
Abstract
Breast cancer resistance protein (ABCG2), the newest ABC transporter, was discovered independently by three groups in the late 1990s. ABCG2 is widely distributed in the body with expression in the brain, intestine, and liver, among others. ABCG2 plays an important role by effluxing drugs at the blood–brain, blood–testis, and maternal–fetal barriers and in the efflux of xenobiotics at the small intestine and kidney proximal tubule brush border and liver canalicular membranes. ABCG2 transports a wide variety of substrates including HMG-CoA reductase inhibitors, antibiotics, and many anticancer agents and is one contributor to multidrug resistance in cancer cells. Quantitative structure–activity relationship (QSAR) models and structure–activity relationships (SARs) are often employed to predict ABCG2 substrates and inhibitors prior to in vitro and in vivo studies. QSAR models correlate in vivo biological activity to physicochemical properties of compounds while SARs attempt to explain chemical moieties or structural features that contribute to or are detrimental to the biological activity. Most ABCG2 datasets available for in silico modeling are comprised of congeneric series of compounds; the results from one series usually cannot be applied to another series of compounds. This review will focus on in silico models in the literature used for the prediction of ABCG2 substrates and inhibitors.
Similar content being viewed by others
INTRODUCTION
Breast cancer resistance protein (BCRP/ABCG2) is the newest member of the ATP binding cassette (ABC) superfamily of transporters. It was named BCRP because it was discovered in a breast cancer cell line co-selected for doxorubicin in the presence of verapamil, a P-glycoprotein inhibitor (1). This breast cancer cell line transported doxorubicin and rhodamine 123 in the absence of P-glycoprotein (ABCB1) and multidrug resistance protein 1 (ABCC1). ABCG2 was also cloned and named MXR for mitoxantrone resistance and named ABCP for ABC transporter expressed in the placenta (2,3). Following its discovery, ABCG2 was fully cloned and characterized and added as the second member of the G subfamily of ABC transporters. Other members of this family include two proteins involved in cholesterol transport, the human homolog of the Drosophila white protein, and two sterol transporters, among others (4).
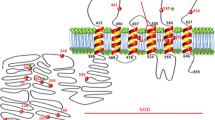
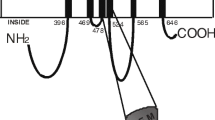
ABCG2 shares limited amino acid sequence similarity with ABCB1 and ABCC1 but has 29.3% identity with the human homolog of Drosophila white protein ABCG1. The difference between ABCG2 and other ABC efflux transporters is evident by examining the structure of the protein. ABCG2 is composed of only 655 amino acids that make up its single nucleotide binding domain and six transmembrane domains, while many other ABC transporters are comprised of two nucleotide binding and 12 transmembrane domains. It is known that eukaryotic ABC transporters require at least two nucleotide binding and 12 transmembrane domains for transporter activity; therefore, ABCG2 is considered a half-transporter and functions by forming homodimers (4).
Molecular mechanisms controlling ABCG2 expression have not been fully identified, but recent studies suggest regulation through sex hormones, hypoxia, peroxisome proliferator activated receptor γ (PPARγ), or the aryl-hydrocarbon (Ahr) receptor. The sex hormones estradiol, progesterone, and testosterone affect ABCG2 regulation at the transcriptional level, but the data are controversial, particularly concerning cell-type-dependent estradiol effects (5). Additionally, histone modification was shown to be important in regulating ABCG2 in resistant cells. ABCG2 induction has also been noted following administration of histone deacetylase inhibitor romidepsin, though the effects are cell type specific (6). Regulation of ABCG2 has been described in normal cells to date, with little known regarding changes in regulation in cancer cells.
Tissue Distribution and Cellular Localization
ABCG2 tissue distribution is ubiquitous, with high mRNA expression in the placenta and lower levels in the brain, prostate, intestine, testis, ovary, and liver. ABCG2 expression in cancer cells has been linked to the multidrug resistance phenomenon, but the data are controversial. ABCG2 expression in breast cancer has been extensively studied, and the consensus is that expression levels are relatively low (5). In polarized cells, ABCG2 is located on the apical membrane and can limit the entry of xenobiotics across the small intestine or the blood–placental barrier (4).
Substrates and Inhibitors
ABCG2 transports a variety of substrates, some shared with ABCB1 and ABCC1 but some are ABCG2 specific. ABCG2 has been shown to transport chemotherapy agents, anti-virals, HMG-CoA reductase inhibitors, carcinogens, fluorescent compounds like rhodamine 123 and Hoechst 33342, flavonoids, phase II metabolites including sulfate and glucuronide conjugates, antibiotics, and many others (4). ABCG2 substrates include mitoxantrone, topotecan, SN-38, and flavopiridol among others. ABCG2 transport, unlike those of ABCC1 and ABCC2, is not dependent on intracellular GSH concentration. ABCG2 inhibitors include some ABCB1 inhibitors such as elacridar (GF120918), cyclosporine A, and tariquidar (XR9576). Other inhibitors include estrone and 17β-estradiol and flavonoids quercetin, biochanin A, and genistein. Since many flavonoids are found in the diet, these compounds can significantly affect the pharmacokinetics and pharmacodynamics of ABCG2 substrates and also lead to diet–drug interactions (7).
Function
ABCG2 plays an important role in drug absorption, distribution, and elimination and in drug efficacy. Based on the tissue distribution of ABCG2, its presumed function includes protecting the body from xenobiotics through its role at the blood–brain, blood–testis, maternal–fetal barriers and in efflux of xenobiotics at the small intestine brush border and liver canalicular membranes. ABCG2 can play a role in multidrug resistance, and many inhibitors have been synthesized to overcome this effect. There is also considerable interest in identifying ABCG2 substrates because ABCG2 substrates can have poor oral bioavailability, extensive first pass elimination via the biliary route, and poor efficacy as cancer therapeutic agents. Supporting evidence has been reported by Jonker et al. (8) in studies performed in Abcb1a/1b (−/−) knockout mice, where the authors found that oral bioavailability of topotecan increased sixfold in knockout mice compared to wild-type mice. Abcb1a/1b (−/−) mice were administered topotecan and the ABCG2 inhibitor GF120918 orally: Topotecan is an ABCG2 and ABCB1 substrate, and GF120918 inhibits both ABCG2 and ABCB1. In these mice, exposure (AUC) of topotecan increased sixfold due to the increased absorption from the intestine and reduced elimination into bile. The role of ABCG2 at the placental barrier was studied in pregnant Abcb1a/1b (−/−) mice. The mice were administered oral GF120918 2 h prior to intravenous topotecan administration. Fetal distribution and maternal plasma distribution were measured, and topotecan levels were threefold higher in the fetus when the parent was treated with GF120918 (8). Overexpression of ABCG2 is one of the mechanisms behind multidrug resistance (MDR), a phenomenon where cancer cells display cross-resistance to structurally unrelated drugs. One approach to circumvent ABCG2-mediated efflux is through inhibition of ABCG2. The role of ABCG2 clinically is controversial where many groups have reported that ABCG2 is predictive of response to chemotherapy, whereas others have reported the opposite (4).
