusing to those working full-time in the field. It broadly follows the gene family nomenclature systems developed for other drug-metabolizing enzyme families such as cytochromes P450, UDP-glucuronosyltransferase, glutathione transferases, and N-acetyltransferase. The nomenclature system is based on family members sharing at least 45% amino acid sequence identity and subfamily members being at least 60% identical. To date, five distinct gene families of SULTs have been identified in mammals: SULT1, SULT2, SULT3, SULT4, and SULT5 (Blanchard et al., 2004). The SULT3 family has only been found in mouse and rabbit, and has been shown to primarily sulfonate amino groups (Yoshinari et al., 1998a). Sult5a1 has only been isolated from mice, and limited information is available on this family (Nagata and Yamazoe, 2000).
Table 1 summarizes the human forms of SULTs that have been characterized to date. These can be divided into three families and collectively account for thirteen distinct members: SULT1—A1, A2, A3, A4, B1, C2, C4, E1; SULT2—A1 and B1 (SULT2B1_v1 and SULT2B1_v2); SULT4A1 (SULT4A1_v1 and SULT4A1_v2) (Blanchard et al., 2004; Hildebrandt et al., 2004; Mammalian Gene Collection (MGC) Program Team, 2002). At least two SULT genes (human SULT2B1 and SULT4A1) encode two SULT isoforms: SULT2B1—SULT2B1_v1 and SULT2B1_v2 differ in their N-terminal amino acid sequences as a result of either alternate transcription initiation or alternate splicing (Blanchard et al., 2004; Fuda et al., 2002); SULT4A1—isoforms v1 and v2 differ in their C-terminal amino acid sequences (Mammalian Gene Collection (MGC) Program Team, 2002). Figure 1 shows a dendogram of these human SULTs. While SULT1D1 has been isolated from the dog (Tsoi et al., 2001), mouse (Sakakibara et al., 1998a), and rat (Herrmann and Stoffel, unpublished), no equivalent human form of this enzyme has been identified.
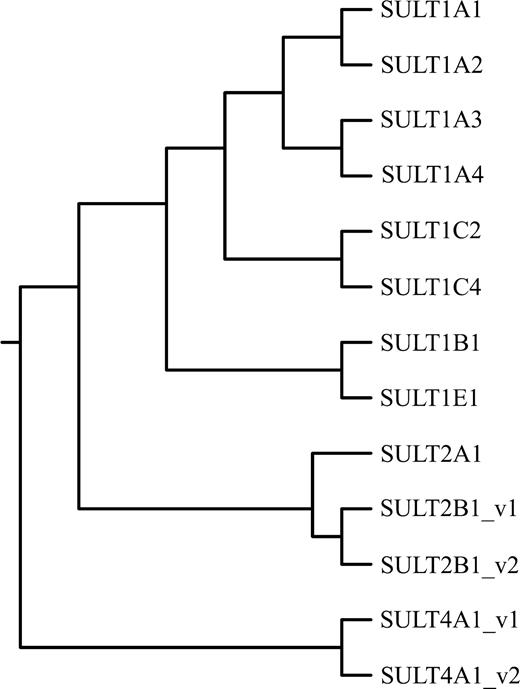
The phylogenetic tree showing the relationship between human cytosolic sulfotransferases. The tree was generated using Clustalw (http://align.genome.jp/). The Pub Med Accession numbers are as follows: SULT1A1 (U26309), SULT1A2 (U28169), SULT1A3 (L19956), SULT1A4 (BK004132), SULT1B1 (U95726), SULT1C2 (U66036), SULT1C4 (AF055584), SULT1E1 (U08098), SULT2A1 (U08024), SULT2B1_v1 (U92314), SULT2B1_v2 (U92315), SULT4A1_v1 (AF188698) and SULT4A1_v2 (AAH28171).
cDNAs and Genes Comprising the Human SULT Superfamily
SULT | Chromosomal location | Name given by author | Accession No. | Reference | |
|---|---|---|---|---|---|
| SULT1A1 | 16p11.2–12.1 | cDNA | P-PST-1 | L19999 | Wilborn et al., 1993 |
| HAST1 | L10819 | Zhu et al., 1993b | |||
| ST1A3 | X78283 | Ozawa et al., 1995 | |||
| P-PST | X84654 | Jones et al., 1995 | |||
| H-PST | U26309 | Hwang et al., 1995 | |||
| HAST2 | L19955 | Zhu et al., 1996 | |||
| U09031 | |||||
| SULT1A1 | NM_001055 | Her et al., 1996 | |||
| SULT1A1 | AJ007418 | Raftogianis et al., 1996 | |||
| Gene | STP1 | U71086 | Dooley and Huang 1996 | ||
| STP | U54701 | Bernier et al., 1996 | |||
| TS- PST1 (STP1) | U52852 | Raftogianis et al., 1996 | |||
| SULT1A2 | 16p11.2–12.1 | cDNA | ST1A2 | X78282 | Ozawa et al., 1995 |
| HAST4v | U28169 | Zhu et al., 1996 | |||
| HAST4 | U28170 | Zhu et al., 1996 | |||
| Gene | STP2 | U76619 | Dooley and Huang, 1996 | ||
| U34804 | Her et al., 1996 | ||||
| U33886 | Gaedigk et al., 1997 | ||||
| SULT1A3 | 16p11.2 | cDNA | HAST3 | L19956 | Zhu et al., 1993a |
| HAST3-intron1 | L19957 | ||||
| TL- PST | U08032 | Wood et al., 1994 | |||
| hEST | L25275 | Bernier et al., 1994a | |||
| m-PST | X84653 | Jones et al., 1995 | |||
| hm-PST | - | Ganguly et al., 1995 | |||
| Gene | STM, | U20499 | Aksoy et al., 1994,1995 | ||
| HAST | U37686 | Dooley et al., 1994 | |||
| L34160 | Bernier et al., 1994b | ||||
| SULT1A4 | 16p12.1 | cDNA | SULT1A4 | MGC5178 | Hildebrandt et al., 2004 |
| Gene | SULT1A4 | Hildebrandt et al., 2004 | |||
| SULT1B1 | 4q11–13 | cDNA | ST1B2 | D89479 | Fujita et al., 1997 |
| U95726 | Wang et al., 1998 | ||||
| Gene | SULT1B2 | AF184894 | Wang et al., 1998 | ||
| SULT1C2 | 2q11.2 | cDNA | SULT1C1 | U66036 | Her et al., 1997 |
| ST1C2 | AB008164 | Yoshinari et al., 1998b | |||
| HAST5 | AF026303 | Hehonah et al., 1999 | |||
| Gene | SULT1C1 | AF186257 | Freimuth et al., 2000 | ||
| SULT1C4 | 2q11.2 | cDNA | hSULT1C | AF055584 | Sakakibara et al., 1998b |
| Gene | SULT1C2 | AF186263 | Freimuth et al., 2000 | ||
| SULT1E1 | 4q13.1 | cDNA | hEST (STE) | U08098 | Aksoy et al., 1994 |
| hEST-1 | S77383 | Falany et al., 1995 | |||
| SULT1E1 | Y11195 | Rubin et al., 1999 | |||
| Gene | STE | U20514-21 | Her et al., 1995 | ||
| SULT2A1 | 19q13.3 | cDNA | DHEA-ST | U08024 | Otterness et al., 1992 |
| U08025 | |||||
| hSTa | S43859 | Kong et al., 1992 | |||
| L02337 | |||||
| DHEA-ST8 | X70222 | Comer et al., 1993 | |||
| Gene | STD | U13056-61 | Otterness et al., 1995 | ||
| L36191-196 | Luu –The et al., 1995 | ||||
| SULT2B1_v1 | 19q13.3 | cDNA | hSULT2B1a | U92314 | Her et al., 1998 |
| SULT2B1_v2 | hSULT2B1b | U92315 | Her et al., 1998 | ||
| Gene | SULT21B1 | U92316-22 | Her et al., 1998 | ||
| SULT4A1_v1 | 22q13.1–13.2 | cDNA | hBR-STL | AF188698 | Falany et al., 2000 |
| SULT4a1_v2 | hSULT4A1 | AF251263 | Walther et al., 1999 | ||
| SULTX3 | AF115311 | Sakakibara et al., 2002 | |||
| AAH28171 | MGC Program Team | ||||
| Gene | SULT4A1 | Z97055 | Dunham et al., 1999 |
SULT | Chromosomal location | Name given by author | Accession No. | Reference | |
|---|---|---|---|---|---|
| SULT1A1 | 16p11.2–12.1 | cDNA | P-PST-1 | L19999 | Wilborn et al., 1993 |
| HAST1 | L10819 | Zhu et al., 1993b | |||
| ST1A3 | X78283 | Ozawa et al., 1995 | |||
| P-PST | X84654 | Jones et al., 1995 | |||
| H-PST | U26309 | Hwang et al., 1995 | |||
| HAST2 | L19955 | Zhu et al., 1996 | |||
| U09031 | |||||
| SULT1A1 | NM_001055 | Her et al., 1996 | |||
| SULT1A1 | AJ007418 | Raftogianis et al., 1996 | |||
| Gene | STP1 | U71086 | Dooley and Huang 1996 | ||
| STP | U54701 | Bernier et al., 1996 | |||
| TS- PST1 (STP1) | U52852 | Raftogianis et al., 1996 | |||
| SULT1A2 | 16p11.2–12.1 | cDNA | ST1A2 | X78282 | Ozawa et al., 1995 |
| HAST4v | U28169 | Zhu et al., 1996 | |||
| HAST4 | U28170 | Zhu et al., 1996 | |||
| Gene | STP2 | U76619 | Dooley and Huang, 1996 | ||
| U34804 | Her et al., 1996 | ||||
| U33886 | Gaedigk et al., 1997 | ||||
| SULT1A3 | 16p11.2 | cDNA | HAST3 | L19956 | Zhu et al., 1993a |
| HAST3-intron1 | L19957 | ||||
| TL- PST | U08032 | Wood et al., 1994 | |||
| hEST | L25275 | Bernier et al., 1994a | |||
| m-PST | X84653 | Jones et al., 1995 | |||
| hm-PST | - | Ganguly et al., 1995 | |||
| Gene | STM, | U20499 | Aksoy et al., 1994,1995 | ||
| HAST | U37686 | Dooley et al., 1994 | |||
| L34160 | Bernier et al., 1994b | ||||
| SULT1A4 | 16p12.1 | cDNA | SULT1A4 | MGC5178 | Hildebrandt et al., 2004 |
| Gene | SULT1A4 | Hildebrandt et al., 2004 | |||
| SULT1B1 | 4q11–13 | cDNA | ST1B2 | D89479 | Fujita et al., 1997 |
| U95726 | Wang et al., 1998 | ||||
| Gene | SULT1B2 | AF184894 | Wang et al., 1998 | ||
| SULT1C2 | 2q11.2 | cDNA | SULT1C1 | U66036 | Her et al., 1997 |
| ST1C2 | AB008164 | Yoshinari et al., 1998b | |||
| HAST5 | AF026303 | Hehonah et al., 1999 | |||
| Gene | SULT1C1 | AF186257 | Freimuth et al., 2000 | ||
| SULT1C4 | 2q11.2 | cDNA | hSULT1C | AF055584 | Sakakibara et al., 1998b |
| Gene | SULT1C2 | AF186263 | Freimuth et al., 2000 | ||
| SULT1E1 | 4q13.1 | cDNA | hEST (STE) | U08098 | Aksoy et al., 1994 |
| hEST-1 | S77383 | Falany et al., 1995 | |||
| SULT1E1 | Y11195 | Rubin et al., 1999 | |||
| Gene | STE | U20514-21 | Her et al., 1995 | ||
| SULT2A1 | 19q13.3 | cDNA | DHEA-ST | U08024 | Otterness et al., 1992 |
| U08025 | |||||
| hSTa | S43859 | Kong et al., 1992 | |||
| L02337 | |||||
| DHEA-ST8 | X70222 | Comer et al., 1993 | |||
| Gene | STD | U13056-61 | Otterness et al., 1995 | ||
| L36191-196 | Luu –The et al., 1995 | ||||
| SULT2B1_v1 | 19q13.3 | cDNA | hSULT2B1a | U92314 | Her et al., 1998 |
| SULT2B1_v2 | hSULT2B1b | U92315 | Her et al., 1998 | ||
| Gene | SULT21B1 | U92316-22 | Her et al., 1998 | ||
| SULT4A1_v1 | 22q13.1–13.2 | cDNA | hBR-STL | AF188698 | Falany et al., 2000 |
| SULT4a1_v2 | hSULT4A1 | AF251263 | Walther et al., 1999 | ||
| SULTX3 | AF115311 | Sakakibara et al., 2002 | |||
| AAH28171 | MGC Program Team | ||||
| Gene | SULT4A1 | Z97055 | Dunham et al., 1999 |
cDNAs and Genes Comprising the Human SULT Superfamily
SULT | Chromosomal location | Name given by author | Accession No. | Reference | |
|---|---|---|---|---|---|
| SULT1A1 | 16p11.2–12.1 | cDNA | P-PST-1 | L19999 | Wilborn et al., 1993 |
| HAST1 | L10819 | Zhu et al., 1993b | |||
| ST1A3 | X78283 | Ozawa et al., 1995 | |||
| P-PST | X84654 | Jones et al., 1995 | |||
| H-PST | U26309 | Hwang et al., 1995 | |||
| HAST2 | L19955 | Zhu et al., 1996 | |||
| U09031 | |||||
| SULT1A1 | NM_001055 | Her et al., 1996 | |||
| SULT1A1 | AJ007418 | Raftogianis et al., 1996 | |||
| Gene | STP1 | U71086 | Dooley and Huang 1996 | ||
| STP | U54701 | Bernier et al., 1996 | |||
| TS- PST1 (STP1) | U52852 | Raftogianis et al., 1996 | |||
| SULT1A2 | 16p11.2–12.1 | cDNA | ST1A2 | X78282 | Ozawa et al., 1995 |
| HAST4v | U28169 | Zhu et al., 1996 | |||
| HAST4 | U28170 | Zhu et al., 1996 | |||
| Gene | STP2 | U76619 | Dooley and Huang, 1996 | ||
| U34804 | Her et al., 1996 | ||||
| U33886 | Gaedigk et al., 1997 | ||||
| SULT1A3 | 16p11.2 | cDNA | HAST3 | L19956 | Zhu et al., 1993a |
| HAST3-intron1 | L19957 | ||||
| TL- PST | U08032 | Wood et al., 1994 | |||
| hEST | L25275 | Bernier et al., 1994a | |||
| m-PST | X84653 | Jones et al., 1995 | |||
| hm-PST | - | Ganguly et al., 1995 | |||
| Gene | STM, | U20499 | Aksoy et al., 1994,1995 | ||
| HAST | U37686 | Dooley et al., 1994 | |||
| L34160 | Bernier et al., 1994b | ||||
| SULT1A4 | 16p12.1 | cDNA | SULT1A4 | MGC5178 | Hildebrandt et al., 2004 |
| Gene | SULT1A4 | Hildebrandt et al., 2004 | |||
| SULT1B1 | 4q11–13 | cDNA | ST1B2 | D89479 | Fujita et al., 1997 |
| U95726 | Wang et al., 1998 | ||||
| Gene | SULT1B2 | AF184894 | Wang et al., 1998 | ||
| SULT1C2 | 2q11.2 | cDNA | SULT1C1 | U66036 | Her et al., 1997 |
| ST1C2 | AB008164 | Yoshinari et al., 1998b | |||
| HAST5 | AF026303 | Hehonah et al., 1999 | |||
| Gene | SULT1C1 | AF186257 | Freimuth et al., 2000 | ||
| SULT1C4 | 2q11.2 | cDNA | hSULT1C | AF055584 | Sakakibara et al., 1998b |
| Gene | SULT1C2 | AF186263 | Freimuth et al., 2000 | ||
| SULT1E1 | 4q13.1 | cDNA | hEST (STE) | U08098 | Aksoy et al., 1994 |
| hEST-1 | S77383 | Falany et al., 1995 | |||
| SULT1E1 | Y11195 | Rubin et al., 1999 | |||
| Gene | STE | U20514-21 | Her et al., 1995 | ||
| SULT2A1 | 19q13.3 | cDNA | DHEA-ST | U08024 | Otterness et al., 1992 |
| U08025 | |||||
| hSTa | S43859 | Kong et al., 1992 | |||
| L02337 | |||||
| DHEA-ST8 | X70222 | Comer et al., 1993 | |||
| Gene | STD | U13056-61 | Otterness et al., 1995 | ||
| L36191-196 | Luu –The et al., 1995 | ||||
| SULT2B1_v1 | 19q13.3 | cDNA | hSULT2B1a | U92314 | Her et al., 1998 |
| SULT2B1_v2 | hSULT2B1b | U92315 | Her et al., 1998 | ||
| Gene | SULT21B1 | U92316-22 | Her et al., 1998 | ||
| SULT4A1_v1 | 22q13.1–13.2 | cDNA | hBR-STL | AF188698 | Falany et al., 2000 |
| SULT4a1_v2 | hSULT4A1 | AF251263 | Walther et al., 1999 | ||
| SULTX3 | AF115311 | Sakakibara et al., 2002 | |||
| AAH28171 | MGC Program Team | ||||
| Gene | SULT4A1 | Z97055 | Dunham et al., 1999 |
SULT | Chromosomal location | Name given by author | Accession No. | Reference | |
|---|---|---|---|---|---|
| SULT1A1 | 16p11.2–12.1 | cDNA | P-PST-1 | L19999 | Wilborn et al., 1993 |
| HAST1 | L10819 | Zhu et al., 1993b | |||
| ST1A3 | X78283 | Ozawa et al., 1995 | |||
| P-PST | X84654 | Jones et al., 1995 | |||
| H-PST | U26309 | Hwang et al., 1995 | |||
| HAST2 | L19955 | Zhu et al., 1996 | |||
| U09031 | |||||
| SULT1A1 | NM_001055 | Her et al., 1996 | |||
| SULT1A1 | AJ007418 | Raftogianis et al., 1996 | |||
| Gene | STP1 | U71086 | Dooley and Huang 1996 | ||
| STP | U54701 | Bernier et al., 1996 | |||
| TS- PST1 (STP1) | U52852 | Raftogianis et al., 1996 | |||
| SULT1A2 | 16p11.2–12.1 | cDNA | ST1A2 | X78282 | Ozawa et al., 1995 |
| HAST4v | U28169 | Zhu et al., 1996 | |||
| HAST4 | U28170 | Zhu et al., 1996 | |||
| Gene | STP2 | U76619 | Dooley and Huang, 1996 | ||
| U34804 | Her et al., 1996 | ||||
| U33886 | Gaedigk et al., 1997 | ||||
| SULT1A3 | 16p11.2 | cDNA | HAST3 | L19956 | Zhu et al., 1993a |
| HAST3-intron1 | L19957 | ||||
| TL- PST | U08032 | Wood et al., 1994 | |||
| hEST | L25275 | Bernier et al., 1994a | |||
| m-PST | X84653 | Jones et al., 1995 | |||
| hm-PST | - | Ganguly et al., 1995 | |||
| Gene | STM, | U20499 | Aksoy et al., 1994,1995 | ||
| HAST | U37686 | Dooley et al., 1994 | |||
| L34160 | Bernier et al., 1994b | ||||
| SULT1A4 | 16p12.1 | cDNA | SULT1A4 | MGC5178 | Hildebrandt et al., 2004 |
| Gene | SULT1A4 | Hildebrandt et al., 2004 | |||
| SULT1B1 | 4q11–13 | cDNA | ST1B2 | D89479 | Fujita et al., 1997 |
| U95726 | Wang et al., 1998 | ||||
| Gene | SULT1B2 | AF184894 | Wang et al., 1998 | ||
| SULT1C2 | 2q11.2 | cDNA | SULT1C1 | U66036 | Her et al., 1997 |
| ST1C2 | AB008164 | Yoshinari et al., 1998b | |||
| HAST5 | AF026303 | Hehonah et al., 1999 | |||
| Gene | SULT1C1 | AF186257 | Freimuth et al., 2000 | ||
| SULT1C4 | 2q11.2 | cDNA | hSULT1C | AF055584 | Sakakibara et al., 1998b |
| Gene | SULT1C2 | AF186263 | Freimuth et al., 2000 | ||
| SULT1E1 | 4q13.1 | cDNA | hEST (STE) | U08098 | Aksoy et al., 1994 |
| hEST-1 | S77383 | Falany et al., 1995 | |||
| SULT1E1 | Y11195 | Rubin et al., 1999 | |||
| Gene | STE | U20514-21 | Her et al., 1995 | ||
| SULT2A1 | 19q13.3 | cDNA | DHEA-ST | U08024 | Otterness et al., 1992 |
| U08025 | |||||
| hSTa | S43859 | Kong et al., 1992 | |||
| L02337 | |||||
| DHEA-ST8 | X70222 | Comer et al., 1993 | |||
| Gene | STD | U13056-61 | Otterness et al., 1995 | ||
| L36191-196 | Luu –The et al., 1995 | ||||
| SULT2B1_v1 | 19q13.3 | cDNA | hSULT2B1a | U92314 | Her et al., 1998 |
| SULT2B1_v2 | hSULT2B1b | U92315 | Her et al., 1998 | ||
| Gene | SULT21B1 | U92316-22 | Her et al., 1998 | ||
| SULT4A1_v1 | 22q13.1–13.2 | cDNA | hBR-STL | AF188698 | Falany et al., 2000 |
| SULT4a1_v2 | hSULT4A1 | AF251263 | Walther et al., 1999 | ||
| SULTX3 | AF115311 | Sakakibara et al., 2002 | |||
| AAH28171 | MGC Program Team | ||||
| Gene | SULT4A1 | Z97055 | Dunham et al., 1999 |
SULT1 Family
SULT1A1.
Members of the SULT1A1 subfamily have been identified in the largest range of species including the rat, mouse, cow, dog, rabbit (refer Blanchard et al., 2004 for references), monkey (Ogura et al. unpublished), pig (Lin et al., 2004), and platypus (Bolton-Grob and McManus, unpublished). A solitary SULT1A1 isoform has been characterized in all the above-mentioned species, but in humans, four SULT1A subfamily members have been identified (SULT1-A1, A2, A3, and A4; Blanchard et al., 2004; Hildebrandt et al., 2004). Their genes are all clustered on the short arm of chromosome 16 and are thought to have arisen following gene duplication or gene duplication plus recombination events (Aksoy et al., 1994; Dooley, 1998a; Rikke and Roy, 1996). The gene sequences of SULT1A1 and SULT1A2 are 93% similar, whereas SULT1A3 shares approximately 60% identity with the other two genes. The differences are most apparent in the 5′ promoter and intron sequences, as the coding exons of all three genes are >90% identical. Based on the available data, the human SULT1A1 is most probably the ortholog of the equivalent animal isoforms (Blanchard et al., 2004).
Wilborn et al. (1993) were the first to isolate a SULT1A1 cDNA from a human liver library. The full-length cDNA was shown to encode a protein of 295 amino acids, which had high activity toward the model substrate p-nitrophenol (pNP). The same protein also had activity toward minoxidil as a substrate (Meisheri et al., 1993). The general fidelity of the Wilborn et al. sequence has now been confirmed by a number of groups (Table 1), and allelic variants of SULT1A1 have been shown to exist in the human population. SULT1A1 is by far the dominant SULT1A protein expressed in human liver and has an estimated molecular weight on SDS–PAGE of 32 kDa. This form was initially labelled by Weinshilboum's group as the TS or P (thermostable or phenol, TS-PST) form of sulfotransferase, and SULT1A3 as the TL or M (thermolabile or monoamine, TL-PST) sulfonating form, and collectively they were shown to be responsible for the metabolism of most phenolic compounds (Hempel et al., 2005; Reiter et al., 1983). In relation to their thermostabilities, SULT1A3 shows no activity toward dopamine as a substrate after treatment at 45°C for 15 min, but SULT1A1 retains approximately 90% of its activity toward pNP following similar treatment. It is also possible to differentiate between SULT1A1 and SULT1A3 activity in tissue fractions using the inhibitor 2,6-dichloro-4-nitrophenol (DCNP), as the sulfonation of pNP by the former is highly sensitive to this compound (Veronese et al., 1994). Unlike SULT1A1 or SULT1A3, SULT1A2 exhibits no activity toward dopamine as a substrate and possesses a Km for pNP sulfonation (∼70 μM) between that of SULT1A1 and SULT1A3 (Zhu et al., 1996).
SULT1A2.
As indicated above, no equivalent form of SULT1A2 has been identified in species other than humans. Ozawa et al. (1995) were the first to clone a SULT1A2 cDNA from a human liver library, and it was originally given the name ST1A2. These authors showed that COS [cells derived from the kidney of an adult male African green monkey (Cercopithecus aethiops)] cell-expressed SULT1A2 sulfonated pNP, minoxidil, and β-naphthol, but at a lower rate than SULT1A1 (ST1A3). In another study, Zhu et al. (1996) isolated from a human liver library two forms of this cDNA (HAST4 and HAST4v) that differ by two amino acids (Thr7 to Ile and Thr235 to Asn). In their coding domains HAST4 and HAST4v were 97% and 94% identical to SULT1A1 and SULT1A3, respectively. On expression of these cDNAs in COS cells, the encoded proteins exhibited markedly different Km values for the sulfonation of pNP: Km values for HAST4 and HAST4v being 74 and 8 μM, respectively. However, unlike SULT1A1 or SULT1A3, SULT1A2 exhibits no activity toward dopamine as a substrate, even though it shared >93% amino acid identity with these proteins. At least 13 different allelic variants of human SULT1A2 have been identified that encode four different amino acid changes resulting in six different SULT1A2 allozymes (Raftogianis et al., 1999).
SULT1A3.
To date, a SULT1A3 gene has only been identified in humans, and it appears that through evolutionary pressures we have acquired a SULT1A3 gene whose expressed protein fulfils a specific role in sulfonating catecholamines such as dopamine (Coughtrie, 1998; Dooley, 1998b; Hempel et al., 2005). SULT1A3 was initially isolated from a human brain cDNA library and was called HAST3 (Zhu et al., 1993). The cDNA isolated was shown to encode a 34 kDa protein that was 93% similar to SULT1A1 and under the new nomenclature was termed SULT1A3 (Blanchard et al., 2004, Zhu et al., 1993). Based on its substrate preference for dopamine, thermal stability, and sensitivity to DCNP inhibition, it was shown to be the thermolabile or M-form of sulfotransferase initially identified by Reiter et al. in 1983 (Veronese et al., 1994). The fidelity of the original sequence was subsequently confirmed (Table 1). An identical cDNA was also isolated by Bernier et al. (1994a), who originally reported it as an estrogen sulfotransferase (hEST). A recent study by Hildebrandt et al. (2004) showed that two copies of SULT1A3 exist in the human genome (SULT1A3 and SULT1A4), and they appear to be transcriptionally active. At least four nonsynonymous single nucleotide polymorphisms (cSNPs) were reported by the above authors for these genes, which show different enzyme activity.
SULT1B1.
The first member of this SULT subfamily was cloned from a rat liver cDNA library and was described as the 3,4-dihydroxyphenylalanine (dopa)/tyrosine sulfotransferase, having activity toward tyrosine and dopa (Sakakibara et al., 1995). The same authors also showed that SULT1B1 had activity toward various thyroid hormone substrates, pNP and dopamine. Since then, SULT1B1 enzymes have been isolated from mouse, dog (Blanchard et al., 2004), and brush-tailed possum (Bolton-Grob and McManus, unpublished). The human form of SULT1B1 was isolated and characterized by Fujita et al. (1997) and shown to be the major thyroid hormone sulfotransferase, having slightly higher affinity for the tri-iodothyronine than SULT1A1.