The focus of this article is to review the literature for in silico models to predict ABCG2 substrates and inhibitors and to determine if any structural features are necessary for transport or inhibition of ABCG2.
SAR AND QSAR OF ABCG2 INHIBITORS
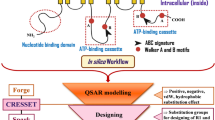
In silico modeling is a powerful tool often employed to predict drug properties prior to in vitro and in vivo studies (9,10). Many different in silico modeling techniques have been employed to discover ABCG2 substrates and inhibitors, including quantitative structure–activity relationships (QSAR), 3D-molecular models, pharmacophore models, and structure based approaches. The most common approaches are development of QSAR models and structure-activity relationships (SARs). The goal of QSAR modeling is to construct a mathematical relationship between descriptors and pharmacological activities of compounds. The model can then be used to predict the activity for an untested compound. The goal of SARs is usually to discern the structural features or side groups that directly lead to the desired activity under investigation. In order to use these in silico modeling techniques, compounds need to be screened to find the degree of substrate binding or inhibition. The two most commonly used assays include whole-cell-based assays screened using flow cytometry or cytotoxicity measurements and using membrane-based assays with inverted vesicles of plasma membranes for high throughput screening (11). So far, there are many inhibitor datasets available for ABCG2 modeling, while very few substrate datasets exist in the literature. Studies examining SARs and QSARs for ABCG2 are presented below, with the SAR studies summarized in Table I and the QSAR studies summarized in Table II.
Taxanes
MDR modulators were designed based on the structure of paclitaxel and tested for their ability to inhibit ABCB1, ABCC1, and ABCG2. While paclitaxel and docetaxel are ABCB1 substrates, they are not ABCG2 substrates. On the other hand, ortataxel is an ABCB1, ABCC1, and ABCG2 inhibitor (12). Eighteen taxane analogues with variations at one of six positions on the baccatin backbone of paclitaxel were generated (Fig. 1a). The ability of these taxane analogues to inhibit ABCG2 was tested in an ABCG2 overexpressing whole-cell-based system with mitoxantrone as the test drug. Multiple linear regression (MLR) was used to establish which side chain was contributing to or subtracting from the ABCG2 inhibition. It was found that an O-methyl group at position 1 provided a positive contribution, while a cyclic carbonate at position 4 was detrimental to the activity of the taxane analogue toward ABCG2 inhibition. Removal of the cyclic carbonate at position 4 resulted in a significant improvement of ABCG2 inhibition. In addition, a tertiary amine in the substituent at position 2 resulted in increased ABCG2 inhibition, while a methyl group at the same position was a strong detriment. Based on the SAR, the authors have proposed the structure for a broad spectrum MDR modulator (Fig. 1b), which remains to be synthesized and tested (13).

Propafenone Analogues
Propafenone and its analogues have been identified as ABCB1 substrates and inhibitors. Twenty-three compounds modified from propafenone in the phenone substructure and in the vicinity of the nitrogen atom were synthesized and their potencies evaluated in mitoxantrone accumulation studies in ABCG2 overexpressing cell lines (Fig. 2). The potencies of the propafenone analogues were calculated, and the EC50 values ranged from ~1 to 600 μM with six compounds having EC50 values less than 5 μM. SARs showed that the charge of the nitrogen atom, a previously accepted descriptor for ABCB1 inhibition (14), does not contribute to the interaction of these compounds with ABCG2, because the activities of the aniline and amide propafenone analogues are similar to that of tertiary amines. Another commonly used in silico approach is the development of a pharmacophore model: A pharmacophore was first defined by Paul Ehrlich in the early 1900s as a “molecular framework that carries the essential features responsible for a drug’s biological activity” (15). Pharmacophore models were generated for ABCB1 showing that the nitrogen atom and carbonyl group act as H-bond acceptors, but when the pharmacophore was applied to ABCG2, it further showed that the nitrogen atom does not contribute to the activity. Hydrogen bond acceptor strength in the vicinity of the nitrogen atom, a previous determinant for ABCB1 activity, does not influence ABCG2 activity. A QSAR model was generated for ABCG2 interaction with propafenone analogues and was described by four descriptors, namely, number of H-bond acceptors, molar refractivity, number of rotatable bonds, and total hydrophobic van der Waals surface area. This model performed satisfactorily with a cross-validated correlation coefficient of 0.780 (16).

Chemical structure of propafenone analogues with two substituent groups R1 and R2 (16)
Flavonoids
Flavonoids, a large class of polyphenolic compounds, are present in the diet in vegetables and fruits and in herbal medicines (Fig. 3). Many studies have suggested that flavonoids have beneficial effects in coronary heart disease, bone loss, prevention of cancer, and others (17,18). Flavonoids are known ABCB1 and ABCG2 modulators with low toxicity and, due to their abundance in the diet, may alter the pharmacokinetics of ABCB1 and ABCG2 substrates (7,19). Our laboratory has derived SARs and QSAR models to predict flavonoid–ABCG2 interactions. The structural features of flavonoids significant to ABCG2 inhibition were elucidated by comparing the EC50 values (concentration that increases the accumulation of mitoxantrone by 50%) of flavonoids with and without a structural element. A genetic algorithm coupled to MLR was used to derive a QSAR model that could predict the ABCG2 inhibition of untested flavonoids. The EC50 values of 25 flavonoids were calculated and ranged from 0.07 ± 0.02 μM for 7,8-benzoflavone to 183 ± 22 μM for silybin. The SAR analysis showed that a 2,3-double bond in ring C led to an increase in ABCG2 inhibition activity. The mean EC50 value of the flavones and flavonols was 1.15 ± 0.38 μM, significantly lower than the mean EC50 value of the flavanones (15.3 ± 2.9 μM). Another structural feature that led to an increase in ABCG2 inhibition activity was the substitution of a methyl group or addition of a benzene ring at position 7 or 8 of the A ring. A QSAR model was also derived for ABCG2 inhibition by the 25 flavonoids. The model consisted of three descriptors; log P and two other 3D structural descriptors Dy and SdssC_acnt. Lipophilicity, calculated as either log P or log D, the octanol–water partition coefficient, is commonly accepted as a descriptor for membrane partitioning. The model predicted the EC50 values of the training set well (r 2 = 0.852) and also produced a satisfactory cross-validation correlation coefficient of 0.784. The model was used to predict the EC50 values for six compounds in a test set and resulted in a correlation coefficient of 0.922 (19). The SAR and QSAR model complement each other in that an increase in log P increases ABCG2 inhibition, and increasing hydrophobicity at certain positions on the structure of the flavonoid also increases ABCG2 inhibition.