SULT1C.
SULT1C subfamily members have been isolated from a variety of species including Sult1c1, 1c2, and 1c3 from the rat, 1c1 from the mouse, and 1c2 from the rabbit (see Blanchard et al., 2004 for references). Weinshilboum's group was the first to isolate a human 1C2 cDNA from a fetal liver-spleen cDNA library (Her et al., 1997) and demonstrate that the gene was located on chromosome 2 at 2q11.2. The identical construct was also cloned and characterized by Yoshinari et al. (1998b) and Hehonah et al. (1999). Another member of the human 1C subfamily, 1C4, was identified by Sakakibara et al. (1998b). To date, no endogenous substrates have been identified for members of the IC subfamily.
SULT1E.
The SULT1E enzymes have been widely studied due to their important role in steroid homeostasis. The bovine SULT1E1 was the first cDNA cloned as a known sulfotransferase (Nash et al., 1988). Since this initial study, members of the 1E subfamily have been isolated from a number of species including the guinea pig, rat, mouse, and pig (Blanchard et al., 2004). Aksoy et al. (1994) were the first to isolate a SULT1E1 cDNA from a human liver library. The protein encoded by this cDNA was subsequently shown to have high affinity (nM range) for E2 and estrone and a variety of synthetic estrogens, including diethylstilbestrol and tamoxifen (Falany, 1997; Falany et al., 1995). While other SULTs such as SULT1A1 and SULT2A1 exhibit high activity toward E2 and estrone, this only occurs at nonphysiological concentrations (Falany, 1997; Falany et al., 1995). Iodothyronines are also good substrates for SULT1E1 (Kester et al., 1999).
SULT2 Family
The SULT2 family contains the hydroxysteroid sulfotransferases, which have been subdivided into two subfamilies based on their amino acid sequence identities. Generally, all members of the SULT2 family display overlapping substrate specificities toward an array of hydroxysteroids and related compounds (Table 2).
Endogenous and Xenobiotic Prototypic Substrates of Human Cytosolic SULTs
Subfamily | Endogenous substrates | Xenobiotic substrates |
|---|---|---|
| SULT1A1 | Iodothyronines: 3,3′diiodothyronine (T2), 3,3′,5-triiodothyronine (T3), (Anderson et al., 1995; Li et al., 2001); Estrogens: β-estradiol (E2) (Falany, 1997) | Simple phenolic compounds: p-nitrophenol, m-nitrophenol, p-ethylphenol, p-cresol (Wilborn et al., 1993; Brix et al., 1999b); Drugs: paracetamol (Lewis et al., 1996), minoxidil (Meisheri et al., 1993); Carcinogens: N-Hydroxy -PhIP (Ozawa et al., 1994). |
| SULT1A2 | Not known | Simple phenolic compounds: p-nitrophenol (Zhu et al., 1996); Carcinogens: N-Hydroxy −2-AAF (Glatt, 2000) |
| SULT1A3 | Catecholamines: dopamine, (Brix et al., 1999b; Dajani et al., 1999b), norepinephrine (noradrenaline) (Ganguly et al., 1995) | Simple Phenols: p-nitrophenol (Brix et al., 1999b); Carcinogens: 1-Hydroxymethylpyrene (Glatt, 2000) |
| SULT1B1 | Iodothyronines: 3,3′diiodothyronine (T2), 3,3′,5-triiodothyronine (T3), 3,3′,5′-reverse triiodothyronine (r-T3), and thyroxine (T4) (Wang et al., 1998) | Simple Phenol: 1-Naphthol (Wang et al., 1998) |
| SULT1C2 | Not known | Simple Phenols: p-nitrophenol (Sakakibara, 1998b); Carcinogens: N-Hydroxy-2-AAF (Yoshinari et al., 1998b) |
| SULT1C4 | Not known | Simple Phenols: p-nitrophenol (Yoshinari et al., 1998a); Carcinogens: N-Hydroxy-2-AAF (Sakakibara et al., 1998b) |
| SULT1E1 | Estrogens: E2, estrone (E1) (Falany et al., 1995) | Estrogens: 17-ethinyl-E2, equilenin (Falany et al., 1995); Catechol estrogens: 2-hydroxyestrone, 2-hydroxyestradiol, 4-hydroxyestrone; 4-hydroxyestradiol (Adjei and Weinshilboum, 2002) |
| SULT2A1 | Steroids: DHEA (Comer and Falany, 1992) | Carcinogens: 1- Hydroxymethylpyrene (Meinl et al., 2002), 6 Hydroxymethylbenzo[a]-pyrene, hycanthone (Glatt, 2000) |
| SULT2B1_v1 | DHEA (Her et al., 1998), pregnenolone (Meloche and Falany, 2001) | Not known |
| SULT2B1_v2 | DHEA (Her et al., 1998), pregnenolone (Meloche and Falany, 2001), cholesterol and oxysterols (Geese and Raftogianis, 2001) | Not known |
| SULT4A1 | Not known | Not known |
Subfamily | Endogenous substrates | Xenobiotic substrates |
|---|---|---|
| SULT1A1 | Iodothyronines: 3,3′diiodothyronine (T2), 3,3′,5-triiodothyronine (T3), (Anderson et al., 1995; Li et al., 2001); Estrogens: β-estradiol (E2) (Falany, 1997) | Simple phenolic compounds: p-nitrophenol, m-nitrophenol, p-ethylphenol, p-cresol (Wilborn et al., 1993; Brix et al., 1999b); Drugs: paracetamol (Lewis et al., 1996), minoxidil (Meisheri et al., 1993); Carcinogens: N-Hydroxy -PhIP (Ozawa et al., 1994). |
| SULT1A2 | Not known | Simple phenolic compounds: p-nitrophenol (Zhu et al., 1996); Carcinogens: N-Hydroxy −2-AAF (Glatt, 2000) |
| SULT1A3 | Catecholamines: dopamine, (Brix et al., 1999b; Dajani et al., 1999b), norepinephrine (noradrenaline) (Ganguly et al., 1995) | Simple Phenols: p-nitrophenol (Brix et al., 1999b); Carcinogens: 1-Hydroxymethylpyrene (Glatt, 2000) |
| SULT1B1 | Iodothyronines: 3,3′diiodothyronine (T2), 3,3′,5-triiodothyronine (T3), 3,3′,5′-reverse triiodothyronine (r-T3), and thyroxine (T4) (Wang et al., 1998) | Simple Phenol: 1-Naphthol (Wang et al., 1998) |
| SULT1C2 | Not known | Simple Phenols: p-nitrophenol (Sakakibara, 1998b); Carcinogens: N-Hydroxy-2-AAF (Yoshinari et al., 1998b) |
| SULT1C4 | Not known | Simple Phenols: p-nitrophenol (Yoshinari et al., 1998a); Carcinogens: N-Hydroxy-2-AAF (Sakakibara et al., 1998b) |
| SULT1E1 | Estrogens: E2, estrone (E1) (Falany et al., 1995) | Estrogens: 17-ethinyl-E2, equilenin (Falany et al., 1995); Catechol estrogens: 2-hydroxyestrone, 2-hydroxyestradiol, 4-hydroxyestrone; 4-hydroxyestradiol (Adjei and Weinshilboum, 2002) |
| SULT2A1 | Steroids: DHEA (Comer and Falany, 1992) | Carcinogens: 1- Hydroxymethylpyrene (Meinl et al., 2002), 6 Hydroxymethylbenzo[a]-pyrene, hycanthone (Glatt, 2000) |
| SULT2B1_v1 | DHEA (Her et al., 1998), pregnenolone (Meloche and Falany, 2001) | Not known |
| SULT2B1_v2 | DHEA (Her et al., 1998), pregnenolone (Meloche and Falany, 2001), cholesterol and oxysterols (Geese and Raftogianis, 2001) | Not known |
| SULT4A1 | Not known | Not known |
Endogenous and Xenobiotic Prototypic Substrates of Human Cytosolic SULTs
Subfamily | Endogenous substrates | Xenobiotic substrates |
|---|---|---|
| SULT1A1 | Iodothyronines: 3,3′diiodothyronine (T2), 3,3′,5-triiodothyronine (T3), (Anderson et al., 1995; Li et al., 2001); Estrogens: β-estradiol (E2) (Falany, 1997) | Simple phenolic compounds: p-nitrophenol, m-nitrophenol, p-ethylphenol, p-cresol (Wilborn et al., 1993; Brix et al., 1999b); Drugs: paracetamol (Lewis et al., 1996), minoxidil (Meisheri et al., 1993); Carcinogens: N-Hydroxy -PhIP (Ozawa et al., 1994). |
| SULT1A2 | Not known | Simple phenolic compounds: p-nitrophenol (Zhu et al., 1996); Carcinogens: N-Hydroxy −2-AAF (Glatt, 2000) |
| SULT1A3 | Catecholamines: dopamine, (Brix et al., 1999b; Dajani et al., 1999b), norepinephrine (noradrenaline) (Ganguly et al., 1995) | Simple Phenols: p-nitrophenol (Brix et al., 1999b); Carcinogens: 1-Hydroxymethylpyrene (Glatt, 2000) |
| SULT1B1 | Iodothyronines: 3,3′diiodothyronine (T2), 3,3′,5-triiodothyronine (T3), 3,3′,5′-reverse triiodothyronine (r-T3), and thyroxine (T4) (Wang et al., 1998) | Simple Phenol: 1-Naphthol (Wang et al., 1998) |
| SULT1C2 | Not known | Simple Phenols: p-nitrophenol (Sakakibara, 1998b); Carcinogens: N-Hydroxy-2-AAF (Yoshinari et al., 1998b) |
| SULT1C4 | Not known | Simple Phenols: p-nitrophenol (Yoshinari et al., 1998a); Carcinogens: N-Hydroxy-2-AAF (Sakakibara et al., 1998b) |
| SULT1E1 | Estrogens: E2, estrone (E1) (Falany et al., 1995) | Estrogens: 17-ethinyl-E2, equilenin (Falany et al., 1995); Catechol estrogens: 2-hydroxyestrone, 2-hydroxyestradiol, 4-hydroxyestrone; 4-hydroxyestradiol (Adjei and Weinshilboum, 2002) |
| SULT2A1 | Steroids: DHEA (Comer and Falany, 1992) | Carcinogens: 1- Hydroxymethylpyrene (Meinl et al., 2002), 6 Hydroxymethylbenzo[a]-pyrene, hycanthone (Glatt, 2000) |
| SULT2B1_v1 | DHEA (Her et al., 1998), pregnenolone (Meloche and Falany, 2001) | Not known |
| SULT2B1_v2 | DHEA (Her et al., 1998), pregnenolone (Meloche and Falany, 2001), cholesterol and oxysterols (Geese and Raftogianis, 2001) | Not known |
| SULT4A1 | Not known | Not known |
Subfamily | Endogenous substrates | Xenobiotic substrates |
|---|---|---|
| SULT1A1 | Iodothyronines: 3,3′diiodothyronine (T2), 3,3′,5-triiodothyronine (T3), (Anderson et al., 1995; Li et al., 2001); Estrogens: β-estradiol (E2) (Falany, 1997) | Simple phenolic compounds: p-nitrophenol, m-nitrophenol, p-ethylphenol, p-cresol (Wilborn et al., 1993; Brix et al., 1999b); Drugs: paracetamol (Lewis et al., 1996), minoxidil (Meisheri et al., 1993); Carcinogens: N-Hydroxy -PhIP (Ozawa et al., 1994). |
| SULT1A2 | Not known | Simple phenolic compounds: p-nitrophenol (Zhu et al., 1996); Carcinogens: N-Hydroxy −2-AAF (Glatt, 2000) |
| SULT1A3 | Catecholamines: dopamine, (Brix et al., 1999b; Dajani et al., 1999b), norepinephrine (noradrenaline) (Ganguly et al., 1995) | Simple Phenols: p-nitrophenol (Brix et al., 1999b); Carcinogens: 1-Hydroxymethylpyrene (Glatt, 2000) |
| SULT1B1 | Iodothyronines: 3,3′diiodothyronine (T2), 3,3′,5-triiodothyronine (T3), 3,3′,5′-reverse triiodothyronine (r-T3), and thyroxine (T4) (Wang et al., 1998) | Simple Phenol: 1-Naphthol (Wang et al., 1998) |
| SULT1C2 | Not known | Simple Phenols: p-nitrophenol (Sakakibara, 1998b); Carcinogens: N-Hydroxy-2-AAF (Yoshinari et al., 1998b) |
| SULT1C4 | Not known | Simple Phenols: p-nitrophenol (Yoshinari et al., 1998a); Carcinogens: N-Hydroxy-2-AAF (Sakakibara et al., 1998b) |
| SULT1E1 | Estrogens: E2, estrone (E1) (Falany et al., 1995) | Estrogens: 17-ethinyl-E2, equilenin (Falany et al., 1995); Catechol estrogens: 2-hydroxyestrone, 2-hydroxyestradiol, 4-hydroxyestrone; 4-hydroxyestradiol (Adjei and Weinshilboum, 2002) |
| SULT2A1 | Steroids: DHEA (Comer and Falany, 1992) | Carcinogens: 1- Hydroxymethylpyrene (Meinl et al., 2002), 6 Hydroxymethylbenzo[a]-pyrene, hycanthone (Glatt, 2000) |
| SULT2B1_v1 | DHEA (Her et al., 1998), pregnenolone (Meloche and Falany, 2001) | Not known |
| SULT2B1_v2 | DHEA (Her et al., 1998), pregnenolone (Meloche and Falany, 2001), cholesterol and oxysterols (Geese and Raftogianis, 2001) | Not known |
| SULT4A1 | Not known | Not known |
SULT2A.