Representative structures of the six different classes of flavonoids (19)
The SARs for ABCG2 inhibition by flavonoids were also explored in a second study (20), with mitoxantrone accumulation studies performed using four different flavonoids from four different classes, each compound having three hydroxyl groups. The flavone apigenin was most efficient with an EC50 of 16 μM, followed by the flavonol galangin (19 μM), and isoflavone genistein (24 μM), and the flavanone naringenin (37 μM) was the least efficient. Based on the EC50 values, a series of flavones based on the structure of apigenin (N = 12) were screened with different hydroxyl groups and hydrophobic substituents and mitoxantrone accumulation studies performed. GF120918 was used as a positive control in the experiments. SARs showed that hydroxyl groups at positions 3, 7, and 4′ had a negative impact on ABCG2 inhibition, while a hydroxyl position at position 5 produced a positive effect. In addition, C-isoprenylation at positions 6 or 8 had a positive effect on ABCG2 inhibition. 6-Prenylchrysin was identified as the best wild-type ABCG2 inhibitor with an EC50 of 0.29 ± 0.06 μM, while tectochrysin was identified as the best mutant ABCG2-T482 inhibitor with an EC50 of 1.9 ± 0.3 μM. Both compounds were further tested and found to be ABCG2 selective with little effect on ABCB1 and ABCC1. These compounds had no effects on ABCG2 ATPase activity, suggesting that the mechanism of inhibition is different from GF120918 and fumitremorgin C (FTC), which both strongly inhibited ABCG2 ATPase activity (20). The positive and negative contributions of different substituents on ABCG2 inhibition are shown in Fig. 4.

Summary of structure–activity relationships for apigenin analogues. Thin arrows show unfavorable substitutions, while thick arrows show favorable substitutions (20). Reproduced with permission with American Association for Cancer Research in the format journal via copyright clearance center. Copyright 2005 by American Association for Cancer Research
Based on the results from the previous study, GF120918 and tectochrysin were selected as lead compounds to potentially inhibit ABCG2, and two series of structural analogues were synthesized. Mitoxantrone accumulation studies were undertaken to determine the extent of inhibition of ABCG2. Nine GF120918 analogues with substitution patterns similar to tectochrysin at three positions were tested resulting in EC50 values ranging from 0.77 ± 0.07 to >20 μM (Fig. 5, series I). SARs showed that a single methoxy group at R1 or R2 improved the inhibition, while hydroxylation at any position was detrimental to the activity. The best inhibitor had methoxy and trifluoromethyl substitution and resulted in an EC50 of 0.77 ± 0.07 μM, though all analogues had EC50 values less than that of GF120918. Four GF120918 analogues with substituents at two positions were tested, resulting in EC50 values ranging from 0.35 ± 0.16 to 1.38 ± 0.22 μM (Fig. 5, series II). SAR studies revealed that single methoxy substitutions on the phenethyl group led to better inhibitors than two methoxy groups (0.35 ± 0.16 vs 0.45 ± 0.07 μM). The best GF120918 derivative was more potent than GF120918 (0.35 ± 0.16 vs 0.41 ± 0.21 μM) (21).

Structural analogues of tectochrysin with substitutions at positions R1, R2, and R3 and of GF120918 with substitutions at position R and Y (21)
Estrone and 17-β-estradiol were recently shown to be ABCG2 inhibitors in ABCG2-overexpressing K562 cells. The structure of flavonoids resembles estrogen, and flavonoids have been shown to have estrogenic activities (22). The inhibitory potencies of 19 flavonoids from different classes and eight glycosylated flavonoids were investigated in ABCG2-overexpressing K562 cells. Intracellular accumulation of SN-38 and mitoxantrone was measured with or without the flavonoids and drug reversal indexes (ratios of EC50 measurements in the absence of reversing agents divided by levels in presence of reversing agents) calculated. The isoflavone genistein, flavanones naringenin and hesperetin, flavonols galangin, kaempferide, and kaempferol and all flavones tested displayed potent reversing effects. Only naringenin-7-glucoside and luteolin-4′-O-glucoside displayed moderate reversal activity, while all other glycosylated flavonoids were poor ABCG2 inhibitors. A SAR suggested the importance of the 3-hydroxyl group of the C ring for ABCG2 inhibitory activity. The 3-hydroxyl group is present on the structures of not only potent ABCG2 inhibitors genistein, naringenin, and kaempferol but also on weak ABCG2 inhibitors daidzein and quercetin (23). A more detailed SAR was proposed in another study by the same authors (24). Potent ABCG2 inhibition was observed in flavonoids with a double bond between position 2 and 3 on the C ring and 4′-O-methoxylation or 4′-hydroxylation of the B ring. Flavonoids with 3-hydroxylation of the C ring or 3′-hydroxylation of the B ring were poor ABCG2 inhibitors. These detrimental effects of 3-hydroxylation seen in this study seems to contradict earlier work by the same group (24). A consensus on the SARs of flavonoids is clearly lacking with different groups presenting contradicting results on which substituents have positive and negative effects on ABCG2 inhibition.
The plant Boerhaavioa diffusa is often used as herbal remedy to improve liver and kidney function. Eleven rotenoids, isoflavonoid derivatives found in plants as secondary metabolites, were isolated from the B. diffusa root extract. These 11 compounds, differing in structure at seven positions (Fig. 6), were assayed for their ABCG2 inhibition activity in HEK293 ABCG2 overexpressing cells. ABCG2 was almost completely inhibited by compound 1 (percent maximal mitoxantrone accumulation = 92 ± 7%) and compound 2 moderately inhibited ABCG2 (68 ± 6%). The remaining compounds were weak inhibitors of ABCG2, with GF120918 used as a reference. Concentration-dependent studies were performed for the two most potent inhibitors, and EC50 values were determined. The EC50 for compound 1 was 0.7 ± 0.1 μM and for compound 2 was 2.5 ± 0.5 μM. A SAR showed that a five-membered B ring was detrimental to ABCG2 inhibition compared to a six-membered ring (compound 11 vs compound 6). A planar molecular structure was deemed necessary for flavonoid-mediated ABCG2 inhibition, and the same effect is seen for these rotenoid derivatives. A loss of the planar structure in compounds 8 and 9 leads to decreased potency. The same effect is seen for compound 10 where a tetra-substitution in ring D leads to loss of the planar structure and reduced activity of the compound. Comparison of the structures of tectochrysin and compound 1 shows that both compounds have similar C and D rings but differ in rings A and B. The EC50 of compound 1 was 0.7 μM as compared to tectochrysin with an EC50 of 3.0 μM. Further structure adjustments are being studied to improve the potency of the rotenoid derivatives (25).

Rotenoid derivatives isolated from the plant Boerhaavia diffusa with substitutions at positions 3, 4, 6, 8, 9, and 10 and the presence or absence of a double bond between positions 6a and 12a (25)
Tamoxifen Analogues
Another set of estrogen-like compounds were tested for their ability to inhibit ABCG2. It was also discovered that tamoxifen and toremifene, a structural analogue of tamoxifen, slightly increased topotecan accumulation in ABCG2-overexpressing K562 cells. Based on this, 14 structural analogues of tamoxifen were synthesized and their ABCG2 inhibition potential tested. The tamoxifen analogue 11 (TAG-11) was identified as the lead structure, and 25 structural analogues of TAG-11 were synthesized. Structures of the 12 representative compounds are shown in Fig. 7. Topotecan accumulation was measured in the presence or absence of the tamoxifen analogues in K562 ABCG2 overexpressing cells. Tag-5, Tag-8, Tag-11, Tag-72, Tag-126, and Tag-139 were identified as the most potent ABCG2 inhibitors. Cytotoxicity studies using SN-38 and mitoxantrone in K562/ABCG2 cells demonstrated Tag-139 at a concentration of 3 μM as the most potent compound. The six lead compounds were also tested for their interactions with the estrogen receptor. Tag-139 showed moderate estrogen receptor alpha inhibition and no estrogen receptor beta inhibition. Based on these results, Tag-139 was selected as the lead compound for further studies on ABCG2 inhibition (26).

Structure of tamoxifen analogues designed to inhibit ABCG2 (26)
Tariquidar Analogues
SARs were utilized to design the third-generation ABCB1 inhibitor tariquidar (XR9576) (27). Recently, it was discovered that tariquidar also inhibits ABCG2. Based on this observation, six anthranilamide derivatives (Fig. 8, template 1) and 24 tetrahydroisoquinoline-ethyl-phenyl-amide derivatives were synthesized and tested for their interaction with ABCG2 (Fig. 8, template 2). The breast cancer cell line MCF/MX100 cells overexpressing ABCG2 were cultured, and different concentrations of test compounds were incubated with 1 μM pheophorbide A. Accumulation of pheophorbide A was determined at 5-min intervals for up to 60 min. pIC50 values were calculated and ranged from 3.58 ± 0.17 to 6.00 ± 0.23. A Free–Wilson analysis was performed on 15 compounds to determine the structural elements that influence ABCG2 function. Methoxy groups at positions 6 and 7 of the structurally related analogues had a negative impact on ABCG2 inhibition. A para nitro group or a 3,4-dimethoxyphenyl at substituent position R2 and an amide linker had positive effects on ABCG2 inhibition. Log P and log D values showed no correlation with pIC50 values or ABCG2 inhibition. 3D-QSARs were also calculated using comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA). CoMFA aims to derive a correlation between biological activity and their 3D shape using steric (s), electrostatic (e), and hydrogen bonding (h-bnd) fields. CoMSIA has similar aims as CoMFA except the 3D-QSAR models are generated using steric (s), electrostatic (e), hydrophobic (h), hydrogen bond donor (d), and hydrogen bond acceptor (a) fields. The 3D-QSAR models were generated using partial least squares regression and evaluated using the leave-one-out cross-validation method. The best CoMFA models were generated using the s, e, and h-bnd fields and s and hbnd fields with cross-validated correlation coefficients of 0.757 and 0.760. The best CoMSIA models were generated with the s, e, and d fields, e and d fields, and the s, e, h, and d fields with cross-validated correlation coefficients of 0.803, 0.801, and 0.785, respectively. The best CoMSIA models were further subjected to leave-many-out cross-validation where the dataset was split into groups of the same size (number of groups = 5, 4, 3, 2). For all models, a significant drop in the q 2 was seen from the leave-one-out method to the leave-many-out method with the dataset split into five groups. As more compounds were left out of the dataset (number of groups = 4, 3, 2), the q 2 decreased slightly for all models. For example, the q 2 generated using the leave-many-out method with five groups for the e and d CoMSIA model dropped to 0.702 from 0.801 to the leave-one-out method. The q 2 further dropped to 0.681, 0.598, and 0.507 for the leave-many-out method with 4, 3, and 2 groups, respectively. Contour plots were generated by the authors to help visualize the contribution of the different fields to the biological activity of compounds toward ABCG2 inhibition (27).