The rat senescence marker protein (SULT2A2) that is predominantly expressed in aging male rats and mentioned above (Chatterjee et al., 1987) was subsequently shown to be a hydroxysteroid sulfotransferase. This became apparent on the cloning of three additional rat SULT2A isoforms, SULT2-A1, A3, A4 (see Blanchard et al., 2004 for references). SULT2A isoforms have also been identified in a variety of species, with SULT2A1 being isolated from the mouse, rabbit (Blanchard et al., 2004), and monkey (Ogura et al. unpublished). Unlike the rat, which has multiple forms of these 2A enzymes, humans have only a solitary isoform, SULT2A1 (Comer et al., 1993; Forbes et al., 1995; Kong et al., 1992; Otterness et al., 1992). Human SULT2A1 is responsible for the sulfonation of hydroxysteroids including DHEA, androgens, pregnenolone, and bile acids and was initially cloned using liver and fetal adrenal RNA and termed DHEA sulfotransferase for its preferred substrate (Comer et al., 1993; Forbes et al., 1995; Kong et al., 1992; Otterness et al., 1992; Radominska et al., 1990).
SULT2B.
While the SULT2A and SULT2B subfamilies are capable of metabolizing a range of similar substrates, it nonetheless appears that SULT2B subfamily members are predominantly cholesterol sulfotransferases (Javitt et al., 2001). To date, three members of this subfamily have been identified, Sult2b1 from the mouse (Sakakibara et al., 1998a) and two human isoforms (SULT2B1_v1 and v2; Her et al., 1998). Human SULT2B1_v1 is 15 amino acids shorter than SULT2B1_v2 at the amino terminus, which imparts a functional distinction. For example, SULT2B1_v1 preferentially sulfonates pregnenolone, whereas SULT2B1_v2 catalyses the sulfonation of cholesterol (Fuda et al., 2002).
SULT4 Family
Falany's group cloned the first members of the SULT4 family from both human and rat brain cDNA libraries (Falany et al., 2000). These authors termed the cDNAs “brain sulfotransferase-like” (BR-STL), because of their structural similarity to published SULTs and their selective expression in brain tissue. Liyou et al. (2003) cloned an identical cDNA from a human brain cDNA library, and Sakakibara et al. (2002) have identified the equivalent mouse brain isoform. At the amino acid level the human, rat, and mouse isoforms are 97% similar, and based on the Blanchard et al. (2004) nomenclature, they have been classified as SULT4A1. These proteins are orphan enzymes, as no substantial activity toward endogenous or xenobiotic substrates has been demonstrated (Sakakibara et al., 2002). The predominant brain localization of SULT4A1 and the high degree of sequence identity across species is suggestive of an important, yet unidentified physiological function.
LOCALIZATION OF HUMAN SULFOTRANSFERASES
SULT1A Subfamily
Much of the early work on the specific cellular localization of human SULT1A members is clouded by the fact that it was not until, 1995 that we definitively knew this subfamily consisted of three closely related members that shared >93% identity at the amino acid level (Blanchard et al., 2004; Hempel et al., 2005). The recent finding of Hildebrandt et al. (2004), which shows that SULT1A3 has undergone a gene duplication and both SULT1A3 and 1A4 appear to be transcriptionally, active requires a more cautious interpretation of SULT1A3 data. However, the available data obtained using an array of methods including metabolic probes, immunohistochemistry, hybridization histochemistry, immunoblotting, and RT-PCR analysis have shown that SULT1A members exhibit probably the widest tissue distribution of any cytosolic SULT subfamily (Blanchard et al., 2004; Dooley et al., 2000; Hempel et al., 2005). SULT1A1 is by far the major adult liver SULT1A subfamily member and has also been identified in brain (Richard et al., 2001; Whittemore et al., 1986; Young et al., 1985), breast (Windmill et al., 1998), intestine (Teubner et al., 1998), endometrium (Falany et al., 1998), adrenal gland, platelets, and placenta (Abenhaim et al., 1981; Hart et al., 1979; Heroux et al., 1989), kidney and lung (Vietri et al., 2003), and jejunum (Sundaram et al., 1989). SULT1A3, as indicated above, is barely detectable in the adult human liver (Eisenhofer et al., 1999; Heroux et al., 1989) but is highly expressed in jejunum and intestine (Eisenhofer et al., 1999; Richard et al., 2001; Sundaram et al., 1989; Teubner et al., 1998) and also present in platelets and placenta (Heroux et al., 1989) and brain (Whittemore et al., 1985; Young et al., 1985). Using hybridization histochemistry, employing a general SULT1A ribo-probe, a specific SULT1A3 ribo-probe, and a SULT1A antibody, a positive signal was observed in epithelial cells lining the lumen of the stomach and the gastric pits, and in the epithelial cells lining the lumen surface of the crypts of Lieberkuhn of the small intestine and colon. In human lung cytosol, SULT1A1 and SULT1A3 proteins are detectable, and histological studies have shown these proteins present in epithelial cells of the respiratory bronchioles (Hempel et al., 2005; Windmill et al., 1998). Since both the intestine and lungs are major portals of entry of drugs and xenobiotics into the body, the above localization pattern suggests that both SULT1A1 and SULT1A3 may play a significant role in the extrahepatic detoxification and metabolic activation of these chemicals. From a developmental perspective, both SULT1A1 and SULT1A3 are abundantly expressed in the fetal liver, but SULT1A3 almost disappears in adult liver and kidney (Cappiello et al., 1991; Hempel et al., 2005; Pacifici et al., 1993; Richard et al., 2001). These results are suggestive of a role for SULT1A members in protecting the fetus from exogenous toxins and in the homeostasis of hormones such as dopamine and iodothyronines. Further, immunoblotting of placenta cytosol showed the presence of both SULT1A1 and SULT1A3, indicating they may have a potential role in the metabolism of xenobiotics entering the fetal circulation from the maternal side (Heroux et al., 1989; Stanley et al., 2001). The fact that placental UDP-glucuronosyltransferases are relatively low and variable in humans (Collier et al., 2002; Pacifici et al., 1998) suggests that SULT1A members may play a significant role in phase II metabolism in this tissue.
The localization and physiological function of SULT1A2 is the least understood of the SULT1A subfamily members. cDNAs of SULT1A2 have been isolated from both human liver and colon libraries (Blanchard et al., 2004; Ozawa et al., 1995; Zhu et al., 1996), and lower mRNA levels than other SULT1A members have been found in liver, kidney, brain, lung, ovary, and some sections of the gastrointestinal tract (Dooley et al., 2000; Glatt et al., 2001). However, it appears that SULT1A2 mRNA expression does not translate into the formation of protein. For example, Nowell et al. (2005), using a specific anti-peptide antibody for SULT1A2, screened more than 200 cytosolic fractions from 10 different human tissues and found no evidence of immunoreactive protein. Dooley et al. (2000) have suggested that the SULT1A2 gene is a quasi-effective pseudogene of SULT1A1 that is occasionally expressed at the RNA level. While SULT1A2 has been shown to be more efficient than SULT1A1 in the metabolic activation of several aromatic amines (Glatt and Meinl, 2004; Meinl et al., 2002) and capable of activating 3-nitrobenzanthrone and its metabolites in model in vitro systems, the current weight of evidence suggests this enzyme is not expressed in vivo in humans. Because of this data, Nowell et al. (2005) have cautioned the interpretation of data from SULT1A2 genotype/disease association studies.
SULT1B Subfamily
The major physiological role for human SULT1B1 appears to be in thyroid hormone metabolism (Fujita et al., 1997; Wang et al., 1998). To date, SULT1B1 (hST1B2) mRNA has been detected in liver, small intestine, colon, and blood leukocytes (Wang et al., 1998). In the same study, it was also demonstrated that SULT1B1 protein was clearly detectable on immunoblots in cytosol from liver, small intestine, and colon.
SULT1C Subfamily
The physiological role of SULT1C members is currently unknown. Her et al. (1997) performed dot blot analysis and obtained positive signal for SULT1C2 (called SULT1C1 in paper) in the adult human stomach, kidney, and thyroid, as well as fetal liver and kidney. Both Yoshinari et al. (1998b) and Hehonah et al. (1999) isolated identical human sequences from fetal liver and adult stomach cDNA libraries, respectively. Sakakibara et al. (1998b) identified another member of this subfamily from human fetal lung that has been termed SULT1C4. At the RNA level, SULT1C4 was shown to be expressed at higher levels in fetal lung and kidney and at lower levels in fetal heart. Positive signal was also found in the adult kidney, ovary, and spinal cord (Sakakibara et al., 1998b).
SULT1E Subfamily
SULT1E1 is present on immunoblots in both human liver and jejunum cytosol (Forbes-Bamforth and Coughtrie, 1994; Falany et al., 1995). Falany et al. (1998) showed that, on immunoblots, SULT1E1 was not detectable in proliferative endometrial cytosol, but was consistently found in the secretory endometrial cytosols. Coughtrie (2002) has also reported significant levels of SULT1E1 in human fetal liver, lung, and kidney, using E2 activity as a diagnostic indicator of this protein.
SULT2A Subfamily
Northern analysis has shown SULT2A1 to be present in the human liver, adrenal, and small intestine (Luu-The et al., 1995; Otterness et al., 1992; Tashiro et al., 2000). In a more extensive investigation using RT-PCR, Javitt et al. (2001) showed that SULT2A1 mRNA was highly expressed in steroidogenic organs (adrenal and ovary), androgen-dependent tissue (prostate), and in the liver, stomach, small intestine, and colon. Barker et al. (1994) have used immunohistochemistry to demonstrate SULT2A1 (DHEA-ST) expression in embryonic human hepatocytes and that this pattern continues into adulthood, when immunostaining is localized around the central vein. The same authors also reported that SULT2A1 expression was detected in fetal zone of the fetal adrenal, and in the adult, staining was localized in the zona reticularis. Further, kidney SULT2A1 immunostaining was present in the proximal and distal tubules, loops of Henle, collecting ducts, and their progenitors.
SULT2B Subfamily
An initial study using Northern analysis showed SULT2B1 to be localized to the human prostate, placenta, small intestine, and trachea (Tashiro et al., 2000). Javitt et al. (2001) extended this study to both isoforms of SULT2B1 using RT-PCR and demonstrated that SUL2B1_v2 was more widely expressed than its counterpart, particularly in a variety of hormone-responsive tissues including the placenta and prostate, which is suggestive of a differential regulatory mechanism of the two transcripts (Geese and Raftogianis, 2001). For example, SULT2B1_v2 mRNA was present in adrenal gland, placenta, ovary, prostate, lung, kidney, colon, stomach, small intestine, spleen, thymus, thyroid, and liver, whereas SULT2B1_v1 mRNA was present in all these tissues except the last six. Interestingly, SULT2A1 mRNA is present in brain and bone marrow but absent from the skin, where the mRNA of both isoforms of SULT2B are expressed.
SULT4A Subfamily
The current data shows an exclusive brain localization of SULT4A1 in rats, mice, and humans (Falany et al., 2000; Liyou et al., 2003; Sakakibara et al., 2002). However, recent microarray data is suggestive of a wider tissue distribution than the brain for the localization of this SULT (Mm.248796 at <http://www.ncbi.nlm.nih.gov/Unigene/>).
STRUCTURAL STUDIES AND ENZYMATIC MECHANISM IN CYTOSOLIC SULTS
SULTs can exhibit quite broad, overlapping substrate specificities; however, individual enzymes often demonstrate strict regio-specificity toward a particular substrate (Falany, 1997). Understanding the structural basis of such specificity has been crucial for elucidating the catalytic mechanism and function of these enzymes. It will also aid in predicting the metabolic fate of drugs and chemical carcinogens that are sulfonated and could provide a more rational approach to drug design and chemical risk assessment.
The major catecholamine SULT, SULT1A3, was the first human cytosolic SULT to be structurally characterized (Bidwell et al., 1999; Dajani et al., 1999a). Prior to this, the mouse cytosolic estrogen SULT (SULT1E1) (Kakuta et al., 1997) and the SULT domain of the human Golgi membrane-bound heparan sulfate N-deacetylase/N-sulfotransferase-1 (HSNST; Kakuta et al., 1999) were published. Since then, the number of crystal structures of cytosolic and membrane-bound SULTs have grown rapidly. Despite this progress, only a few crystal structures have been determined with both cofactor and substrate bound (Table 3). This relative lack of information has hindered elucidation of the structural principles underlying the recognition and utilization of a given substrate.