Template 1 derived from anthranilamide and template two based on the tetrahydroisoquinoline-ethyl-phenyl-amide backbone (27)
Fumitremorgin Analogues
FTC is a potent and specific ABCG2 inhibitor but exhibits in vivo toxicities (28). A group of 42 FTC analogues synthesized as diastereoisomers were screened for their ABCG2 inhibitory activity (Fig. 9). The analogues differed from FTC at two substituent positions, R1 and R2 with six possible substituents at R1 (A-F) and seven possible substituents at R2 (1–7). Mitoxantrone accumulation assays were performed in a T6400 mouse cell line overexpressing mouse ABCG2 but lacking both ABCB1 and ABCC1 activity and in a human T8 cell line overexpressing human ABCG2 and lacking human ABCB1 and ABCC1. Fluorescence of mitoxantrone was measured in the cells using flow cytometry; the higher the mitoxantrone fluorescence inside the cells, the greater the inhibition of ABCG2. GF120918 was used as a positive control and gave 438 ± 17 and 722 ± 27 fluorescence units in the mouse and human cell lines, respectively. SARs were performed on the effect of substituents on human and mouse ABCG2 inhibition, and it was found that lipophilic substituents increase the inhibition of ABCG2. Substituents C, D, and E at the R1 position and 4, 5, and 6 at the R2 position were most beneficial with efficacy approaching that of GF120918. The two most potent analogues, Ko132 and Ko134, both with an isobutyl substitution at the R1 position, were chosen for further in vitro and in vivo studies (29).

Ko134, Ko132, and Ko143 (a newly synthesized methoxy analogue of Ko134) were selected for detailed in vitro and in vivo studies. Mitoxantrone accumulation studies showed that the three compounds were more potent than FTC and that Ko143 was twice as potent as GF120918. All three compounds had low activity against ABCB1 and ABCC1, with Ko143 being the most specific ABCG2 inhibitor. Ko143 was selected for in vivo studies in Abcb1a/1b (−/−) knockout mice. Topotecan plasma concentrations increased four- to sixfold at 30 and 60 min post-administration of the drug, and this was speculated to be the direct result of ABCG2 inhibition in the small intestine (30).
Cyclin-Dependant Kinases
Cyclin-dependant kinases (CDKs) regulate cell cycle progression and gene transcription necessary for survival of cancer cells (31), and CDK inhibitors are being pursued as potential drug targets in drug discovery and development. Based on a previous QSAR model by Saito et al. (32), compounds with an amine bonded to a heterocyclic carbon, a fused heterocyclic ring(s), and two substituents on a carboxylic ring of the fused heterocyclic ring(s) are chemical moieties important for inhibition of ABCG2. This QSAR model predicted five CDK inhibitors, purvalanol A, WHI-P180, bohemine, roscovitine, and olomoucine to be ABCG2 inhibitors. Hematoporphyrin transport was measured in ATP-dependent plasma membrane vesicles isolated from ABCG2 overexpressing cells. Most CDK inhibitors (except olomoucine) inhibited hematoporphyrin transport by ABCG2, and purvalanol A was the strongest ABCG2 inhibitor with an IC50 of 3 μM. Photosensitivity of pheophorbide A was measured in ABCG2-expressing Flp-In-293 cells with or without the CDK inhibitors. Only purvalanol A increased the photosensitivity of the ABCG2-expressing Flp-In-293 cell line, suggesting that purvalanol A plays a significant role in the inhibition of ABCG2. SARs showed that only two CDK inhibitors, purvalanol A and WHI-P180, have a planar structure and are also the two strongest ABCG2 inhibitors. WHI-P180 did not enhance the photosensitivity of ABCG2-expressing Flp-In-293 cells. Energy of the highest occupied molecular orbital (HOMO) profile was calculated using molecular orbital calculations. The HOMO profile of WHI-P180 was similar to that of gefitinib, an ABCG2 substrate. The molecular structure and HOMO profile suggest that WHI-P180 is also an ABCG2 substrate; on the other hand, purvalanol A, a potent inhibitor, exhibits a different HOMO profile and molecular structure (33).
Structurally Unrelated Compounds
A disadvantage of most SAR and QSAR models is the use of structurally related compounds in the dataset. In this case, the generated model does not apply to a different structural set of compounds. Matsson et al. (34) generated 2D and 3D models to predict ABCG2 inhibition using 123 structurally unrelated compounds. Mitoxantrone accumulation was measured in Saos-2 ABCG2 overexpressing cells incubated with different compounds. The increase in mitoxantrone accumulation was normalized to the accumulation measured using Ko143, a potent ABCG2 inhibitor. Molecular descriptors (N = 152) representing molecular size, flexibility, connectivity, polarity, charge, hydrogen bonding potential, and log P were calculated and a model generated for ABCG2 inhibition using orthogonal partial least squares regression. The dataset was split into training and test sets of 80 and 43 compounds, respectively. A final model was generated with two descriptors, log D pH=7.4 and molecular polarizability. The model correctly predicted 93% of ABCG2 inhibitors and 92% of non-interactors for the training set and 83% of ABCG2 inhibitors and 76% of non-interactors for the test set. The authors reasoned that lipophilicity represents the need for partitioning of the compound into the cell membrane as a first step before interacting with ABCG2. To model the binding of compounds to ABCG2, a lipophilicity-independent dataset (N = 22) was used with each inhibitor (N = 11) and non-inhibitor (N = 11) paired for lipophilicity. The final model was described by hydrogen bonding and nitrogen atoms and correctly classified 91% of inhibitors and 82% of non-interactors. A molecular polarizability-independent dataset (N = 10) was used to generate a model that correctly classified 100% of inhibitors and non-interactors. The descriptors in the final model represented lipophilicity, the surface area of non-polar atoms, and the surface area of nitrogen atoms. A pharmacophore model was generated using 28 inhibitors in the training set. The pharmacophore was described by two hydrophobic features and one hydrogen bond acceptor, confirming the QSAR results (34).
A second study utilizing a non-congeneric dataset was performed to determine a QSAR model to predict ABCG2 inhibition (32). In this study, 49 compounds were selected to represent nine different classes: neurotransmitters, calcium channel blockers, potassium channel modulators, steroids, non-steroidal anti-inflammatory drugs, anti-cancer, antibiotics, other drugs like tacrolimus and quinidine, and ABCG2 inhibitors. ATP-dependent inhibition of mitoxantrone transport by the 49 test compounds was determined in ATP-dependent plasma membrane vesicles. A QSAR model was generated using chemical fragmentation codes calculated using the program Markush Topfrag (Derwent Information Limited, London, UK). A chemical fragmentation code consists of alphanumeric symbols that represent a fragment of the chemical structure of the molecule of interest. A QSAR model describing the extent of ABCG2 inhibition was derived:
where i designated a specific chemical fragmentation code and the score represents the presence or absence of that code in the structure of the molecule of interest. C is a constant, and c is the chemical fragmentation code coefficients that are calculated by MLR. A QSAR model was derived, which could explain the ABCG2 inhibition using one set of chemical fragmentation codes. The final model consisted of eight descriptors representing 29 different chemical fragmentation codes and was able to predict the inhibition of ABCG2 reasonably well (r 2 = 0.920). The observed ABCG2 inhibition values ranged from 0% to 94.5%. The model predictions ranged from −11.3% to 93.8%. The model better predicted the higher values with significant deviations in the lower range. For example, the observed ABCG2 inhibition for bepridil, a calcium channel blocker, is 20.7, while the predicted value is 1.74%. The predictability of the method was tested on gefitinib, a compound not included in the original analysis. The percent of ABCG2 inhibition was calculated experimentally to be 95% and was predicted well by the model (percent ABCG2 inhibition=104%). The model predicts that an amine bonded to a carbon in a heterocyclic ring, a fused heterocyclic ring(s), and two substituents on a carboxylic ring of the fused heterocyclic ring(s) are important positive contributors to ABCG2 inhibition. These structural features are present on gefitinib, and thus, it is correctly predicted to fully inhibit ABCG2 (32). One disadvantage of this approach is that the model cannot predict inhibition of ABCG2 for steroids because chemical fragmentation codes cannot be calculated for steroids.
A pharmacophore model was derived by Chang et al. (35) using four ABCG2 inhibitors: GF120918, Ko143, nelfinavir, and nicardipine. The pharmacophore model contained three hydrogen bond acceptors and three hydrophobic features and was tested on 500 commercially available drugs. The search resulted in 37 ABCG2 ligands with six already known inhibitors: digoxin, nicardipine, indinavir, lopinavir, ritonavir, and saquinavir (35).
SAR AND QSAR OF ABCG2 SUBSTRATES
Very few studies have examined the SARs and QSAR models for ABCG2 substrates. Irinotecan is a wide spectrum anticancer agent and is a standard therapy for many cancers including colon cancer. ABCG2 overexpression in tumor cells is associated with poor outcome because ABCG2 actively transports anticancer agents out of tumor cells (36), including SN-38, the active metabolite of irinotecan. Plasma membrane vesicles prepared from an ABCG2 overexpressing cell line transported SN-38 and its glucuronide conjugate. To circumvent efflux by ABCG2, 14 analogues of SN-38 were synthesized and studied for their substrate specificity toward ABCG2 (Fig. 10). The 14 structural analogues differed from SN-38 at positions 10 and 11 on the A ring. Accumulation and cytotoxicity of SN-38 and its analogues was measured in SN-38-sensitive PC-6 and SN-38-resistant PC-6/SN2-5H2 cell lines and the IC50 values calculated. Drug resistance ratios were also calculated as the ratio of the IC50 in the resistant cell line to the IC50 in the sensitive cell line. The IC50 for SN-38 in the sensitive cell line was 1.00 ± 0.10 μM and in the resistant cell line was 262 ± 6 μM, corresponding to a drug resistance ratio of 262, indicating that SN-38 is effluxed out of the SN-38-resistant cell line by ABCG2. Three SN-38 analogues had IC50 values similar to that of SN-38 in the resistant cell line. The drug resistance ratios for SN-355, SN-392, and SN-398 were 105, 55, and 213, suggesting that they are also substrates for ABCG2. The accumulation of these three compounds and SN-38 was much lower in the resistant cell line confirming the results of the cytotoxicity assay. A SAR analysis showed similarities in substituents in SN-38 and SN-355, SN-392, and SN-398. SN-38 and SN-398 have a hydroxyl group at position 10 on the A ring, SN-355 has a hydroxyl group on position 11 of the A ring, while SN-392 has an amino group at position 10. No other structural analogues have a hydroxyl or amino substitution at positions 10 or 11 on the A ring. A hydroxyl or amino group can form hydrogen bonds and might be an important step in the substrate recognition pattern of ABCG2. It was also suggested that polarity of the compound might be an important feature for transport by ABCG2. The high-performance liquid chromatography retention time of the compounds, as measured using a reverse phase column, correlated inversely with the drug resistance ratio where compounds with a short retention time had higher ratios and vice versa. The SAR showed that SN-355, SN-392, and SN-398 have similar substituents on the 10 and 11 positions making these compounds susceptible to transport by ABCG2 like SN-38 (37). Based on the SAR, it was speculated that the remaining SN-38 analogues would be able to circumvent efflux by ABCG2.

Chemical structure of SN-38 analogues with substitutions at position X and Y (37). Reprinted with permission of John Wiley & Sons, Inc from Novel camptothecin analogues that circumvent ABCG2-associated drug resistance in human tumor cells, Vol. 110, No. 6, 2004, pp 921–927. Copyright 2009
The initial SARs for SN-38 analogues were followed up with quantum mechanical calculations and derivation of neural network QSARs. The solvation free energy (ΔG) was calculated as:
where E COSMO, E invacuo, and SASA are the energies of SN-38 and its analogues as calculated by AM1/COSMO, AM1/invacuo, and the solvation accessible surface area, respectively. A plot of the drug resistance ratios vs the solvation energy separated the SN-38 analogues into two distinct groups: substrates and non-substrates of ABCG2. Electrostatic properties were also calculated for the analogues, and a negative electrostatic potential area was seen at position 10 in the A ring for SN-38 and SN-398. On the other hand, this negative electrostatic potential area was missing for SN-22 (drug resistance ratio = 3.5). This suggests that the negative electrostatic potential at position 10 in the A ring is crucial for hydrogen bond formation with the active site of ABCG2 and SN-38 analogues with this feature are potential substrates for ABCG2 (38).
CONCLUSION
In silico models can be used to predict pharmacokinetic and pharmacodynamic properties during the drug discovery and development phases and can be used to synthesize drugs with favorable physicochemical properties. SAR and QSAR models are commonly applied in silico models that have been extensively used to predict drug distribution properties like volume of distribution and clearance (9,39). QSAR models for ABCB1 substrates and inhibitors have been comprehensively studied (40,41), but so far, a model that can predict substrates and inhibitors accurately has not been discovered. Similarly, QSAR models to predict ABCG2 substrate and inhibitors are missing. One common structural descriptor among the SARs and QSAR models is lipophilicity. Lipophilicity, calculated as either log P or log D, has been shown to be a predictor of inhibition of ABCG2 by a few groups (19,29,34). In our laboratory, we have derived a QSAR model to describe the pEC50(-log EC50) values of ABCG2 inhibitors using 19 flavonoids in the training set. The final model consisted of three descriptors including log P and was able to accurately predict the pEC50(-log EC50) values for the six flavonoids in the test set (19). Lipophilicity, expressed as log D 7.4 was present in another model to predict ABCG2 inhibitors. The authors suggest a log D cutoff of 0.5, above which there is an increased likelihood of a compound-inhibiting ABCG2 (34). Though lipophilicity has been shown to be a good predictor of ABCG2 inhibition in some studies, other investigators have not found this descriptor to be significant (16,27). No correlation was found between log IC50 and log P or log D for 30 tariquidar analogues (27). In another study, ten descriptors, including log P, were used to construct a QSAR model for 23 propafenone analogues. The final model consisted of four descriptors but did not include log P (16). Finally, no correlation was found between percent ABCG2 inhibition observed and log P for 49 structurally unrelated compounds (32).
A second possible common feature among ABCG2 inhibitors is the existence of a planar structure required for binding to the protein and inhibiting its function. Proof of this comes from multiple studies looking at different structural series of compounds. We have proposed that the 2,3-double bond in ring C in flavonoids increases the inhibition potency for flavonoids, and this is clearly observed in the low EC50 values for flavones (1.15 ± 0.38 μM) vs flavanones (15.3 ± 2.87 μM) (19). A similar observation was made by Ahmed-Belkacem et al. (20,25) with regards to flavonoids and rotenoid derivatives. The loss of the 2,3-double bond in ring C of flavonoids and the 6a,12a-double bond between rings B and C of rotenoid derivatives led to a decrease in inhibition of ABCG2 (20,25). A similar conclusion was drawn for CDK inhibitors with regards to their ABCG2 inhibition potency. Purvalanol A and WHI-P180, the two most potent ABCG2 inhibitors in the class had a planar structure, whereas the other compounds do not have a planar structure and are weak ABCG2 inhibitors (33). Nakagawa et al. (38) examined SARs for SN-38 analogues in an attempt to discover analogues with similar anticancer properties as SN-38 but without the drug resistance of SN-38. It was suggested that the planar structure of SN-38 is critical for binding to the active site of the protein.
QSAR models for ABCG2 inhibitors and substrates have mostly been evaluated using congeneric series of compounds. A lead compound with experimental data on the interaction with ABCG2 is used to derive a series of compounds with good pharmacokinetic, pharmacodynamic, or toxicokinetic properties. ABCG2 inhibitors have been tested in vitro, and a model is derived. This model has limited applicability because it cannot be applied to another congeneric series, presenting a major disadvantage of most QSAR models. Another disadvantage of QSAR studies is the dependence of the relationships on the experimental systems used to generate the data. Activity measurements (EC50 values) in cellular systems may be influenced by permeability and cellular uptake of the substrate or inhibitor. A high resolution crystallized structure of ABCG2 has not been discovered, and thus, interactions of compounds with ABCG2 cannot be accurately modeled.
In conclusion, QSAR models have been used to predict potent and specific ABCG2 inhibitors that have been successfully tested in vitro. In a study of non-congeneric compounds, log D and molecular polarizability emerged as significant determinants of ABCG2 inhibition. Studies with congeneric series of compounds indicated the importance of log P or lipophilicity, planar structure, and hydrogen bonding potential for ABCG2 inhibition.
References
Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95:15665–70.
Allikmets R, Schriml LM, Hutchinson A, Romano-Spica V, Dean M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998;58:5337–9.
Miyake K, Mickley L, Litman T, Zhan Z, Robey R, Cristensen B, et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes. Cancer Res. 1999;59:8–13.
Robey R, Polgar O, Deeken J, To KK, Bates SE. Breast cancer resistance protein. In: You G, Morris ME, editors. Drug transporters: molecular characterization and role in drug disposition. Hoboken: Wiley; 2007. p. 319–58.
Robey RW, To KK, Polgar O, Dohse M, Fetsch P, Dean M, et al. ABCG2: a perspective. Adv Drug Deliv Rev. 2009;61:3–13.
To KK, Polgar O, Huff LM, Morisaki K, Bates SE. Histone modifications at the ABCG2 promoter following treatment with histone deacetylase inhibitor mirror those in multidrug-resistant cells. Mol Cancer Res. 2008;6:151–64.
Zhang S, Yang X, Morris ME. Flavonoids are inhibitors of breast cancer resistance protein (ABCG2)-mediated transport. Mol Pharmacol. 2004;65:1208–16.
Jonker JW, Smit JW, Brinkhuis RF, Maliepaard M, Beijnen JH, Schellens JH, et al. Role of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan. J Natl Cancer Inst. 2000;92:1651–6.
Mager DE, Jusko WJ. Quantitative structure-pharmacokinetic/pharmacodynamic relationships of corticosteroids in man. J Pharm Sci. 2002;91:2441–51.
Mager DE. Quantitative structure-pharmacokinetic/pharmacodynamic relationships. Adv Drug Deliv Rev. 2006;58:1326–56.
Nicolle E, Boumendjel A, Macalou S, Genoux E, Ahmed-Belkacem A, Carrupt PA, et al. QSAR analysis and molecular modeling of ABCG2-specific inhibitors. Adv Drug Deliv Rev. 2009;61:34–46.
Minderman H, Brooks T, O’Loughlin KL, Ojima I, Bernacki RJ, Baer MR. Multidrug resistance (MDR) modulation by the taxane derivatives IDN-5109 and tRA 96023: effects on P-glycoprotein (Pgp)-, multidrug resistance protein (MRP-1)-, and breast cancer resistance protein (BCRP)-mediated drug transport. Proc Am Assoc Cancer Res. 2002;43:950.
Brooks TA, Kennedy DR, Gruol DJ, Ojima I, Baer MR, Bernacki RJ. Structure-activity analysis of taxane-based broad-spectrum multidrug resistance modulators. Anticancer Res. 2004;24:409–15.
Ecker G, Huber M, Schmid D, Chiba P. The importance of a nitrogen atom in modulators of multidrug resistance. Mol Pharmacol. 1999;56:791–6.
Ehrlich P. Present status of chemotherapy. Ber Dtsch Chem Ges. 1909;42:17–47.
Cramer J, Kopp S, Bates SE, Chiba P, Ecker GF. Multispecificity of drug transporters: probing inhibitor selectivity for the human drug efflux transporters ABCB1 and ABCG2. ChemMedChem. 2007;2:1783–8.
Havsteen BH. The biochemistry and medical significance of the flavonoids. Pharmacol Ther. 2002;96:67–202.
Middleton E Jr, Kandaswami C, Theoharides TC. The effects of plant flavonoids on mammalian cells: implications for inflammation, heart disease, and cancer. Pharmacol Rev. 2000;52:673–751.
Zhang S, Yang X, Coburn RA, Morris ME. Structure activity relationships and quantitative structure activity relationships for the flavonoid-mediated inhibition of breast cancer resistance protein. Biochem Pharmacol. 2005;70:627–39.
Ahmed-Belkacem A, Pozza A, Munoz-Martinez F, Bates SE, Castanys S, Gamarro F, et al. Flavonoid structure-activity studies identify 6-prenylchrysin and tectochrysin as potent and specific inhibitors of breast cancer resistance protein ABCG2. Cancer Res. 2005;65:4852–60.
Boumendjel A, Macalou S, Ahmed-Belkacem A, Blanc M, Di Pietro A. Acridone derivatives: design, synthesis, and inhibition of breast cancer resistance protein ABCG2. Bioorg Med Chem. 2007;15:2892–7.
Imai Y, Tsukahara S, Ishikawa E, Tsuruo T, Sugimoto Y. Estrone and 17beta-estradiol reverse breast cancer resistance protein-mediated multidrug resistance. Jpn J Cancer Res. 2002;93:231–5.
Imai Y, Tsukahara S, Asada S, Sugimoto Y. Phytoestrogens/flavonoids reverse breast cancer resistance protein/ABCG2-mediated multidrug resistance. Cancer Res. 2004;64:4346–52.
Katayama K, Masuyama K, Yoshioka S, Hasegawa H, Mitsuhashi J, Sugimoto Y. Flavonoids inhibit breast cancer resistance protein-mediated drug resistance: transporter specificity and structure-activity relationship. Cancer Chemother Pharmacol. 2007;60:789–97.
Ahmed-Belkacem A, Macalou S, Borrelli F, Capasso R, Fattorusso E, Taglialatela-Scafati O, et al. Nonprenylated rotenoids, a new class of potent breast cancer resistance protein inhibitors. J Med Chem. 2007;50:1933–8.
Sugimoto Y, Tsukahara S, Imai Y, Sugimoto Y, Ueda K, Tsuruo T. Reversal of breast cancer resistance protein-mediated drug resistance by estrogen antagonists and agonists. Mol Cancer Ther. 2003;2:105–12.
Pick A, Muller H, Wiese M. Structure-activity relationships of new inhibitors of breast cancer resistance protein (ABCG2). Bioorg Med Chem. 2008;16:8224–36.
Cui C, Kakeya H, Osada H. Novel mammalian cell cycle inhibitors, cyclotryprostatins A-D, produced by Aspergillus fumigatus, which inhibit mammalian cell cycle at G2/M phase. Tetrahedron. 1997;53:59–72.
van Loevezijn A, Allen JD, Schinkel AH, Koomen GJ. Inhibition of BCRP-mediated drug efflux by fumitremorgin-type indolyl diketopiperazines. Bioorg Med Chem Lett. 2001;11:29–32.
Allen JD, van Loevezijn A, Lakhai JM, van der Valk M, van Tellingen O, Reid G, et al. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol Cancer Ther. 2002;1:417–25.
Nurse PM. Nobel Lecture. Cyclin dependent kinases and cell cycle control. Biosci Rep. 2002;22:487–99.
Saito H, Hirano H, Nakagawa H, Fukami T, Oosumi K, Murakami K, et al. A new strategy of high-speed screening and quantitative structure-activity relationship analysis to evaluate human ATP-binding cassette transporter ABCG2-drug interactions. J Pharmacol Exp Ther. 2006;317:1114–24.
An R, Hagiya Y, Tamura A, Li S, Saito H, Tokushima D, et al. Cellular phototoxicity evoked through the inhibition of human ABC transporter ABCG2 by cyclin-dependent kinase inhibitors in vitro. Pharm Res. 2009;26:449–58.
Matsson P, Englund G, Ahlin G, Bergstrom CA, Norinder U, Artursson P. A global drug inhibition pattern for the human ATP-binding cassette transporter breast cancer resistance protein (ABCG2). J Pharmacol Exp Ther. 2007;323:19–30.
Chang C, Ekins S, Bahadduri P, Swaan PW. Pharmacophore-based discovery of ligands for drug transporters. Adv Drug Deliv Rev. 2006;58:1431–50.
Xu Y, Villalona-Calero MA. Irinotecan: mechanisms of tumor resistance and novel strategies for modulating its activity. Ann Oncol. 2002;13:1841–51.
Yoshikawa M, Ikegami Y, Hayasaka S, Ishii K, Ito A, Sano K, et al. Novel camptothecin analogues that circumvent ABCG2-associated drug resistance in human tumor cells. Int J Cancer. 2004;110:921–7.
Nakagawa H, Saito H, Ikegami Y, Aida-Hyugaji S, Sawada S, Ishikawa T. Molecular modeling of new camptothecin analogues to circumvent ABCG2-mediated drug resistance in cancer. Cancer Lett. 2006;234:81–9.
Zhou XF, Shao Q, Coburn RA, Morris ME. Quantitative structure-activity relationship and quantitative structure-pharmacokinetics relationship of 1, 4-dihydropyridines and pyridines as multidrug resistance modulators. Pharm Res. 2005;22:1989–96.
Gombar VK, Polli JW, Humphreys JE, Wring SA, Serabjit-Singh CS. Predicting P-glycoprotein substrates by a quantitative structure-activity relationship model. J Pharm Sci. 2004;93:957–68.
Ha SN, Hochman J, Sheridan RP. Mini review on molecular modeling of P-glycoprotein (Pgp). Curr Top Med Chem. 2007;7:1525–9.
Acknowledgements
Support was provided by the Susan G. Komen Foundation BCTR0601385 and from Pfizer Inc.
Author information
Authors and Affiliations
Corresponding author
Additional information
Guest Editor: Marilyn E. Morris
Rights and permissions
About this article
Cite this article
Gandhi, Y.A., Morris, M.E. Structure–Activity Relationships and Quantitative Structure–Activity Relationships for Breast Cancer Resistance Protein (ABCG2). AAPS J 11, 541–552 (2009). https://doi.org/10.1208/s12248-009-9132-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-009-9132-1