Crystal Structures of Human SULTs as of Mid-2005
SULT Structure | Cofactor bound | Substrate/inhibitor bound | Resolution (Å) | PDB code | Publication |
|---|---|---|---|---|---|
| SULT1A1*2 | PAP | p-nitrophenol | 1.9 | 1LS6 | Gamage et al., 2003 |
| SULT1A1*3 | PAP | estradiol | 2.3 | 2D06 | Gamage et al., 2005 |
| # | # | # | # | Lu et al. unpublished | |
| SULT1A2 | # | # | # | # | Lu et al. unpublished |
| SULT1A3 | SO42– | — | 2.4 | 1CJM | Bidwell et al., 1999 |
| PAP | — | 2.5 | — | Dajani et al., 1999a | |
| PAP | dopamine | 2.6 | 2A3R | Lu et al., 2005 | |
| SULT1B1 | PAP | — | 2.1 | 1XV1 | Dombrovski et al. (unpublished) |
| SULT1C1 | PAP | — | 2.22 | 1ZHE | Dong et al. (unpublished) |
| SULT1E1 | PAP | (OH-PCB)a | 1.7 | 1G3M | Shevtsov et al., 2003 |
| PAPS | — | 1.8 | 1HY3 | Pedersen et al., 2002 | |
| PAP | vanadate | 2.1 | 1BO6 | Kakuta et al., 1998 | |
| SULT2A1 | PAP | — | 2.4 | 1EFH | Pedersen et al., 2000 |
| SULT2A3 | — | DHEAb | 1.99 | 1J99 | Rehse et al., 2002 |
| DHEA-ST | — | ADTc | 2.7 | 1OV4 | Chang et al., 2003 |
| SULT2B1_v1 | PAP | — | 2.91 | 1Q1Q | Lee et al., 2003 |
| SULT2B1_v2 | PAP | — | 2.4 | 1Q1Z | Lee et al., 2003 |
| PAP | pregnenolone | 2.3 | 1Q20 | Lee et al., 2003 | |
| PAP | DHEAb | 2.5 | 1Q22 | Lee et al., 2003 | |
| SULT4A1 | — | — | 2.24 | 1ZD1 | Dong et al. (unpublished) |
| H3-OST-1d | PAP | — | 2.1 | 1ZRH | Dong et al. (unpublished) |
| HSNSTe | PAP | — | 2.3 | 1NST | Kakuta et al., 1999 |
| 3-OST-3f | PAP | — | 1.85 | 1T8T | Moon et al., 2004 |
| PAP | Tetrasaccharide | 1.95 | 1T8U | Moon et al., 2004 |
SULT Structure | Cofactor bound | Substrate/inhibitor bound | Resolution (Å) | PDB code | Publication |
|---|---|---|---|---|---|
| SULT1A1*2 | PAP | p-nitrophenol | 1.9 | 1LS6 | Gamage et al., 2003 |
| SULT1A1*3 | PAP | estradiol | 2.3 | 2D06 | Gamage et al., 2005 |
| # | # | # | # | Lu et al. unpublished | |
| SULT1A2 | # | # | # | # | Lu et al. unpublished |
| SULT1A3 | SO42– | — | 2.4 | 1CJM | Bidwell et al., 1999 |
| PAP | — | 2.5 | — | Dajani et al., 1999a | |
| PAP | dopamine | 2.6 | 2A3R | Lu et al., 2005 | |
| SULT1B1 | PAP | — | 2.1 | 1XV1 | Dombrovski et al. (unpublished) |
| SULT1C1 | PAP | — | 2.22 | 1ZHE | Dong et al. (unpublished) |
| SULT1E1 | PAP | (OH-PCB)a | 1.7 | 1G3M | Shevtsov et al., 2003 |
| PAPS | — | 1.8 | 1HY3 | Pedersen et al., 2002 | |
| PAP | vanadate | 2.1 | 1BO6 | Kakuta et al., 1998 | |
| SULT2A1 | PAP | — | 2.4 | 1EFH | Pedersen et al., 2000 |
| SULT2A3 | — | DHEAb | 1.99 | 1J99 | Rehse et al., 2002 |
| DHEA-ST | — | ADTc | 2.7 | 1OV4 | Chang et al., 2003 |
| SULT2B1_v1 | PAP | — | 2.91 | 1Q1Q | Lee et al., 2003 |
| SULT2B1_v2 | PAP | — | 2.4 | 1Q1Z | Lee et al., 2003 |
| PAP | pregnenolone | 2.3 | 1Q20 | Lee et al., 2003 | |
| PAP | DHEAb | 2.5 | 1Q22 | Lee et al., 2003 | |
| SULT4A1 | — | — | 2.24 | 1ZD1 | Dong et al. (unpublished) |
| H3-OST-1d | PAP | — | 2.1 | 1ZRH | Dong et al. (unpublished) |
| HSNSTe | PAP | — | 2.3 | 1NST | Kakuta et al., 1999 |
| 3-OST-3f | PAP | — | 1.85 | 1T8T | Moon et al., 2004 |
| PAP | Tetrasaccharide | 1.95 | 1T8U | Moon et al., 2004 |
Note. # indicates structure deposited with the Protein Data Bank but not yet released; details not available.
3,5,3′,5′-Tetrachloro-Biphenyl-4,4′-Diol.
Dehydroepiandrosterone.
Androsterone.
Heparan sulfate glucosamine 3-O-sulfotransferase-1.
SULT domain of heparin sulfate-N-deacetylase sulfotransferase.
3-O-sulfotransferase.
Crystal Structures of Human SULTs as of Mid-2005
SULT Structure | Cofactor bound | Substrate/inhibitor bound | Resolution (Å) | PDB code | Publication |
|---|---|---|---|---|---|
| SULT1A1*2 | PAP | p-nitrophenol | 1.9 | 1LS6 | Gamage et al., 2003 |
| SULT1A1*3 | PAP | estradiol | 2.3 | 2D06 | Gamage et al., 2005 |
| # | # | # | # | Lu et al. unpublished | |
| SULT1A2 | # | # | # | # | Lu et al. unpublished |
| SULT1A3 | SO42– | — | 2.4 | 1CJM | Bidwell et al., 1999 |
| PAP | — | 2.5 | — | Dajani et al., 1999a | |
| PAP | dopamine | 2.6 | 2A3R | Lu et al., 2005 | |
| SULT1B1 | PAP | — | 2.1 | 1XV1 | Dombrovski et al. (unpublished) |
| SULT1C1 | PAP | — | 2.22 | 1ZHE | Dong et al. (unpublished) |
| SULT1E1 | PAP | (OH-PCB)a | 1.7 | 1G3M | Shevtsov et al., 2003 |
| PAPS | — | 1.8 | 1HY3 | Pedersen et al., 2002 | |
| PAP | vanadate | 2.1 | 1BO6 | Kakuta et al., 1998 | |
| SULT2A1 | PAP | — | 2.4 | 1EFH | Pedersen et al., 2000 |
| SULT2A3 | — | DHEAb | 1.99 | 1J99 | Rehse et al., 2002 |
| DHEA-ST | — | ADTc | 2.7 | 1OV4 | Chang et al., 2003 |
| SULT2B1_v1 | PAP | — | 2.91 | 1Q1Q | Lee et al., 2003 |
| SULT2B1_v2 | PAP | — | 2.4 | 1Q1Z | Lee et al., 2003 |
| PAP | pregnenolone | 2.3 | 1Q20 | Lee et al., 2003 | |
| PAP | DHEAb | 2.5 | 1Q22 | Lee et al., 2003 | |
| SULT4A1 | — | — | 2.24 | 1ZD1 | Dong et al. (unpublished) |
| H3-OST-1d | PAP | — | 2.1 | 1ZRH | Dong et al. (unpublished) |
| HSNSTe | PAP | — | 2.3 | 1NST | Kakuta et al., 1999 |
| 3-OST-3f | PAP | — | 1.85 | 1T8T | Moon et al., 2004 |
| PAP | Tetrasaccharide | 1.95 | 1T8U | Moon et al., 2004 |
SULT Structure | Cofactor bound | Substrate/inhibitor bound | Resolution (Å) | PDB code | Publication |
|---|---|---|---|---|---|
| SULT1A1*2 | PAP | p-nitrophenol | 1.9 | 1LS6 | Gamage et al., 2003 |
| SULT1A1*3 | PAP | estradiol | 2.3 | 2D06 | Gamage et al., 2005 |
| # | # | # | # | Lu et al. unpublished | |
| SULT1A2 | # | # | # | # | Lu et al. unpublished |
| SULT1A3 | SO42– | — | 2.4 | 1CJM | Bidwell et al., 1999 |
| PAP | — | 2.5 | — | Dajani et al., 1999a | |
| PAP | dopamine | 2.6 | 2A3R | Lu et al., 2005 | |
| SULT1B1 | PAP | — | 2.1 | 1XV1 | Dombrovski et al. (unpublished) |
| SULT1C1 | PAP | — | 2.22 | 1ZHE | Dong et al. (unpublished) |
| SULT1E1 | PAP | (OH-PCB)a | 1.7 | 1G3M | Shevtsov et al., 2003 |
| PAPS | — | 1.8 | 1HY3 | Pedersen et al., 2002 | |
| PAP | vanadate | 2.1 | 1BO6 | Kakuta et al., 1998 | |
| SULT2A1 | PAP | — | 2.4 | 1EFH | Pedersen et al., 2000 |
| SULT2A3 | — | DHEAb | 1.99 | 1J99 | Rehse et al., 2002 |
| DHEA-ST | — | ADTc | 2.7 | 1OV4 | Chang et al., 2003 |
| SULT2B1_v1 | PAP | — | 2.91 | 1Q1Q | Lee et al., 2003 |
| SULT2B1_v2 | PAP | — | 2.4 | 1Q1Z | Lee et al., 2003 |
| PAP | pregnenolone | 2.3 | 1Q20 | Lee et al., 2003 | |
| PAP | DHEAb | 2.5 | 1Q22 | Lee et al., 2003 | |
| SULT4A1 | — | — | 2.24 | 1ZD1 | Dong et al. (unpublished) |
| H3-OST-1d | PAP | — | 2.1 | 1ZRH | Dong et al. (unpublished) |
| HSNSTe | PAP | — | 2.3 | 1NST | Kakuta et al., 1999 |
| 3-OST-3f | PAP | — | 1.85 | 1T8T | Moon et al., 2004 |
| PAP | Tetrasaccharide | 1.95 | 1T8U | Moon et al., 2004 |
Note. # indicates structure deposited with the Protein Data Bank but not yet released; details not available.
3,5,3′,5′-Tetrachloro-Biphenyl-4,4′-Diol.
Dehydroepiandrosterone.
Androsterone.
Heparan sulfate glucosamine 3-O-sulfotransferase-1.
SULT domain of heparin sulfate-N-deacetylase sulfotransferase.
3-O-sulfotransferase.
Overall SULT Structure
The SULT crystal structures have shown that the enzymes are generally globular proteins with a single α/β domain that forms characteristic five-stranded parallel β-sheet surrounded on either side with α-helices (Fig. 2). The β-sheets contribute the PAPS-binding site and the core catalytic residues. These catalytic residues have been shown to be conserved across the cytosolic and membrane-bound SULTs. Interestingly, SULTs share similarities in their structural fold with nucleotide kinases such as uridylate kinase, adenylate kinase, and guanylate kinase (Kakuta et al., 1997).
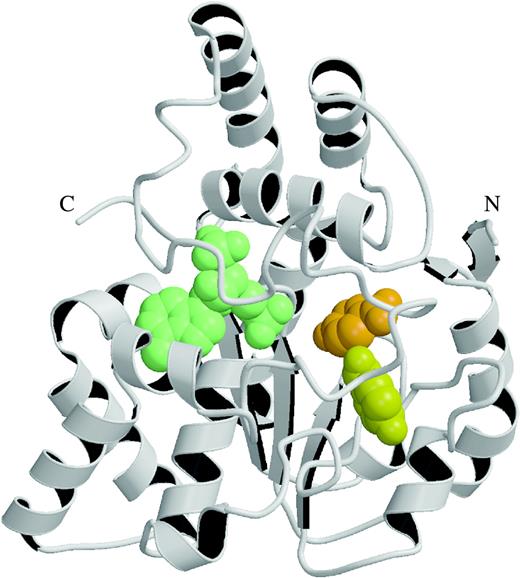
Overall structure of a cytosolic SULT: structure of human SULT1A1 complex with 3′-phosphoadenosine-5′-phosphate (PAP) and p-nitrophenol (pNP; Gamage et al., 2003). Secondary structural elements are depicted as coils for helices and arrows for strands. The bound ligands are shown as spherical atomic models; PAP, green; pNP1, orange; pNP2, yellow.
PAP Binding Site
Early sequence analysis suggested that the sequence motif GxxGxxK, present in nearly all cytosolic SULTs, and shown to be important for PAPS binding, was equivalent to the P-loop found in ATP- and GTP-binding proteins (Chiba et al., 1995; Driscoll et al., 1995). However, structural studies on mouse SULT1E1 revealed that the “P-loop” equivalent in SULTs actually corresponds to another loop region, comprising residues 45-TYPKSGT-51 of SULT1E1 (Kakuta et al., 1997). This loop, termed the phosphosulfate binding (PSB) loop, of SULTs provides the major binding site for the 5′-phosphate group of PAP (Kakuta et al., 1997) and is thought to be important for orienting the cofactor for in-line sulfuryl transfer to the acceptor substrate (Dajani et al., 1999a; Kakuta et al., 1997; Pedersen et al., 2002). Please note that the term PAP is used because mouse SULT1E1 was crystallized in the presence of this molecule and not with PAPS.
The 3′-phosphate of PAP interacts with two conserved regions of sequence, residues 257–259 located at the beginning of the GxxGxxK region, and two additional residues, Arg130 and Ser138 in mouse SULT1E1 (Kakuta et al., 1997). The positioning of the adenine ring of PAP is determined by the residues Trp53, Thr227, and Phe229. These general features identified in SULT1E1 for binding PAP are conserved in other SULT structures, suggesting that the cytosolic and membrane-bound SULTs evolved from a common ancestor (Yoshinari et al., 2001).
Substrate Binding Site
Generally, cytosolic SULTs have a covered hydrophobic substrate binding site, whereas the presumed substrate binding pocket of the membrane-bound HSNST is a large open cleft to allow sulfonation of carbohydrates, glucosaminylglycans, and proteins. As mentioned before, SULTs display broad substrate specificity, though a given enzyme can often be characterized by having a preference for a specific substrate. The underlying principles that regulate this specificity most probably reside in the substrate binding sites of these enzymes. Thus, in contrast to the PAPS binding site, which is characterized by conserved residues across all the SULTs, the substrate binding pocket of SULTs shows a great deal of variability. Despite the elucidation of several SULT crystal structures, the structural principles that underpin substrate specificity are still not fully understood. As indicated above, this is due to the fact that only a few crystal structures have been solved with bound substrate and cofactor present.
In the SULT1A family, the crystal structures of SULT1A1*2, SULT1A1*3, SULT1A2, and SULT1A3 (Table 3) have been determined. The Bidwell et al. (1999) structure of SULT1A3 was solved with a sulfate ion in the cofactor site, whereas the Dajani et al. (1999a) SULT1A3 structure was complexed with PAP. Both structures show large stretches of disordered regions, and this was thought to be a consequence of the lack of a bound substrate. In contrast, the crystal structure of SULT1A1 was crystallized in the presence of both PAP and a model xenobiotic substrate pNP that revealed an L-shaped and very hydrophobic substrate-binding pocket (Gamage et al., 2003). Indeed, Gamage et al. (2003) has shown that the binding site of SULT1A1 is plastic, allowing this enzyme to adopt varying architectures so that it can interact with small aromatics (pNP), L-shaped aromatics (diiodothyronine), and fused ring compounds (E2; Gamage et al., 2005).
Human SULT1A1 and 1A3 share 90% sequence identity, though they exhibit distinct substrate preferences. SULT1A1 prefers uncharged simple phenolic compounds such as pNP, p-cresol, or p-ethylphenol, whereas 1A3 prefers positively charged substrates such as dopamine or tyramine (Brix et al., 1999b). The crystal structure of SULT1A1 revealed the hydrophobic substrate binding pocket mentioned above, which clearly favors binding of uncharged substrates. By contrast, the SULT1A3 substrate binding site includes acidic residues such as Glu146 and Glu89, which favors binding of positively charged substrates. Indeed, site-directed mutagenesis and molecular modeling studies identified Glu146 as a critical residue for the recognition of dopamine by SULT1A3 (Brix et al., 1999a ; Dajani et al., 1998).
Mouse SULT1E1 was the first cytosolic SULT to be structurally characterized (Kakuta et al., 1997); its structure showed the enzyme in a complex with both the substrate E2 and PAP. In the active site, His108 is directly coordinated to the 3-phenolic group of E2 and acts as the catalytic base in the sulfuryl transfer mechanism. This histidine residue is conserved in all cytosolic SULTs, and its mutation has been shown to abolish activity of mouse SULT1E1 (Kakuta et al., 1998). Thus, this structure provided the basis for understanding β-estradiol binding in the active site and for catalysis of sulfonation by the proposed SN2 in-line displacement mechanism. The crystal structures of SULT1E1:PAP:vanadate (Kakuta et al., 1998) and human SULT1E1:PAPS (Pedersen et al., 2002) provided further evidence as to the structure of the transition state during sulfuryl transfer and gave supporting evidence for the proposed mechanism.
In the SULT2A subfamily, the crystal structures of the human dehydroepiandrosterone sulfotransferase enzymes (SULT2A1, SULT2A3), which sulfonate steroids such as DHEA, androsterone, E2, and pregnenolone, have been solved in complex with PAP (Pedersen et al., 2000), DHEA (Rehse et al., 2002), and androsterone (Chang et al., 2003). In the DHEA-bound structure, two alternative substrate-binding orientations were identified for DHEA, and the authors suggested that the second orientation may reflect a binding mode associated with substrate inhibition. The work of Chang et al. (2003) demonstrated that this enzyme recognizes androsterone as a cognate substrate, with similar kinetics but higher specificity and stronger substrate inhibition than DHEA.
In the SULT2B subfamily, SULT2B1_v1 (SULT2B1a) and 2B1_v2 (2B1b), as outlined above, are splice variants (Her et al., 1998) and have different substrate specificities (Fuda et al., 2002). The crystal structures of SULT2B1_v1 and 2B1_v2 bound with PAP and that of SULT2B1_v2 with its substrate pregnenolone have been determined (Lee et al., 2003). These structures reveal a different catalytic binding orientation for the acceptor substrate, pregnenolone, than that observed for the related steroid DHEA in SULT2A1. It was shown that the amino-terminal helix comprising residues19–26 determines the substrate specificity between the two isoforms. The residues 19-Asp-Ile-Ser-Glu-Ile-23 are responsible for the ability of SULT2b1_v2 to sulfonate cholesterol (Fuda et al., 2002; Lee et al., 2003). Thus, the substrate specificity difference between the two SULT2B1 isoforms appears to lie at the unique amino terminus.
The crystal structures of SULT1B1:PAP, SULT1C1:PAP, and SULT4A1 have also been determined and submitted to the Protein Data bank (PDB, http://www.rcsb.org/pdb), but are not yet described in the literature (Table 3).
Sulfuryl Transfer Mechanism
Duffel and Jakoby (1981) reported that pNP sulfonation by rat aryl sulfotransferase IV has a random Bi Bi mechanism in which PAPS and pNP bind to the enzyme independently. Kinetic studies on the catalytic mechanism of recombinant mouse SULT1E1 suggested that sulfonation follows a random Bi Bi mechanism with dead-end complexes (Zhang et al., 1998). On the other hand, studies using purified human brain aryl sulfotransferase (Whittemore et al., 1986) and flavonol sulfotransferase (Varin and Ibrahim, 1992) suggested that sulfonation occurs via an ordered Bi Bi mechanism. All these studies agree that the sulfonate transfer reaction occurs without formation of intermediates. When the crystal structure of mouse SULT1E1 was solved in the presence of PAP and E2, it became clear that the core structure resembles that of uridylate kinase, with striking similarities between the PAP and ADP binding sites (Kakuta et al., 1997). These structural features of SULT1E1 suggest that the sulfotransferase reaction takes place via an SN2 in-line displacement, a mechanism similar to phosphoryl transfer (Kakuta et al., 1997, 1998). The active site and transition state mimicked by SULT1E1:PAP:vanadate (Kakuta et al., 1998) and human SULT1E1:PAPS complexes (Pedersen et al., 2002) provide further supporting evidence for this mechanism.
Substrate Inhibition
Substrate inhibition, observed at high concentrations of their preferred substrates, is a characteristic feature of SULTs (Raftogianis et al., 1999; Reiter et al., 1983). However, several of the published studies have assumed a Michaelis–Menten model to analyse the kinetics of these enzymes using limited substrate concentration ranges below the overtly inhibitory range (Brix et al., 1999b; Lewis et al., 1996). The recent studies on the crystal structure of SULT1A1 from our laboratory gave the first clues to the molecular basis of substrate inhibition that takes place with small planar substrates such as pNP (Gamage et al., 2003). In the SULT1A1:PAP:pNP structure (Gamage et al., 2003), we observed two pNP molecules bound in the L-shaped active site. When the kinetic implications of this observation were investigated using a wide array of pNP concentrations, we found that there was slight positive cooperativity at low substrate concentrations and substrate inhibition at higher pNP concentrations (above 2 μM; Fig. 3A). From these data, a general kinetic model was constructed (Fig. 3B). The model fits well to the experimental data, and we proposed that impeded catalysis results when both binding sites are occupied, and this gives rise to the observed substrate inhibition with pNP in SULT1A1. Furthermore, we have also shown by molecular modeling and site-directed mutagenesis that the SULT1A3 active site could accommodate two molecules of dopamine (Barnett et al., 2004). From these studies, we have been able to conclude that the substrate inhibition at high concentrations of the substrate is due to impeded catalysis when both binding sites are occupied.
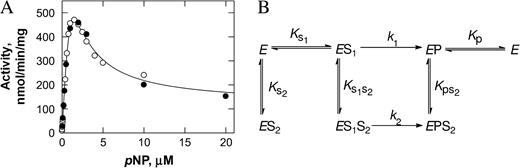
Kinetic implications of p-nitrophenol (pNP) sulfonation in SULT1A1. (A) Substrate inhibition is observed above 2 μM of pNP. Each data point is a mean of duplicate or triplicate assays. (B) Kinetic model to explain the observed substrate inhibition. The enzyme (E) binds at site 1 to give ES1 (dissociation constant KS1) or at site 2 to give ES2 (dissociation constant KS2). If site 2 is occupied, pNP will not bind to site 1. The binding of pNP at site 1 will not prevent binding of pNP at site 2 to give ES1S2. ES1 and ES1S2 are catalytically competent species, and they form EP and EPS2 enzyme product complexes with rate constants k1 and k2, respectively. Product is released directly from EP (dissociation constant Kp), and EPS2 requires prior release of pNP from site 2 (dissociation constant KpS2), (Gamage et al., 2003; with permission from J. Biol.Chem.).
The mechanism of substrate inhibition that takes place with multi-ring substrates such as E2 is not well understood. SULT1A1 and SULT1E1 both show strong substrate inhibition at high substrate concentrations of E2 (Adjei and Weinshilboum, 2002; Falany and Falany, 1997; Fig. 4A). In our recent crystal structure of SULT1A1, there is one molecule each of PAP and E2 in the active site, but the latter is bound in a nonproductive mode (Gamage et al., 2005). This has led us to propose a model (Fig. 4B) that gives an excellent quantitative explanation of the observed substrate inhibition by E2. In this model, a dead-end complex is formed during catalysis, as deduced previously from kinetic studies (Duffel and Jakoby, 1981; Yang et al., 1998). The SULT:PAP:E2 complex that we crystallized is a direct demonstration of this type of dead-end complex, and it provides a ready explanation for the substrate inhibition. A similar type of model may also explain the substrate inhibition that is observed for other SULTs.
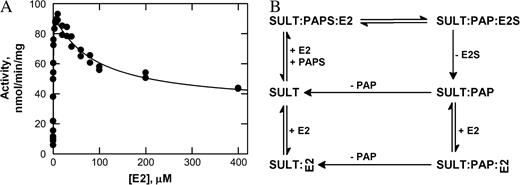
Kinetic implications of 17β-estradiol (E2) sulfonation in SULT1A1. (A) Substrate inhibition is observed above 1.5 μM of E2. Each data point is an average of duplicate assays. (B) Kinetic model to explain the observed substrate inhibition. The enzyme binds to E2 and 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to give rise to SULT:PAPS:E2, which undergoes catalysis, and E2S and 3′-phosphoadenosine-5′-phosphate (PAP) are released. Alternatively, the SULT:PAP complex can bind E2 in a nonproductive mode (E2 is drawn sideways) to give a dead-end complex. The enzyme can reenter catalysis by releasing PAP followed by E2. The crystal structure of SULT1A1:PAP:E2 demonstrates the dead-end complex mentioned above (Gamage et al., 2005; with permission from J. Biol.Chem.).
Dimerization
Most of the cytosolic SULTs generally exist as dimers in solution, and it appears that they are capable of forming not only homodimers but also heterodimers (Petrotchenko et al., 2001). A conserved dimerization motif was identified by Petrotchenko et al. (2001) in human SULT1E1 consisting of 10 residues near the C-terminus and represented by the consensus sequence KXXXTVXXXE (the so-called KTVE motif). The KTVE motif is conserved in nearly all SULTs (Petrotchenko et al., 2001), though the physiological significance of dimerization for the function of SULTs has not yet been identified. However, biophysical studies carried on dimerization and activation of epidermal growth factor receptor (EGFR) by ligand binding may provide future guidance in understanding the role of dimerization in SULT activity (Schlessinger, 2002).
Molecular Modeling of N-Hydroxy Metabolites of 2AAF and PhIP into SULT1A Isoforms
At the time of writing this review, no structural studies have been published that demonstrate the binding of N-hydroxy aromatic and heterocyclic amines or hydroxy methyl polycyclic aromatic hydrocarbons to human SULTs. However, using the published structures of SULT1A1 (Gamage et al., 2003), SULT1A3 (Bidwell et al., 1999), and the computer model of SULT1A2, we have investigated their ability to accommodate these carcinogens.
Specifically, we have modeled the binding of the N-hydroxylated metabolites of the model carcinogen 2-AAF and the major food-derived mutagen PhIP into the active sites of the three enzymes to investigate their binding interactions. The structures of SULT1A2*1 (unpublished data) and 1A3 (Barnett et al., 2004) were modeled based on the crystal structure of SULT1A1 (Gamage et al., 2003). PAPS was modeled into each of the three structures, based on the crystal structure of the SULT1E1:PAPS complex (Pedersen et al., 2002). The two ligands were docked into each of the three enzyme structures using the GOLD software (Jones et al., 1997, Fig. 5). The allozyme of SULT1A2 (SULT1A2*1 (HAST4v)) and 1A3 we chose to model with is the wild type. The crystal structure of SULT1A1 is that of SULT1A1*2, which has similar binding characteristics to wildtype SULT1A1*1 (with respect to PAPS and pNP) and has slightly lower thermal stability (Rafogianis et al., 1999).
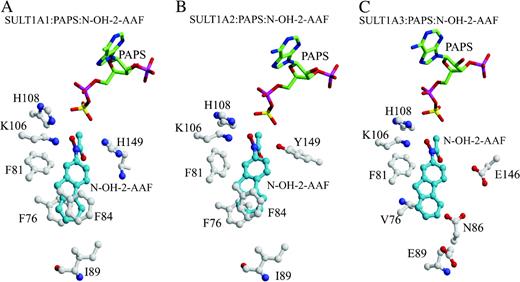
Models of N-hydroxy 2-acetylaminofluorene (N-OH-2-AAF) bound in the active sites of (A) SULT1A1, (B) 1A2, and (C) 1A3. The cofactor 3′-phosphoadenosine 5′-phosphosulfate (PAPS) is shown as a stick model. N-OH-2-AAF (blue) and enzyme residues (white) are represented as ball-and-stick models. Atom colouring is red for oxygen, blue for nitrogen, yellow for sulfur, and pink for phosphorous.
Our results showed that, in all three enzymes, the hydroxyl group of N-OH-2-AAF is within hydrogen bonding distance of the donor sulfonate group of PAPS and of the catalytic residue H108 (Fig. 5), consistent with the possibility of catalysis occurring. In the SULT1A1 and 1A2 models, residues F84 and F76 form stacking interactions with the ligand (Figs. 5A and 5B). However, in the 1A1 model the side chain of F81, the gate residue at the active site, forms an unfavorable interaction with the ligand (2.2 Å). In this case, it seems that a conformational change is necessary in F81 to accommodate the ligand. This is not seen in the SULT1A2 model. This observation could perhaps explain the higher activation of N-OH-2-AAF by SULT1A2 compared with SULT1A1 (Glatt, 2000; Meinl et al., 2002). In our SULT1A3 model (Fig. 5C), the ligand makes unfavorable interactions with both F81 (2.8Å; gate residue) and V84 (2.9 Å) at the enzyme active site. In addition, the active site is relatively acidic as a consequence of residues including E146 and E89; such an environment would be less favorable than the uncharged active sites of SULT1A1 and SULT1A2 for the binding of hydrophobic substrates. Once again, this finding is consistent with the reported lower activation of N-OH-2-AAF by SULT1A3 (Glatt, 2000; Meinl et al., 2002).
The modeling of N-OH-PhIP into the active sites of the enzymes revealed catalytically competent orientations for binding to all three SULT1A structures. In the SULT1A1 and 1A2 models, F76 and F84 form favorable stacking interactions with the benzyl ring of PhIP, as shown in Figures 6A and 6B. Positioning of the residues in the SULT1A1 and 1A2 active sites are similar except for Y149 in SULT1A2. This residue is a histidine in SULT1A1. In the SULT1A2 model, the phenolic hydroxyl of Y149 forms an interaction with the nitrogen of the N-OH group of PhIP. It is not clear whether such an interaction is favorable or unfavorable for binding and catalysis, but given that the interaction is absent in the SULT1A1:N-OH-PhIP model and the two models are otherwise similar, this specific interaction could perhaps explain the lowered metabolic activation of PhIP by SULT1A2 compared with SULT1A1 (Ozawa et al., 1994). By comparison with the very hydrophobic active sites of SULT1A1 and SULT1A2, the SULT1A3 active site is highly charged (Brix et al., 1999a; Dajani et al., 1999a). In our SULT1A3:PAPS:N-OH-PhIP model, we find that the acidic residues (E146 and D86) in the SULT1A3 active site form unfavorable interactions with N-OH-PhIP (Fig. 6C). This could, at least in part, explain the lowered activation of this compound observed with SULT1A3 (Glatt, 2000).
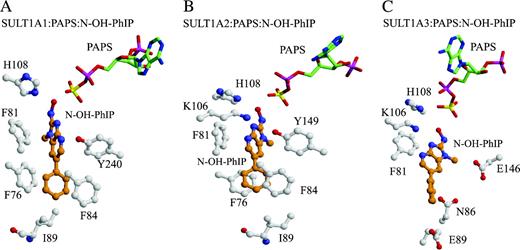
Docking of N-OH-PhIP (N-hydroxy-2-amino-1-methyl-6-phenylimidazo (4,5b-bipyridine)) into (A) SULT1A1, (B) 1A2, and (C) 1A3 structures. The cofactor 3′-phosphoadenosine 5′-phosphosulfate (PAPS) is shown as a stick model. N-OH-PhIP (orange) and enzyme residues (white) are represented as ball-and-stick models. Atom colouring is oxygen (red), nitrogen (blue), sulfur (yellow), and phosphorous (pink).
Human SULT1E1 and Hydroxylated Polychlorinated Biphenyls (OH-PCBs)
It is reported that certain environmentally relevant PCBs (polychlorinated biphenyls) such as 4,4′-OH-3,5,3′,5′-tetraCB inhibit human SULT1E1 at subnanomolar concentrations (Kester et al., 2000). PCBs are man-made pollutants that persist in the environment and exert a variety of toxic effects on experimental animals by inducing estrogenic activities through increased availability of estradiol (Kester et al., 2000). These compounds and their metabolites have been shown to cause endocrine-disrupting effects such as disturbance of sexual development and reproductive function in animals and humans (Cheek et al., 1998). To address the structural basis of inhibition, the crystal structure of human SULT1E1 was determined with the above environmental pollutant and the cofactor product PAP (Shevtsov et al., 2003). The structure reveals that this compound binds in the active site of SULT1E1 in a very similar way to estradiol (E2) but with a 30° twist between the phenyl rings. This suggests that certain OH-PCBs can act as competitive inhibitors by mimicking E2 binding to SULT1E1 (Shevtsov et al., 2003).
BIOACTIVATION
The early work of the Millers (Millers, 1970, 1978) on the model carcinogen 2-AAF plus other aromatic amines led to the hypothesis that most chemicals require metabolism before being mutagenic or carcinogenic. The first step in the activation of AAF is CYP1A2-mediated N-hydroxylation to N-hydroxy-2-acetylaminofluorene, which is then a substrate for sulfonation, N,O-acyltransfer or deacetylation (McManus et al., 1984; Thorgeirsson et al., 1983). The influential studies by King and Phillips (1968) and the Millers (DeBaun et al., 1968, 1970) were the first to demonstrate the importance of sulfonation as one of the pathways involved in the activation of 2-AAF. These studies followed the seminal finding of Cramer et al. (1960), which showed that AAF undergoes N-hydroxylation to N-hydroxy-AAF. This metabolite was found to be more carcinogenic than the parent amide, was often active locally, and was active in the guinea pig that had been shown to be resistant to AAF-induced carcinogenicity (Miller, 1970; Miller, 1978). Sulfonation has now been shown to be important in the activation of a range of compounds such as aminoazo dyes, benzidines, heterocyclic amines, hydroxymethyl polycyclic aromatic hydrocarbons, terpenes, β-aminoethyl alcohols, and 2-nitropropane (Michejda et al., 1994).
Recently, this area has been the subject of a range of reviews (Banoglu, 2000; Glatt, 1997, 2000, 2005; Glatt et al., 1998, 2001; Kauffman, 2004; Surh, 1998), and much of the focus has been on determining the specificity of individual sulfotransferase isoforms for the above substrates using model in vitro systems. These studies have shown that certain sulfotransferases are very selective in their activation of promutagens (Glatt, 1997, 2000, 2005), and since they also display tissue-specific expression, it is highly likely that these factors play a role in the organ-selective toxicity of their substrates (Glatt, 2001; Thorgeirsson et al., 1983).
Polymorphisms have been shown in a number of sulfotransferases, and this area has been expertly reviewed by a number of investigators (Glatt, 2000; Glatt and Meinl, 2004; Meinl et al., 2002; Weinshilboum and Adjei, 2005). It was shown that the human SULT1A*Arg (*1) alloenzyme expressed in Salmonella typhimurium cells was 12–350 times more active in metabolizing 2-nitropropane, 2,4-dinitrobenzylalcohol, 1-HMP, (−)-1-(α-hydroxyethyl) pyrene, and 2-acetylamino-4-hydroxylaminotoluene to mutagens compared to cells expressing SULT1A1*His (Glatt, 2000). Enantioselectivity has also been demonstrated, with human SULT1E1 and SULT2A1 exhibiting 160-fold preference for the (−)-enantiomer and a 15-fold preference for the (+) enantiomer of 1-(α-hydroxyethyl)pyrene, respectively. Our understanding of such differences in the ability of different sulfotransferases to metabolize chemicals has recently been enhanced with the resolution of a number of the crystal structures of these enzymes (Table 3).
REGULATION OF SULFOTRANSFERASES
Many xenobiotics are effective inducers of transcription and exert their effects via activation of nuclear receptors. These include receptors that have endogenous hormone ligands and orphan nuclear receptors, for which no endogenous ligand have been identified. The role of xenobiotics, and their corresponding nuclear receptors, in the regulation of the Phase I CYP gene family members has been extensively explored, and investigators have now focused on the role of these in the transcriptional regulation of cytosolic SULTs (Runge-Morris, 1998). Sex-specific regulation of rodent isoforms has further broadened our understanding of the transcriptional regulation of cytosolic SULTs. However, it appears that significant interspecies differences in the regulation of SULTs exist. Although there have been some advances in our knowledge of regulation of the human SULT families in recent years, it remains one of the least-explored areas of research in the field.
Xenobiotics such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and the model carcinogen 2-AAF are able to regulate transcription via interaction with the nuclear arylhydrocarbon receptor (AhR). Metabolites of these compounds are known substrates for several SULTs, in particular the SULT1 family. From studies in rodents, it is believed that AhR agonists have negative effects on sulfotransferase regulation. β-Napthoflavone, TCDD, 2-AAF, and 3- methylcholanthrene (3-MC) have all been shown to markedly reduce phenol (SULT1A1) and hydroxysteroid (SULT2A) sulfotransferase activities and mRNA levels in rat livers (Runge-Morris, 1998). A recent study showed no effects on expression of SULT1A1, SULT2A1, or SULT1E1 in the human colon carcinoma cell line Caco-2 after exposure to a variety of polycyclic aromatic hydrocarbons (Lampen et al., 2004). Similarly, we have seen no change in SULT1A1 and SULT1A3 mRNA levels after treatment of primary human hepatocytes with 3-MC (Hempel et al., 2004). The consequences of transcriptional regulation by these chemicals are not clear, as sulfonation can lead to both the detoxification and bioactivation of some of these xenobiotic compounds.
Orphan nuclear receptors represent a common mechanism by which xenobiotics elicit their transcriptional activation of metabolic enzymes and drug transporters. Phenobarbital and phenobarbital-like compounds, such as polychlorinated biphenyls, regulate transcription by activating the nuclear constitutive androstane receptor (CAR). A functional nuclear response element responsive to CAR ligands has recently been identified on the mouse SULT2A2 promoter (Saini et al., 2004). There appears to be some conflicting evidence on whether CAR ligands influence the levels of human SULTs. One study describes 11-fold induction of SULT1A1 mRNA after treatment of primary human hepatocytes with the human CAR ligand 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl) oxime [Citco; (Maglich et al., 2003)]. On the contrary, we were unable to reproduce this effect on SULT1A1 or SULT1A3 message levels in human primary hepatocytes (Hempel et al., 2004).
Several SULT isoforms exhibit high affinity for the sulfonation of steroids, and these hormones are involved in transcriptional regulation of some SULTs. The estrogen sulfotransferase (SULT1E1) is one human sulfotransferase expressed under hormonal regulation. Progesterone was shown to control the cyclical expression of SULT1E1 in the menstrual cycle (Falany and Falany, 1996). Transcriptional regulation of SULT1E1 may have therapeutic consequences. For example, it is believed that the progestin-derived anti-breast cancer agents medrogestone and tibolone elicit their effects by inducing SULT1E1 in estrogen-responsive breast cancers, which normally express low levels of this sulfotransferase (Chetrite and Pasqualini, 2001). Sulfonation of estrogens inhibits their action at the estrogen receptor, and SULT1E1 has been implicated as an important enzyme in estrogen homeostasis that may be disrupted in breast cancer. For example, reexpression of recombinant SULT1E1 in the MCF-7 estrogen-responsive breast cancer cell line has been shown to inhibit cell growth (Falany et al., 2002).
Although several sulfotransferases show high affinity for estrogen substrates, studies investigating the role of estrogens in the gene regulation of sulfotransferases require further attention. Evidence suggests that the rat liver hydroxysteroid sulfotransferases (SULT2) may be inducible by estrogens and the anti-breast cancer drug tamoxifen (Hellriegel et al., 1996). It is currently unclear whether the estrogen sulfotransferase is regulated by its own ligand. Treatment of human endometrial Ishikawa cells with β-estradiol did not significantly change SULT1E1 activity (Falany and Falany, 1996), and some studies suggest a potential inhibitory effect of estrogens on SULT1E1 expression, such as the observation of significantly reduced SULT1E1 levels in estrogen-receptor-positive breast cancer biopsy samples and cell lines (Deng et al., 2003; Falany and Falany, 1996). SULT1A1, another sulfotransferase displaying high affinity for estrogens, appears to be positively regulated by tamoxifen, which was shown to significantly increase rat liver and intestinal SULT1A1 mRNA levels (Hellriegel et al., 1996). Interestingly, using serial analysis of gene expression, one study found that human SULT1A enzymes represented the only transcripts up-regulated following treatment of the human breast cancer cell line ZR75-1 with tamoxifen (Seth et al., 2000). Further studies are required to investigate the regulation of human cytosolic sulfotransferases by estrogens.
Glucocorticoids act on two nuclear receptors, the glucocorticoid (GR) and the orphan pregnane X receptor (PXR). Induction by glucocorticoids was confirmed for the human and murine SULT2A1 enzymes (Duanmu et al., 2002a,b), and it has been demonstrated that both PXR and GR directly act on the human SULT2A1 promoter (Duanmu et al., 2002a). GR was shown to regulate the rat SULT1A1 promoter at a glucocorticoid response element, which is also shared by other steroid receptors, such as AR and the retinoic acid receptor (RAR). RAR potentially competes with GR for binding at the response element, and retinoic acid was recently shown to positively affect the expression of human SULT1A1, SULT1E1, and SULT2A1 levels (Maiti et al., 2005). Interestingly, there appears to be interspecies variation of SULT1A1 induction in response to glucocorticoids. Contrary to its rat ortholog, human SULT1A1 enzyme levels are unaffected by treatment with these compounds. For example, human SULT1A1 mRNA levels did not change after treatment of primary human hepatocytes with dexamethasone (Duanmu et al., 2002b; Hempel et al., 2004).
Bile acids are known substrates of the hydroxysteroid sulfotransferases (SULT2) and have been shown to regulate their transcription via the nuclear receptor Farnesoid X Receptor (FXR) (Francis et al., 2003). Recently human SULT2A1 expression was shown to be regulated by the peroxisome proliferator-activated receptor (PPAR), another orphan nuclear receptor, involved in hepatic lipid metabolism (Fang et al., 2005). This effect appears to be species specific, as ligands of this receptor failed to induce SULT2A1 levels in rat primary hepatocytes (Fang et al., 2005).
Slowly studies are focusing on the molecular transcriptional mechanisms controlling the individual sulfotransferase promoters, to reveal a more complex picture of sulfotransferase regulation outside the nuclear receptor field. For example, adrenal-specific expression of human SULT2A1 was recently linked to the ability of another orphan nuclear receptor, steroidogenic factor 1 (SF1), to interact with the transcription factor GATA-6 (Saner et al., 2005). We recently identified the mechanisms regulating the ubiquitous expression of the human SULT1A1 enzyme, which does not appear to be regulated by nuclear receptor activators, unlike its rodent homolog. The human SULT1A1 promoter was highly activated by the synergistic interaction between the ubiquitous Ets factor GA binding protein (GABP) and Sp1 (Hempel et al., 2004). On the contrary, the highly homologous SULT1A3 promoter lacks an Ets factor binding repeat, preventing this synergistic interaction (Hempel et al., 2004). This may explain the differences in expression observed between these two human SULT1A members in the adult liver.
CONCLUSION
Until recently, information on SULTs at the molecular, functional, and structural levels has lagged behind other xenobiotic-metabolizing enzyme systems. However, the advances over the last decade have clearly shown this enzyme system to be comprised of a multi-gene family of proteins that differ markedly in their localization, regulation, and metabolic profiles. A recent book edited by Pacifici and Coughtrie has reviewed each of these fields in depth (Pacifici and Coughtrie, 2005). To date, at least 13 different human SULTs have been characterized. Many of the substrates metabolized by sulfotransferases are also substrates for UDP-glururonosyltransferases, and in this context, the former system has generally been considered a high-affinity, low-capacity pathway, whereas the latter system is considered a low-affinity, high-capacity pathway (Burchell and Coughtrie, 1997). This suggests that SULTs may be more important than UDP-glucuronosyltransferases in the metabolism of chemicals via low-level exposure from the environment or through food consumption.
Recent studies have provided new insights into the transcriptional regulation of both the human SULT1A1 (Hempel et al., 2004) and SULT2A1 (Fang et al., 2005) genes. However, this area remains one of the least explored in the human sulfotransferase field. While some of the marked interindividual variation in sulfotransferase activity in the human population (5- to 36-fold) can be explained by polymorphisms in the coding regions of SULT genes (Pacifici and Coughtrie, 2005; Pacifici and De'Santi, 1995), variations in regulatory regions have not been fully explored.
The physiological significance of SULT1C2, 1C4, 4A1_v1, and 4A1_v2 are poorly understood, and at the present time, they are orphan enzymes (Blanchard et al., 2004). We have shown that SULT4A1 is localized in discrete regions of the human brain such as cerebral cortex, cerebellum, pituitary, and brainstem (Liyou et al., 2003). In addition, SULT 1A1, 1A3, 2A1, and 1E1 are also expressed in the human brain (Liyou et al., 2003). Recently, Richard et al. (2001) have shown that both SULT1A1 and SULT1A3 are widely distributed within the developing human fetal brain. Further, the neurotransmitter systems such as GABA, cholinergic, glutaminergic and σ-opioid receptors are modulated by sulfonated neuro-steroids (Hempel et al., 2005; Kauffman, 2004). These data signal the beginning of a brain mosaic in relation to SULTs, and it should help unravel the tentative associations that have previously been made between SULTs and neurodegenerative disorders (Hempel et al., 2005).
To determine the physiological role and examine the consequences of inactivation of SULTs in vivo, the gene knock-out approach has already been applied (Qian et al., 2001, Tong et al., 2005). These investigators have generated mice deficient in Sult1e1 and have shown that these mice had spontaneous fetal loss caused by placental thrombosis. This study provides clear evidence as to the importance of estrogen sulfotransferase in reprouction, and further studies in this area should help evaluate the physiological relevance of other sulfotransferases.
Probably the most confronting difficulty facing toxicologists is the species differences that exist in SULTs, which complicate the extrapolation of animal data to humans. For example, humans have four members of the SULT1A subfamily, where rodents have a single member. In contrast, rats have four members of the SULT2A subfamily, while humans have a single gene. Further, equivalent forms of the mouse Sult3a1 or Sult5a1 have not been identified in humans (Blanchard et al., 2004). In addition, the expression of SULTs in humans has been shown to exhibit a pronounced extra hepatic pattern, whereas in rodents it appears to be predominantly hepatic (Eisenhofer et al., 1999, Hempel et al., 2005). Finally, marked differences in the way in which human and rodent SULT genes are regulated have also been reported; many of the rodent forms exhibit dramatic sexual dimorphisms (Coughtrie and Johnson, 2001). Such data highlight the importance of recent advances in the structural biology of SULTs. Indeed, in silico toxicology modeling of substrates utilizing the crystal structure of human SULTs may be a much more predictive tool in chemical risk assessment than data obtained in animal systems. The brief modeling of N-hydroxy aromatic and heterocyclic amines to human SULT1A subfamily members in this review supports this approach. This advance, together with a bank of model cell and bacterial systems expressing human SULTs, where their toxicity can be determined, may provide the most suitable approach in estimating the importance of sulfonation in chemical risk assessment.
References
Adjei, A. A., and Weinshilboum, R. M. (
Aksoy, I. A., Wood, T. C., and Weinshilboum, R. (
Barnett, A. C., Tsvetanov, S., Gamage, N., Martin, J. L., Duggleby, R. G., and McManus, M. E. (
Bernier, F., Lopez Solache, I., Labrie, F., and Luu-The, V. (
Bidwell, L. M., McManus, M. E., Gaedigk, A., Kakuta, Y., Negishi, M., Pedersen, L., and Martin, J. L. (
Blanchard, R. L., Freimuth, R. R., Buck, J., Weinshilboum, R. M., and Coughtrie, M. W. H. (
Brix, L. A., Barnett, A. C., Duggleby, R. G., Leggett, B., and McManus, M. E. (
Brix, L. A., Duggleby, R. G., Gaedigk, A., and McManus, M. E. (
Chatterjee, B., Majumdar, D., Ozbilen, O., Murty, C. V. R., and Roy, A. K. D. (
Comer, K. A., Falany, J. L., and Falany, C. N (
Coughtrie, M. W. H. (
Dajani, R., Cleasby, A., Neu, M., Wonacott, A. J., Jhoti, H., Hood, A. M., Modi, S., Hersey, Taskinen, A. J., Cooke, R. M., et al. (
Dooley, T. P. (
Dooley T. P. (
Dooley, T. P., Haldeman-Cahill, R., Joiner, J., and Wilborn, T. W. (
Duanmu, Z., Locke, D., Kocarek, T. A., and Runge-Morris, M. (
Duffel, M. W., and W. B. Jakoby (
Falany, C. N., Krasnykh, V., and Falany, J. L. (
Falany, J. L., and Falany, C. N. (
Falany, J. L., Macrina, N., and Falany, C. N. (
Falany, C. N., Xie, X., Wang, J., Ferrer, J., and Falany, J. L. (
Fang, H. L., Abdolalipour, M., Duanmu, Z., Smigelski, J. R., Weckle, A., Kocarek, T. A., and Runge-Morris, M. (
Forbes, K. J., Hagen, M., Glatt, H., Hume, R., and Coughtrie, M. W. H. (
Fuda, H., Lee, Y. C., Shimizu, C., Javitt, N. B., and Strott, C. A. (
Fujita, K., Nagata, K., Ozawa, S., Sasano, H., and Yamazoe, Y. (
Gamage, N. U., Duggleby, R. G., Barnett, A. C., Tresillian, M., Latham, C. F., Liyou, N. E., McManus, M. E., and Martin, J. L. (
Gamage, N. U., Tsvetanov, S., Duggleby, R. G., McManus, M. E., and Martin, J. L. (
Glatt, H. (
Hehonah, N., Zhu, X., Brix, L., Bolton-Grob, R., Barnett, A., Windmill, K., and McManus, M. (
Hellriegel, E. T., Matwyshyn, G. A., Fei, P., Dragnev, K. H., Nims, R. W., Lubet, R. A., and Kong, A.-N. T. (
Hempel, N., Barnett, A., Gamage, N., Duggleby, R. G., Windmill, K. F., Martin, J. M., and McManus, M. E. (
Hempel, N., Wang, H., LeCluyse, E. L., McManus, M. E., and Negishi, M. (
Her, C., Kaur, G. K., Athwal, R. S., and Weinshilboum, R. M. (
Hildebrandt, M. A., Salavaggione, O. E., Martin, Y. N., Flynn, H. C., Jalal, S., Wieben, E. D., and Weinshilboum, R. M. (
Jakoby, W. B., Sekura, R. D., Lyon, E. S., Marcus, C. J., and Wang, J.-L. (
Kakuta, Y., Pedersen, L. G., Carter, C. W., Negishi, M., and Pedersen, L. C. (
Kakuta, Y., Petrotchenko, E. V., Pedersen, L. C., and Negishi, M. (
Kester, M. H., Kaptein, E., Roest, T. J., van Dijk, C. H., Tibboel, D., Meinl, W., Glatt, H., Coughtrie, M. W., and Visser, T. J. (
Klaassen, C. D., and Boles, J. W. (
Kong, A.-N. T., Yang, L., Ma, M., Tao, D., and Bjornsson, T. D. (
Lee, K. A., Fuda, H., Lee, Y. C., Negishi, M., Strott, C. A., and Pedersen, L. C. (
Lin, Z., Lou, Y., and Squires, J. E. (
Maglich, J. M., Parks, D. J., Moore, L. B., Collins, J. L., Goodwin, B., Billin, A. N., Stoltz, C. A., Kliewer, S. A., Lambert, M. H., Willson, T. M., et al. (
Mammalian Gene Collection (MGC) Team. (
Markovich, D. (
McManus, M. E., Minchin, R. F., Schwartz, D., Wirth, P. J., Johnson, E. F., and Thorgeirsson, S. S. (
Meinl, W., Meerman, J. H., and Glatt, H. (
Meisheri, K. D., Johnson, G. A., and Puddington, L. (
Miller, J. A. (
Nagata, K., and Yamazoe, Y. (
Nash, A. R., Glenn, W. K., Moore, S.S., Kerr, J., Thompson, A. R., and Thompson, E. O. (
Negishi, M., Pedersen, L. G., Petrotchenko, E., Shevtsov, S., Gorokhov, A., Kakuta, Y., and Pedersen, L. C. (
Otterness, D. M., Wieben, E. D., Wood, T. C., Watson, W. G., Madden, B. J., McCormick, D. J., and Weinshilboum, R. M. (
Ozawa, S., Nagata, K., Shimada, M., Ueda, M., Tsuzuki, T., Yamazoe, Y., and Kato, R. (
Pacifici, G. M., and Coughtrie, M. W. H. (Eds.) (
Pedersen, L. C., Petrotchenko, E., Shevtsov, S., and Negishi, M. (
Petrotchenko, E. V., Pedersen, L. C., Borchers, C. H.,Tomer, K. B., Negishi, M., and Pedersen L. C. (
Radominska, A., Comer, K. A., Zimniak, P., Falany, J., Iscan, M., and Falany, C. N. (
Raftogianis, R. B., Wood, T. C., and Weinshilboum, R. M. (
Reiter, C., Mwaluko, G., Dunnette, J., Van Loon, J., and Weinshilboum, R. (
Rikke, B. A., and Roy, A. K. (
Runge-Morris, M. (
Sakakibara, Y., Takami, Y., Zwieb, C., Naakayama, T., Suiko, M., Nakajima, H., and Liu, M.-C. (
Sakakibara, Y., Yanagisawa, K., Takami, Y., Nakayama, T., Suiko, M., and Liu, M. C. (
Sakakibara, Y., Yanagisawa, K., Katafuchi, J., Ringer, D. P., Takami, Y., Nakayama, T., Suiko, M., and Liu, M.-C. (
Saner, K. J., Suzuki, T., Sasano, H., Pizzey, J., Ho, C., Strauss, J. F., 3rd, Carr, B. R., and Rainey, W. E. (
Schwartz, N. B. (
Shevtsov, S., Petrotchenko, E. V., Pedersen, L. C., and Negishi, M. (
Tashiro, A., Sasano, H., Nishikawa, T., Yabuki, N., Muramatsu, Y., Coughtrie, M. W., Nagura, H., and Hongo, M. (
Tsoi, C., Falany, C. N., Morgenstern, R. and Swedmark, S. (
Vargas, F., Frerot, O., Brion, F., Trung Tuong, M.D., Lafitte, A., and Gulat-Marnay, C. (
Veronese, M. E., Burgess, W., Zhu, X., and McManus, M. E. (
Wang, J., Falany, J. L., and Falany, C. N. (
Weinshilboum, R. M., Otterness, D. M., Aksoy, I. A., Wood, T. C., Her, C., and Raftogianis, R. B. (
Wilborn, T. W., Comer, K. A., Dooley, T. P., Reardon, I. M., Heinrikson, R. L., and Falany, C. N. (
Windmill, K. F., Christiansen, A., Teusner, J. T., Bhasker, C. R., Birkett, D. J., Zhu, X., and McManus, M. E. (
Yoshinari, K., Nagata, K., Ogino, M., Fujita, K., Shiraga, T., Iwasaki, K., Hata, T., Yamazoe, Y. (
Yoshinari, K., Nagata, K., Shimada, M., and Yamazoe, Y. (
Yoshinari, K., Petrotchenko, E. V., Pedersen, L. C., and Negishi, M. (2001). Crystal structure-based studies of cytosolic sulfotransferase.
Zhu, X., Veronese, M. E., Bernard, C. C., Sansom, L. N., and McManus, M. E. (
Author notes
*School of Biomedical Sciences, ‡School of Molecular and Microbial Sciences, and ¶Institute for Molecular Bioscience, University of Queensland, Queensland 4072, Australia; †Department of Medicine/Oncology, Duke University Medical Center, Durham North Carolina 27710; and §School of Exercise and Health Sciences, Deakin University, Australia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments