





Abstract
To further elucidate the molecular mechanisms underlying the transcriptional regulation of the GHRH receptor (GHRH-R) gene, hormonal regulation of the promoter activity of this gene was examined. An approximately 3-kb genomic fragment spanning the promoter region of the gene was sequenced and the transcription start site was determined by RT-PCR and RNase protection assay. A major start site was localized at −105 (relative to the translation initiation codon, ATG), and a pit-1 binding sequence characteristic of pituitary specific genes was found at −155 to −146. Deletion and mutation studies demonstrated this site to be functional. In the presence of dexamethasone, the GHRH-R promoter (from −2935 to −11) directed luciferase expression in MtT-S cells, a somatotropic cell line, but not in the PC12 cells that normally do not express GHRH-R. While T3, all trans-RA, and 9cis-RA alone weakly enhanced the reporter gene expression, each of these substances was found to act as a synergistic enhancer in the presence of dexamethasone. Additional deletion and mutation analyses demonstrated a functional RA response element at −1090 to −1074. Two functional glucocorticoid response elements and a T3 response element were found in an 80-bp 5′-flanking sequence of the pit-1 site. Interestingly, it is suggested that the 6-bp half-site AGGACA (from −209 to −204) functions as a 3′-half-site of T3 response element as well as a 5′-half-site of one of the glucocorticoid response elements.
GHRH IS A major factor secreted from hypothalamus to regulate pituitary GH secretion and proliferation of GH cells. The plasma membrane receptor for GHRH (GHRH-R) is a member of G protein-coupled receptors with seven transmembrane helix regions and mediates increases in intracellular cAMP levels in response to GHRH stimulation (1, 2). The coordinate expressions of GHRH in the hypothalamus (3, 4) and GHRH-R in the anterior pituitary gland (5, 6) occur at late gestation in rodents to establish central nervous system regulation of GH secretion. The increase in the intracellular cAMP levels, which is normally brought about by the GHRH stimulation, induces proliferation of GH cells (7–9). In the developing pituitary gland, the expansion of the GH cell population is believed to be the result of the GHRH stimulation, because a deleterious genetic mutation in the GHRH-R gene in mice results in a severe reduction of GH cell number (10).
The physiologic changes in GH secretion reflect in part changes in pituitary GHRH-R expression levels. Glucocorticoids (6, 11–15), T3 (14, 16), and GHRH (17, 18) have been shown to up-regulate GHRH-R mRNA expression, whereas estrogens have been shown to down-regulate the expression of the mRNA (13). This down-regulation by estrogen was postulated to account in part for the sexual dimorphic expression of GH. Petersenn et al. (19) analyzed the promoter region of the human GHRH-R gene and found that glucocorticoids stimulate and estrogens inhibit the expression of reporter gene driven by human GHRH-R promoter. In contrast, T3 and forskolin had no effect on expression. The nuclear receptors for glucocorticoids, thyroid hormones, and estrogens are known to exert their physiologic effects by interacting with a specific cis-elements located in the promoter region of the target genes. However, neither the sequences nor locations of hormone response elements in the GHRH-R gene have been determined. Thus, although several hormonal factors have been demonstrated to regulate GHRH-R gene expression, an understanding of the molecular mechanisms underlying the hormonal regulation of the transcription of the GHRH-R gene is still incomplete.
Several lines of experiments have established that pit-1, a pituitary specific transcription factor (20, 21), is required for the GHRH-R gene expression (10, 22, 23). Lin et al. (10) showed that the expression of a mouse GHRH-R promoter/luciferase fusion gene in heterologous cell types was markedly enhanced by the cotransfection of human pit-1 expression plasmid, suggesting that the GHRH-R gene is a target gene of pit-1. The pit-1 dependent transcription of the GHRH-R gene has also demonstrated in the human (23) and the rat (22). Iguchi et al. (23) found a functional pit-1 binding site in the human GHRH-R promoter and showed that the mutation of the pit-1 site resulted in the marked decrease in the promoter activity. Miller et al. (22) recently demonstrated that cotransfection of pit-1 expression construct enhances basal activity of rat GHRH-R promoter in GH3 cells; however, the potential pit-1 binding site has not been specified in this species.
In the present study, the 5′-flanking region of the rat GHRH-R gene was cloned and analyzed to elucidate the mechanisms responsible for the regulation of GHRH-R gene expression by various hormones. The results demonstrate that glucocorticoids transcriptionally up-regulate the expression of the GHRH-R gene and that, in the presence of glucocorticoid, T3, and RAs synergistically enhance the expression of GHRH-R gene in MtT/S cells (24), a clonal cell line of somatotropes that normally expresses GHRH-R. The synergism between T3 and glucocorticoids appears to depend on a composite hormone response element composed of a T3 response element (TRE) and two glucocorticoid response elements (GREs) located at between −230 and −150 relative to a translation initiation codon.
Materials and Methods
Isolation of a rat GHRH-receptor genomic clone
An approximately 3.5-kb genomic DNA fragment 3′ of the translation initiation codon was amplified by PCR using primers correspond to the sequences of 29–48 and 361–380 of the GHRH-R cDNA sequence reported by Mayo (1). After cloning the PCR product in pGEM-T Easy plasmid (Promega Corp., Madison, WI), a 2-kb fragment of the 5′-half of the insert was excised by ApaI and SacI digestion and used as a probe for a screening of cosmid library of rat genomic DNA of Wistar Kyoto strain (Stratagene, La Jolla, CA). Through screening more than 400,000 clones, one clone carrying at least 10 kb of genomic DNA from the 5′-flanking region of the rat GHRH receptor was isolated. A BamHI fragment (4.4 kb) containing approximately 3 kb of 5′-flanking region, the entire first exon, and the 5′ one-third of the first intron was subcloned into BamHI site of pBluescript II SK-plasmid (pBSII). This plasmid was referred to as prGHRH-R-B3–1. A total of 2925 bp (from −2935 to −11, all positions within 5′-flanking sequence have been indexed relative to +1 translation start site in this paper) of the 5′-flanking region was amplified by PCR along with multiple cloning sites using prGHRH-R-B3–1 as a template, using a sense (5′-GTAATACGACTCACTATAGGG-3, T7 primer) and an antisense primer (5′-CCTGTAGTCCGCCCCAAAGA-3′, from −30 to −11). The PCR fragment was blunt ended by T4 polymerase. Then the fragment was cloned into a blunt-ended BglII/HindIII site of pGL2 (Promega Corp.). Finally, the T7 promoter sequence derived from pBSII plasmid was eliminated by digestion with KpnI and self-ligation, and the resultant plasmid was designated as prGHRH-R-2935/-11-Luc. The nucleotide sequence was determined by cycle sequencing using dye-labeled terminators (ABI PRISM Dye Terminator Cycle Sequencing Ready Reaction Kit, Applied Biosystems, Foster City, CA).
Determination of the transcription start site
To determine the transcription start site of the rat GHRH-R gene, two different methods were employed, RT-PCR and RNase protection assay. For RT-PCR, total RNA was extracted from MtT/S cells by the method of Chomczynski and Sacchi (25). One microgram of total RNA was reverse transcribed with murine leukemia virus reverse transcriptase (Applied Biosystems) at 42 C for 15 min using primer A5 (5′-TGCTCTCTCAGCTGAGTGAT-3′—located in the second exon, see below). The PCR was performed using GeneAmp PCR kit (Applied Biosystems). The initial denaturation was at 95 C for 2 min, followed by 35 cycles of 95 C for 1 min and 60 C for 1 min, and final extension at 72 C for 7 min. Seven upstream primers (S1-S7, 20 bases) and a downstream primer A4 (5′-CACATTCTAGGTGGAGGTGA-3′) were used in this experiment. The positions of 5′-end of the primers S1–7 are −326, −286, −243, −190, −153, −116, and −84, respectively. The primers A4 and A5 are located in the second exon, their 5′-end being at +85, and +110, respectively, of the cDNA sequence reported previously (1). The PCR product was fractionated in a 2% agarose gel and visualized by ethidium bromide staining. Then the PCR products were transferred onto a nylon membrane (Hybond N+, Amersham Pharmacia Biotech, Buckinghamshire, UK) by capillary action. The blot was hybridized with 32P-labeled internal fragment (240 bp) that is PCR amplified using primers S4 and A3 (5′-CACAGGTTCAGCAAGCAGAG-3′, from +31 to +50).
For RNase protection assay, total RNA was purified from the anterior pituitary gland and liver of adult male rat at 8 wk of age (Sprague Dawley strain, obtained from CLEA Japan Inc., Tokyo, Japan), and from MtT/S cells using the method described above. A PCR fragment obtained using primers S4 and A3 (see above) was cloned in pBSII. The plasmid was linearized with BamHI digestion and 32P-labeled cRNA probe (322 bases) was transcribed by T7-polymerase using 32P-uridine triphosphate (ICN Biomedicals, Inc., Costa Mesa, CA). The total RNA (10 μg) was ethanol precipitated and dissolved in 20 μl of hybridization buffer (400 mm NaCl; 1 mm EDTA; 40 mm PIPES, pH 6.4; 80% formamide) containing 2 × 104 cpm of cRNA. The hybridization was carried out overnight at 55 C. After the hybridization, 150 μl of RNase digestion buffer (300 mm sodium acetate; 5 mm EDTA; 10 mm Tris-HCl, pH 7.5) containing 10 μg/ml of RNase A and 100 U/ml of RNase T1 (both from Roche Molecular Biochemicals) and incubated for 30 min at 37 C. The mixture was incubated at 37 C for additional 15 min after addition of 2.5 μl of Proteinase K (Roche Molecular Biochemicals, 20 mg/ml) and 10 μl of 10% sodium dodecyl sulfate. The reaction mixture was extracted with the same volume of phenol/chloroform, and the 120 μl of upper aqueous phase was saved and the RNA was ethanol precipitated. The protected RNA fragment was analyzed by electrophoresis on 6% polyacrylamide gel containing 8M urea.
Construction of reporter genes
The prGHRH-R-2935/-11-Luc was digested with KpnI and HindIII, and the vector carrying the remaining insert was blunt ended by T4 polymerase and self-ligated to form prGHRH-R-1167/-11-Luc. Similarly, KpnI/StuI digestion or KpnI/SacI digestion and self-ligation gave prGHRH-R-403/-11-Luc or prGHRH-R-230/-11-Luc, respectively. The prGHRH-R-B3–1 was digested with EcoRI, and after removing EcoRI fragment, the remaining part of the vector was self-ligated. From this plasmid, KpnI/SacI fragment was isolated and inserted into KpnI/SacI site of prGHRH-R-2935/-11-Luc to form prGHRH-R-1469/-11-Luc. Using similar procedure, prGHRH-R-644/-11-Luc was prepared by HincII digestion and self-ligation of the prGHRH-R-B3–1 plasmid and the substitution of the KpnI/SacI fragment for the corresponding fragment of prGHRH-R-2935/-11-Luc. The prGHRH-R-644/-11-Luc was digested with SmaI and PvuII, and after removing excised fragment, the remaining plasmid was self-ligated to form prGHRH-R-145/-11-Luc. The PCR fragments spanning −220/−11, −210/−11, −200/−11, −190/−11, −153/−11 of the GHRH-R promoter were cloned in pCR2.1 plasmid (Invitrogen, Groningen, The Netherlands), and the inserts were excised with KpnI and XhoI digestion. The inserts were purified and ligated to a corresponding site of pGL-2 basic vector to generate prGHRH-R-220/-11-Luc, prGHRH-R-210/-11-Luc, prGHRH-R-200/-11-Luc, prGHRH-R-190/-11-Luc, or prGHRH-R-153/-11-Luc, respectively.
Three reporter constructs bearing mutations in the pit-1 site were prepared. The PCR was performed using prGHRH-R-B3–1 as a template and combinations of an upstream primer F (5′-GCCCTGTGATAGAGACTTAG-3′, from −513 to −492) and one of the mutated downstream primers, pit-1-m1 (5′-CCAGCTGAATCTTGAACAGGATG-3′), pit-1-m2 (5′-CCAGCTGACTATTGAACAGGATG-3′) or pit-1-m3 (5′-CCAGCTGACACTTGAACAGGATG-3′). The PCR products were digested with StuI (at −400) and PvuII (at −145), and the isolated fragments were purified and replaced for the corresponding fragments of prGHRH-R-2935/-11-Luc to generate prGHRH-R-pm-1-Luc, prGHRH-R-pm-2-Luc or prGHRH-R-pm-3-Luc, respectively. The pm-1, pm-2, or pm-3 mutations alter the putative pit-1 binding sequence TTCAATATTC to TTCAAGATTC, TTCAATAGTC or TTCAAGTGTC, respectively (mutated bases are underlined).
Site-directed mutations for GREs and TREs were introduced into a pGHRH-R-230/-11-Luc and that for RA response element (RARE) were introduced into a pGHRH-R-1167/-11-Luc by a PCR-based method using LA-PCR in vitro mutagenesis kit (Takara Shuzo). The oligonucleotides used to generate site-directed mutations were: TRE-m1: 5′-TGTGGACAGCAGGGGTTGGCT-3′; TRE-m2: 5′-CCTCAGCAGGTTGTGGCTGA-3′; TRE-m3: 5′-TGTCCTCAGCCAAGGTTGGC-3′; GRE-1m: 5′-GGGGTTCTGTGTCCTCAGCA-3′; GRE-2m: 5′-GGGGTATTGTGTTACTGCCA-3′; GRE-3m: 5′-TGAGTAGGATGGTGGTACAT-3′; RARE-m1: 5′-CTTGACCACGGAATCGTCGT-3′; RARE-m2: 5′-CTTCGTCACGGAATGACCGT-3′; RARE-m3: 5′-CTTCGTCACGGAATCGTCGT-3′.
Cell culture and transient expression studies
The MtT/S cell was supplied by Dr. K. Inoue (Saitama University, Saitama, Japan) and maintained in DMEM/F12 medium containing 10% horse serum, 2.5% FBS (Life Technologies, Inc., Rockville, MD; heat inactivated before use) and antibiotics (control medium). For the transfection studies, MtT/S cells (1–2 × 105 cells/well) or PC12 cells (5 × 104 cells/well) were grown in a 48-well plate with 0.5 ml of control medium for 24 h and on the day of experiment, the medium was replaced with serum-free DMEM/F12. The plasmids were introduced into the cells by a lipofection reagent (Lipofectamine 2000, Life Technologies, Inc.). One microgram of GHRH-R-luciferase plasmid and 0.1 μg of pRL-TK (Promega Corp.), which was used to monitor the efficacy of transfection, were used per well in conjunction with 1.25 μl of lipofection reagent. The transfection was carried out in a serum-free medium for overnight, and then the medium was replaced with a new serum-free medium containing test substances, followed by additional 24 h incubation. Dexamethasone (DEX), all-trans RA, and 9cis-RA (9cRA) were added to culture at a final concentration of 1 μm, and T3 was used at 1 nm, otherwise stated. Then the cell lysate was prepared and the luciferase activities were determined using dual-luciferase reporter assay system (Promega Corp.). The luciferase activity derived from GHRH-luciferase construct was normalized for that of Renilla luciferase of pRL-TK cotransfected for internal standard.
Construction of receptor expression plasmids and EMSA
The expression plasmid for pit-1 was provided by Dr. Z. Dave Sharp (University of Texas at San Antonio, San Antonio, TX), which produces a fusion protein of rat pit-1 and glutathione S-transferase (GST). The expression plasmids for rat TRα1 and rat RXRα were supplied by Dr. T. Goda, Department of Nutrition, School of Food and Nutritional Sciences, University of Shizuoka (Shizuoka, Japan). A cDNA fragment for rat GR cloned in pBS (pXGR14), was supplied by Dr. K. R. Yamamoto (University of California at San Francisco, San Francisco, CA), that carries a 1158-bp cDNA fragment of rat GR [486 to 1643 of cDNA sequence reported by Miesfeld et al. (26)] The upstream fragment (59–485) and downstream fragment (1644–2480) of pXGR14 were PCR amplified and connected with pXGR14 to make approximately 2.5-kb cDNA that encompasses the entire coding sequence. The rat GR expression plasmid, prGR2.5, was prepared by the insertion of the 2.5-kb rat GR cDNA to downstream of CMV promoter of plasmid pcDNA3 (Invitrogen, San Diego, CA). The cDNA fragments spanning entire cording sequences of TRβ1 or TRβ2 were prepared by PCR and cloned into pcDNA4/His-Max C or B (Invitrogen), respectively. The receptor proteins were in vitro translated using a reticurocyte-lysate translation system (Promega Corp.), and the translation reaction was used to EMSA without protein purification. The translation reaction (4 μl) was mixed with 2 μl of binding buffer (50 mm Tris-HCl; pH 8.0, 750 mm KCl; 0.25% NP-40; 30% glycerol; and 2.5 mm dithiothreitol), 2 μl of poly (deoxyinosine-deoxycytidine) (1 mg/ml), 0.2 μl of sonicated salmon sperm DNA (10 mg/ml), and 0.8 μl of distilled water containing 12.5 μm of DEX and/or 12.5 nm of T3, and incubated at room temperature for 15 min. Then, 2 × 104 cpm of radiolabeled probe (1 μl) were added to the reaction and incubated for additional 20 min. The protein-DNA complexes were separated on 6% polyacrylamide gels in 0.5× TBE buffer. The gels were then dried and exposed to x-ray film at −80 C for 10 d. The sequences of the probes were given (see Fig. 6). For each probe, 5′-end-20mer oligonucleotides of sense and antisense strands were synthesized, annealed, and the 3′ ends were filled in by Klenow fragment in the presence of α32P-deoxy-CTP (ICN Biomedicals). The palindromic TRE probe was prepared with oligonucleotides 5′-TGTCAGGTCATGACCTGAAG-3′ (sense) and 5′-GGGCTTCAGGTCATGACCTG-3′ (antisense), and the consensus GRE probe was prepared with 5′-GGGGGTACAAAATGTTCTGG-3′ (sense) and 5′-GGGCCAGAACATTTTGTACC-3′ (antisense). Synthetic sense and antisense oligonucleotides from −160 to −125 was used for the demonstration of pit-1 binding.
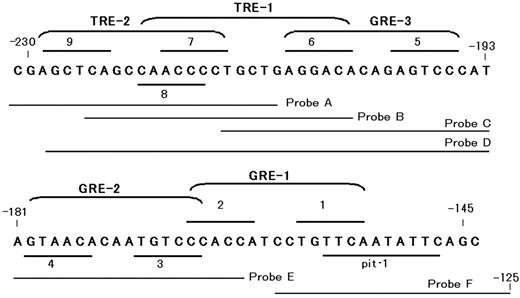
Putative binding elements for hormone receptors and pit-1 in the 5′-flanking region of the rat GHRH-R gene. Localizations of putative GREs, TRE, and pit-1 in the 5′-flanking region of the rat GHRH-R gene are shown. The hexamer half-sites of hormone response elements are tentatively numbered from 1 through 9. GRE-1 (composed of half-sites 1 and 2), GRE-2 (3 and 4), and GRE-3 (5 and 6) contains 7, 8, or 7 of 12 nucleotides identical to the consensus GRE (GGTACAnnnTGTTCT), respectively. TRE-1, consists of half-sites 6 and 8, appears to be an inverted palindrom-like sequence, and TRE-2 consists of half-sites 7 and 9 appears to be a palindromic TRE. Probes A–F represent the sequences of the probes used for EMSA experiment (Fig. 10).
Results
Cloning of rat GHRH-R promoter and determination of the transcription start site
Through the screening of the rat genomic DNA library, one clone carrying the rat GHRH-R gene was isolated, and an approximate 3-kb 5′-flanking region was sequenced in this study. The nucleotide sequence of 1.9-kb 5′-flanking region were identical to that published by Miller et al. (22) (GenBank accession no. AF121969), except for cytosine residues at −1903 and −1553 both of which were found to be thymine in the present study.
The transcription start site was examined by RT-PCR and the RNase protection assay. As shown in Fig. 1, combinations of primers S6 and A4 gave a faint band and primers S7 and A4 gave shorter but intense band. Both PCR products reacted to 32P-labeled internal sequence used in the Southern hybridization. These results suggest the presence of the transcription start site within the sequence of primer S6. In the RNase protection assay (Fig. 2), only mRNAs from the pituitary gland and MtT/S cells were shown to protect the cRNA probe from RNase digestion. Protected fragments formed a single major band of 155-nucleotides in size, and this was aligned with an adenine residue at position −105 of the sequence ladder. Thus, the major transcription start site of the rat GHRH-R gene was assigned to be T, which is 105 bp upstream from the translation initiation codon.
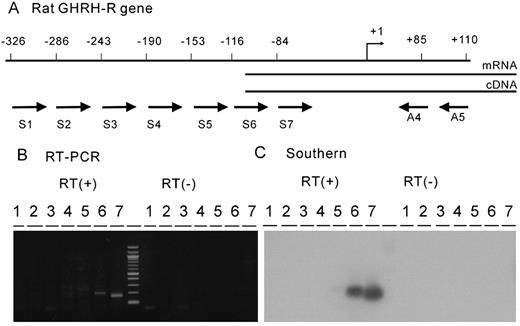
RT-PCR analyses of the transcription start site of the rat GHRH-R gene. A, Schematic diagram showing positions of PCR primers used in this study. Total RNA was prepared from MtT/S cells and reverse transcribed using primer A5, and the PCR was performed with primer combinations of A4 and one of the seven sense primers. Positions of 5′-end of the seven sense primers (S1-S7) and two antisense primers (A4 and A5, set in the second exon) were shown. B, The PCR products were analyzed in a 2% agarose gel. Only S6 and S7 gave distinct PCR products that migrated to the position of the predicted sizes (201 bp and 169 bp, respectively). Any PCR product was not seen in a control experiment where reverse transcriptase was not added to the reaction. C, The PCR products separated in an agarose gel were transferred to a nylon membrane and hybridized with 32P-labeled internal fragment.
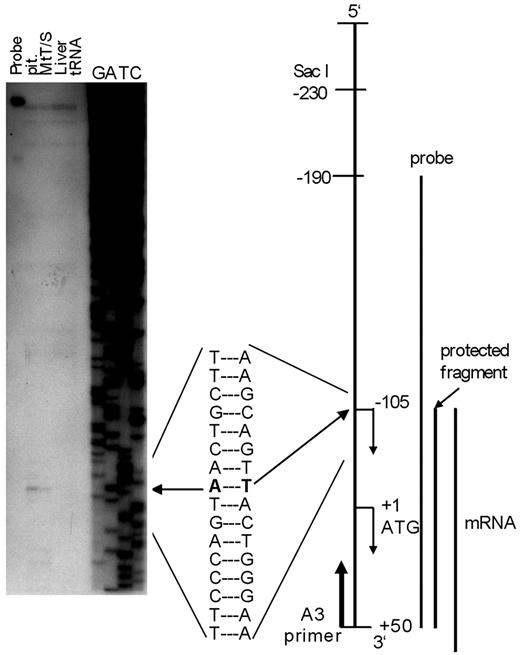
Determination of the transcription start site by RNase protection assay. Total RNA was extracted from anterior pituitary gland (pit) and liver of a normal male rat and from MtT-S cells. Ten micrograms of RNA was used for assay. Yeast tRNA was processed similarly as a negative control. The protected fragment was analyzed in 6% denaturing polyacrylamide gel alongside a sequence ladder, which was generated by a conventional sequence reaction using prGHRH-R-B3–1 as a template and primer A3 (from +31 to +50) as a sequence primer. Only RNAs from anterior pituitary gland and MtT/S cells were able to protect the probe from RNase digestion (left arrow). The assay resulted in only one major protected fragment, which migrated to a position equivalent to an adenine residue at −105 of the ladder, suggesting that a transcription start site for the rat GHRH-R gene can be assigned to the T 105 bp upstream from the translation initiation codon.
Hormonal regulation of promoter activity of the rat GHRH-R gene
The reporter construct prGHRH-R-2935/-11-Luc, which contains the longest 5′-flanking region, was transiently transfected into MtT/S cells, which normally express GHRH-R, or PC12 cells that normally do not (Fig. 3). This experiment demonstrated that a 2925-bp fragment of 5′-flanking region of the gene directed the reporter gene expression in MtT/S cells in the presence of DEX, whereas the expression level in PC12 cells was only 3.3% that in MtT/S cells. In the MtT/S cells transfected with the promoterless pGL2-basic vector, only 0.8% of the reporter gene expression could be detected compared with that with the GHRH-R promoter.
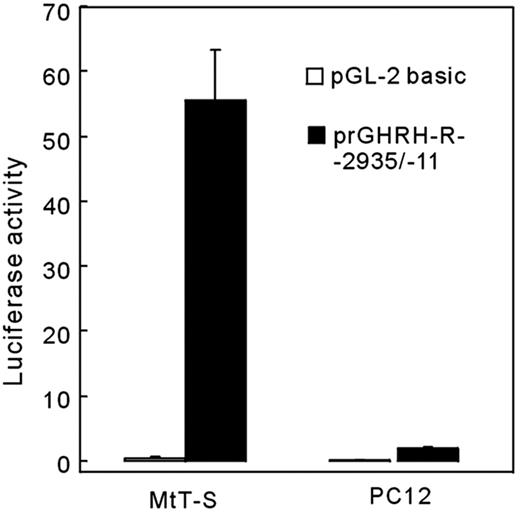
Cell type-specific expression of the reporter gene directed by the rat GHRH-R promoter. A luciferase expression construct (prGHRH-R-2935/-11-Luc, 1 μg) or its vector alone were transfected to MtT/S cells or PC12 cells and incubated for 24 h in a serum-free medium containing 500 nm of DEX. A distinct reporter gene expression was observed in GHRH-R expressing MtT/S cells in the presence of DEX but not in PC12 cells that do not express GHRH-R. In this and in the following figures, values were normalized for the activity of Renilla luciferase that is derived from a cotransfected pRL-TK (0.1 μg).
The hormonal regulation of the rat GHRH-R gene was examined in MtT/S cells. The cells were transfected with prGHRH-R-2935/-11-Luc, followed by 24 h incubation in serum-free medium containing DEX, T3, RA, or 9cRA alone or in combination. A weak reporter gene expression was observed in the serum-free medium, whereas the expression level was increased by 12.5-fold in response to DEX (Fig. 4). T3, RA, and 9cRA alone had a weak stimulatory effect on the promoter activity. T3, RA, and 9cRA each showed synergistic effects with DEX by further increasing reporter gene expression over that seen with DEX only. However, the addition of any two of the hormones (T3, RA, or 9cRA) in the presence of DEX did not statistically enhance promoter expression over that seen with any one in the presence of DEX.
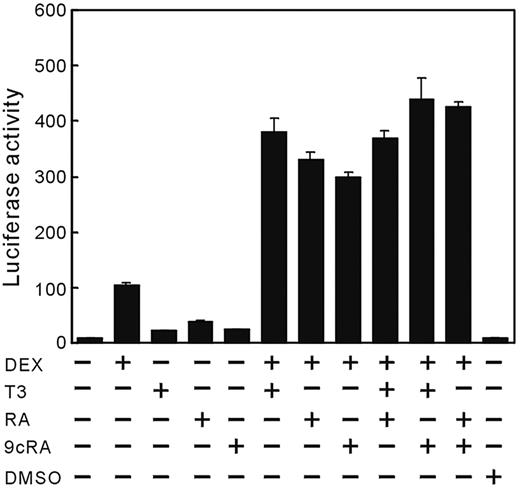
Regulation of the rat GHRH-R gene transcription by DEX, T3, RA, or 9cRA. MtT/S cells were transfected with prGHRH-R-2935/-11-Luc (1 μg), followed by incubation for 24 h in a fresh serum-free medium containing substances indicated. A faint basal expression of the promoter was observed in MtT/S cells incubated in serum-free medium without test substances, and addition of DEX induced a marked increase in the reporter gene expression. T3, RA, and 9cRA induced weak promoter expression. However, when added in conjunction with DEX, T3, RA, and 9cRA equally enhanced effect of DEX synergistically. No synergism was observed between T3 and RAs in the presence of DEX. Values were mean ± sem of the triplicate determinations. The same experiment was repeated three times with similar results and the representative data are shown in this figure. DMSO, Dimethylsulfoxide (0.01%), solvent of RA and 9cRA.
Response elements for pit-1, glucocorticoids, T3, and RAs
Because DEX induced distinct GHRH-R gene transcription by itself, the promoter activity of the deletion constructs were measured in the presence of DEX (Fig. 5). Reduction of promoter region from −2935 bp to −1459 bp resulted in nearly 3-fold increase in the reporter gene expression, suggesting the presence of negative regulatory element(s) in this region. Further progressive deletions revealed that the minimal promoter sequence necessary to sustain DEX-dependent transcription in MtT/S cells is located within 230-bp sequence upstream of translation initiation site. The promoter activity was markedly reduced with further deletion, suggesting that the 5′-flanking region between −230 and −145 contains primary DNA elements required for the regulation of the rat GHRH-R gene transcription by glucocorticoids. The locations of putative hormone response elements in this region are shown in Fig. 6. Within an 80-bp 5′-flanking region of the putative pit-1 element, three GRE-like sequence and two TRE-like sequences were found.
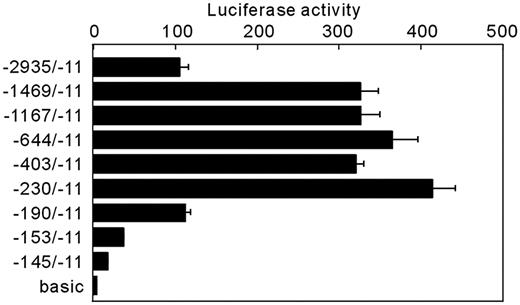
DEX regulation of transiently transfected 5′-deletion mutants of the rat GHRH-R-Luc fusion gene. A series of constructs carrying 5′-deletion mutations of the rat GHRH-R promoter fused to firefly luciferase (1 μg) were transfected into MtT/S cells. Cells were incubated for 24 h in a serum-free medium containing DEX. Values were mean ± sem of the triplicate determinations. Deletion of 5′-half of the promoter region resulted in about 3-fold increase in the promoter activity. The minimum promoter region required for a full DEX-induced activity was 230 bp flanking the translation initiation codon. A stepwise reduction of the promoter activity was observed along with further deletions, suggesting a localization of potential glucocorticoid response elements within a downstream region of −230.basic, pGL2-bacic vector without promoter.
Mutations were introduced to the putative pit-1 response element (−155 to −146) of prGHRH-R-2935/-11-Luc to examine whether this element is functional. Two kinds of mutations (pm-1 and pm-3) resulted in a 70% reduction in promoter activity, whereas the mutation 2 affected little (Fig. 7). However, synergistic effects of T3, RA, or 9cRA with DEX were still observed in the mutated constructs. The results suggest that the element located at −155 to −146 is a functional pit-1 binding element, and that the functional pit-1 is not required for synergistic activation of the promoter activity by T3, RA, or 9cRA.
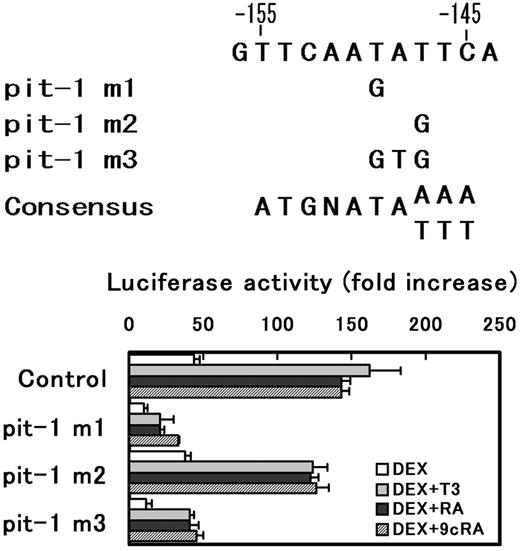
Effects of mutations of putative pit-1 response element in the rat GHRH-R promoter. Three different mutations (pit-1 m1, pit-1 m2, and pit-1 m3) were introduced to a putative pit-1 response elements in prGHRH-R-2935/-11-Luc and the mutated plasmid (1 μg) were transiently transfected into MtT/S cells. Cells were incubated in a serum-free medium with or without substances indicated. The results were expressed as fold increase in the luciferase activity by the hormone addition over the values obtained in hormone free culture. Values are means ± sem of the triplicate determinations. Both pit-1 m1 and m3 mutations reduced promoter activity to about 30% that of control; however, the synergism between DEX and one of T3, RA, or 9cRA was still observed in the mutated plasmid. The result suggests that the sequences between −155 and −146 is a functional pit-1 response element, and T at −150 is an important base as pit-1 response element, but T at −148 is of less importance.
The result from a deletion study (Fig. 5) suggests the presence of at least two functional GREs; one localized between −230 and −190 (containing GRE-3) and the other between −190 and −153 (containing GRE-1 and GRE-2). To examine whether these putative GREs are functional or not, the effect of mutations that disrupt a conserved GRE sequences were examined. A consensus GRE is composed of two hexamer half-sites separated by three nonspecific nucleotides, i.e. GGTACAnnnTGTTCT (27). Because GT in the 3′-half-site of the consensus GRE is of particular importance not only for GR binding but also for GRE activity (28–30) and is conserved in all three putative GREs, this sequence was replaced with AC and the changes in the DEX-induced transcriptional activity were examined (Fig. 8). The promoter activity was significantly reduced by the mutation in GRE-2 and GRE-3, but not in GRE-1, suggesting that GRE-2 and GRE-3 are functional. In these mutated constructs, the synergism between DEX and T3 was still observed.
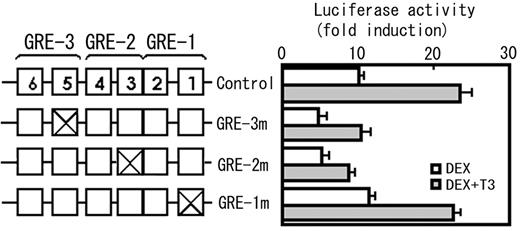
A mutation analysis of the putative GREs in the rat GHRH-R promoter. Because a dinucleotide GT in the 3′-half-site of the consensus GRE (GGTACAnnnTGTTCT) is considered to be important for both GRE activity and GR binding, and this sequence was conserved in all three putative GREs found in the rat GHRH-R promoter, a dinucleotide GT in each GRE was replaced with CA and the changes in the promoter activity were determined in the presence of DEX or DEX + T3. The numbered boxes represent the half-sites (see Fig. 6). The mutations were introduced to pGHRH-R-230/-11-Luc. The plasmid carrying the same promoter fragment without mutation was used as a control. The mutations in GRE-3 (GRE-3m) and GRE-2 (GRE-2m) reduced the promoter activity, whereas that in GRE-1 (GRE-1m) did not, suggesting that GRE-3 and GRE-2 are functional GREs, but GRE-1 is not. The mutations in any GREs did not affect the synergism between glucocorticoids and T3. The results were expressed as fold increase in the luciferase activity by the hormone addition over the values obtained in hormone free culture. Values are means ± sem of the triplicate determinations.
A detailed mapping was carried out to localize functional TRE in 5′-franking region of the pit-1 site (Fig. 9A). The TRE activity was consistently observed in the constructs of −230/−11, −220/−11, reduced in the construct of −210/−11 and not observed in the construct of −200/−11 or −190/−11. This result suggests that TRE-1 (Fig. 6) may be a functional TRE. The TRE-1 (CAACCCctgctgAGGACA, conserved nucleotides are underlined), composed of half-sites 6 and 8 (Fig. 6), has a similarity to an inverted palindromic sequences of AGGTCA motif and the TRE-2 (AGCTCAgccaACCCCT), composed of half-sites 7 and 9, appears to be a palindromic TRE. The mutations were introduced into the conserved sequences of half-sites 6–8 of pGHRH-R-230/-11, and the effect of these mutations in the promoter activity was evaluated (Fig. 9B). The mutations that disrupt TRE-1 (TREm1 and TREm2) abolished the TRE activity, whereas the mutation that disrupts TRE-2 (TREm3) did not affect the synergism between DEX and T3, suggesting that only TRE-1 is a functional element.
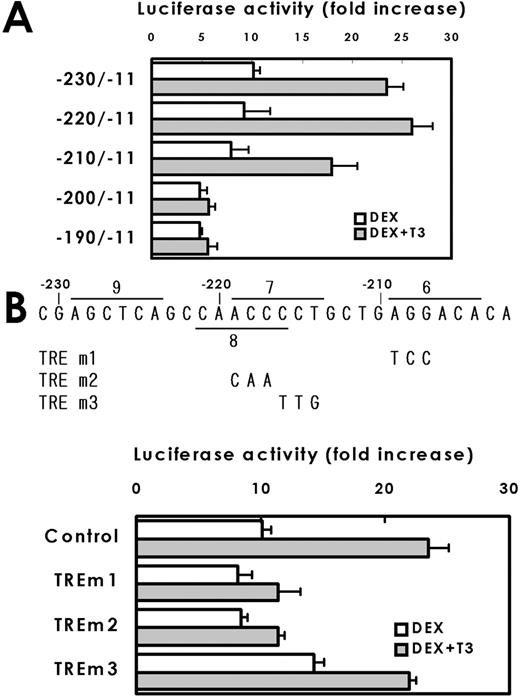
Deletion (A) and mutation (B) analyses of localization of TRE. A, Series of deletion mutants with promoter regions indicated in the figure were prepared and the promoter activity was determined in the presence of DEX or DEX + T3. Values are fold induction of promoter activities by DEX + T3 over DEX alone (means ± sem of five independent assays are shown). The effect of T3 was observed in the construct of −230/−11 and −220/−11, reduced in the construct of −210/−11 and disappeared in −210/−11. B, The mutations shown in the figure were introduced to pGHRH-R-230/-11-Luc and the changes in promoter activity were determined. The results were expressed as fold increase in the luciferase activity by the hormone addition over the values obtained in hormone free culture. The mutation in the half-sites 6 (TREm1) or 8 (TREm2), both of which disrupt TRE-1, abolished the synergistic activation of the promoter activity by T3, but the mutation in half-site 7, which disrupt TRE-2, did not.
The EMSA was carried out to examine whether a pit-1 response element, GRE-2, GRE-3, and TRE-1, which are considered to be functional, really bind the corresponding proteins. The heterodimer complexes comprise TRα1 and RXRα or TRβ1 and RXRα were evident on probe B containing TRE-1 (Fig. 10a, lanes 10 and 11), whereas these complexes were barely detectable on probe A (containing TRE-2, Fig. 10a, lanes 4 and 5) and not observed on probe C or probe E containing GRE-3 or GRE-2, respectively. Binding of GRs was demonstrated only on probe C (Fig. 10a, lane 18) and E (Fig. 10a, lane 24), supporting the results from mutational analyses. These results suggest that the half-site 6 is required for both GR binding as a component of GRE-3 and for TR/RXR binding as a component of TRE-1. To test whether overlapping TRE-1 and GRE-3 can bind both TR/RXR and GR homodimer simultaneously, EMSA was carried out using probe D, which contains entire sequences for both of these elements (Fig. 10, b and c). The incubation of probe D with GR, TRβ1 (or TRβ2) and RXRα resulted in two distinct bands, each of them corresponds to GR homodimer or TR/RXR heterodimer, respectively (Fig. 10b, lane 7, and 10c, lane 4), with no additional bands. These results suggest that GR homodimers and TR/RXR heterodimers bind to the probe independently in this system. The pit-1-GST fusion protein bound to probe F (Fig. 10d, lane 2), whereas control translation reaction generated no band. In the next experiments, we examined the location of the DNA elements responsible for the synergistic activation of the rat GHRH-R gene by RA and 9cRA with DEX. The prGHRH-R-1167/-11-Luc displayed the synergy of RA or 9cRA with DEX, whereas it was absent in prGHRH-R-644/-11-Luc or shorter constructs. These data suggest that RARE may be located between −1167 and −644 (Fig. 11A). In this region, we found a RARE-like element consists of half-site 10 and 11 (Fig. 11B). This putative RARE (CGGTCAttccgTGGTCA, conserved nucleotides are underlined) contains 10 of 12 bases match to the consensus RARE, which is considered to be a direct repeat of AGGTCA motif separated by a 5-base spacer. Although mutations in half-sites 10 or 11 alone did not affect the promoter activity of pGHRH-R-1167/-11, the mutation in both half-sites abolished the synergism of DEX and RA completely, suggesting that this element is a functional RARE.
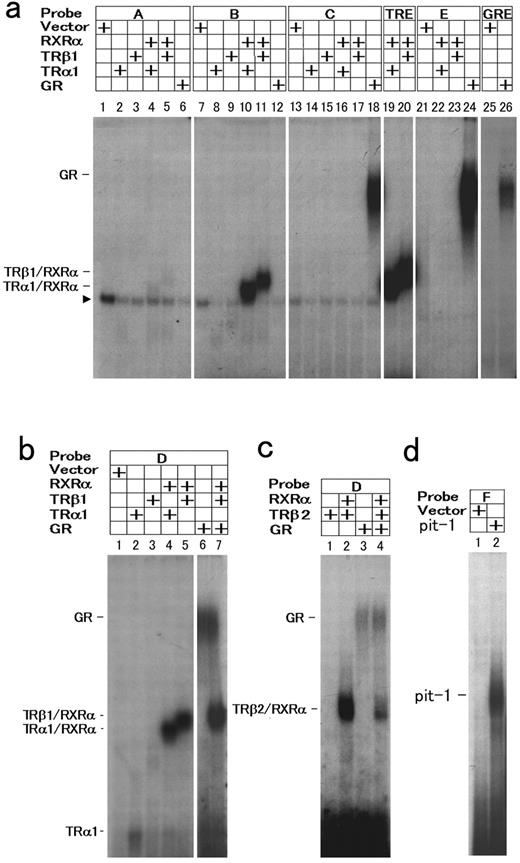
EMSA for the determination of the binding sites of hormone receptor complexes and pit-1 in the promoter of the rat GHRH-R gene. a, Radiolabeled probes, A, B, C, and E (see Fig. 6) were incubated with the in vitro translated receptor proteins in combinations indicated in the figure. The palidromic TRE probe (TRE) and consensus GRE probes (GRE) were included in this experiment as positive controls. The translation reaction with empty vector was used for the negative control (Vector). An arrowhead to the left indicates nonspecific bands. b, An EMSA was performed with Probe D spanning entire sequences of TRE-1, TRE-2, and GRE-3. TRα1 and TRβ1 formed heterodimers with RXRα, but the interaction with these heterodimers with GR was not observed. c, The binding of the pituitary specific TRβ2 to the promoter was examined. TRβ2 also formed heterodimer with RXRα on probe D as other subtypes of TRs, and did not interact with GR. d, A probe F containing presumptive pit-1 response element bound pit-1-GST.
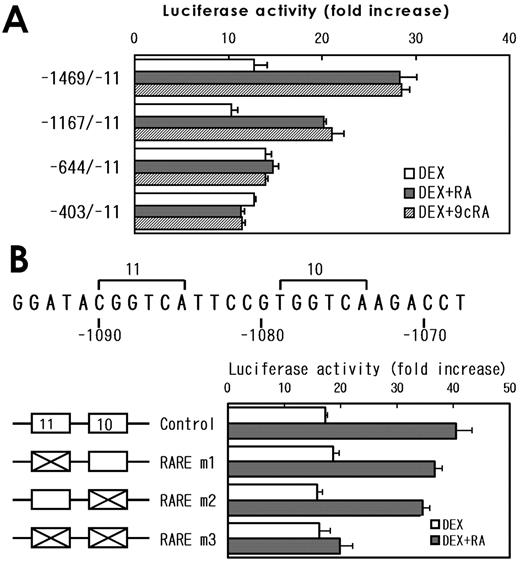
Deletion and mutation analyses of a RARE. A, Series of deletion constructs containing promoter regions of the rat GHRH-R gene as indicated in the figures were transiently transfected into MtT/S cells and the cells were incubated for 24 h in a fresh serum-free medium with or without substances indicated in the figures. The results were expressed as fold increase in the luciferase activity by the hormone addition over the values obtained in hormone-free culture. Values are mean ± sem of the triplicate determinations. The results suggest the presence of a response element for RAs between −1167 and −644. B, The mutation analyses were carried out to clarify whether the putative RARE is functional or not. A putative RARE that localizes between −1090 and −1074 contains two AGGTCA-like half- sites (half-sites 10 and 11 in this figure). Mutations of either half-site 11 (RARE m1, CGGTCA 224 CGACGA, mutated bases are underlined) or half-site 10 (RARE m2, TGGTCA 224 TGACGA) in prGHRH-R-1167/-11-Luc alone did not affect the synergism between DEX and RA. However, the effect of RA was abolished after both half-sites were mutated (RARE m3).
Discussion
Cloning and characterization of the 5′-flanking region of the rat GHRH-R gene have been reported recently by Miller et al. (22). The nucleotide sequence of approximately 1.9 kb region 5′-flanking the translation initiation codon determined in this study were almost identical to those described by Miller et al. (22) (two nucleotides were different and may represent polymorphisms). The transcription start site was determined in this study using RT-PCR and the RNase protection assay. The RNase protection analysis yielded a single major band at a position corresponding to 105 nucleotides upstream from the translation initiation codon using RNA preparations from either an adult rat pituitary or from MtT/S cells. This position falls within the sequence predicted by RT-PCR analyses (between −116 and −97 bp). These combined results suggest that thymine at −105 is a major transcription start site of the rat GHRH-R gene. The start site determined in this study did not coincide with any of the multiple transcription start sites, at −84, −93, −203, and −286 relative to the translation initiation site, determined by Miller et al. (22) using primer extension, Northern blot and RT-PCR. As will be discussed later, the region between −230 and −146 is important for the synergistic activation of the transcription by glucocorticoids and T3. Therefore, the positions −285 and −203 determined by Miller et al. (22) are not likely to be the major start sites because these two sites are localized upstream or within this primary regulatory region of the promoter.
The transient transfection study suggests that 2925 bp upstream region carries most of DNA elements required for a cell type-specific expression of the gene. Because the promoter activity was extremely low when the transfected cells were cultured in a serum free defined medium, the expression of this gene requires additional factors other than just pit-1, which has been demonstrated to be a prerequisite for GHRH-R expression in mice (10), rats (22), and humans (23). The present results are in agreement with the previous studies (6, 11–15, 31) showing that glucocorticoids, T3, and RAs are involved in the GHRH-R gene expression, and the DEX acts synergistically with T3 or RAs with respect to the GHRH-R mRNA accumulation in GH cells. Therefore, it is strongly suggested that the up-regulation of the rat GHRH-R mRNA levels by these hormones is a results of increased transcription rate of this gene.
Pit-1 dependence of the GHRH-R gene transcription was previously demonstrated in mice and humans. Miller et al. (22) showed that the coexpression of pit-1 enhances rat GHRH-R transcription; however, the location of the pit-1 binding site had not been specified. The present results of mutation experiments suggest that a DNA element from −155 to −146 is a functional pit-1 response element. Furthermore, the binding of pit-1 protein to this region was also demonstrated in this study. The results of the mutation study also demonstrated that a thymine at −150, but not at −148, is required for pit-1 activity. The pit-1 element of rat GHRH-R gene has very high similarity to the high affinity pit-1 binding site termed P2 in the human GHRH-receptor gene (23) and to the consensus DNA sequences for pit-1, ATGNATA(A/T) (A/T) (A/T), described recently (32), especially at 3′ located A/T-rich region.
Deletion study predicted the presence of functional GREs in downstream of SacI site at −230 of the rat GHRH-R gene (Fig. 6). As shown in Fig. 6, three putative GREs were found between −230 and −105 of the rat GHRH-R gene, and tentatively designated as GRE-1 (composed of half-sites 1 and 2; Fig. 6), GRE-2 (composed of half-sites 3 and 4) and GRE-3 (composed of half-sites 5 and 6). The GRE-1 matches the consensus (5′-GGTACAnnnTGTTCT-3′, 28) at 7 of 12 positions, the GRE-2 matches at 8 of 12, and the GRE-3 at 7 of 12. Mutations in the conserved sequence of GRE-3 and GRE-2 reduced the promoter activity, suggesting that these two elements are functional GREs. The results of EMSA study supported this result.
We found an 18-bp TRE-like element located from −221 to −204 of the rat GHRH-R gene (composed of half-sites 6 and 8 of Fig. 6), and tentatively designated as TRE-1, that appeared to be an inverted palindromic sequence of AGGTCA motif similar to a TR binding site, termed F2, found in the upstream region of the chicken lysozyme gene (33). Another TRE-like sequence (−229 to −214) is composed of half-sites 7 and 9, and termed as TRE-2. TRE-2 matched the palindromic TRE at 8 of 12 positions. The present results from deletion and mutation studies demonstrated that the TRE-1 but not TRE-2 is a functional TRE. This result was consistent with that of EMSA. Unlike a palindromic TRE, a TRE with an inverted palindromic sequence shows weak affinity for RA receptor (33). This is in favor of the present results showing that the transcription from a promoter construct with −403/−11 containing TRE-1, but not RARE, was not enhanced by RAs.
It should be noted that the sequences spanning TRE-1 and GRE-3 partly overlap each other. The 3′-half-site of TRE-1 (half-site 6 in Fig. 6) is also a component of GRE-3. The mutation in GRE-3 reduced the glucocorticoid induced promoter activity, but the synergism with T3 was not affected. This suggests that the synergism between glucocorticoids and T3 in the transcription of the rat GHRH-R gene is mediated at least in part by TRE-1 and GRE-2. It appears also possible that the synergism is also achieved through GRE-3/TRE-1 region alone, because a mutation in GRE-2 did not abolish the synergism between T3 and DEX. However, the binding of GR and TR/RXR to GRE-3/TRE-1 was mutually exclusive, and no interaction between these two receptor complexes was demonstrated in this study. The mechanism(s) for the synergistic activation of GHRH-R gene transcription by T3 and DEX through GRE-3/TRE-1 complex still remains to be elucidated.
The present data suggest that the effect of glucocorticoids is mediated by the binding of activated GRs to GRE-2 and GRE-3, which appears to be equipotent. A possible mechanism for the synergy between glucocorticoids and T3 involve binding of TR/RXR to TRE-1 and GR to GRE-2, which may allow recruitment of coactivator proteins and a ligand-dependent transcriptional activation. The binding of TR/RXR alone is not enough to promote these processes. It is also suggested that the synergism through TRE-1/GRE-3 region alone is possible, while its molecular mechanism remains to be elucidated. The effect of RAs is considered to be mediated by an RARE located at −1090 to −1074. The present findings provide support for the transcriptional regulation of GHRH-R gene expression by glucocorticoids, T3, and RAs. It is likely that these substances play a role in the developmentally regulated and physiologic changes that occur in GHRH-R transcription in GH cells.
Acknowledgments
The authors thank Dr. Z. Dave Sharp (University of Texas at San Antonio, San Antonio, TX) for providing a pit-1 expression plasmid, Dr. Toshinao Goda (University of Shizuoka, Shizuoka, Japan) for TRα1 and RXRα, and Dr. Keith R. Yamamoto (University of California at San Francisco, San Francisco, CA) for pXGR14. The authors are grateful to Dr. Peter J. Sheridan (NIH, Bethesda, MD) for critical reading of this manuscript.
This work was supported in part by Grant-in-Aid for Scientific Research No. 11670020 and No. 13670003 from the Ministry of Education, Science, Sports and Culture, Japan.
Abbreviations:
- DEX,
Dexamethasone;
- GHRH-R,
GHRH receptor;
- GRE,
glucocorticoid response element;
- GST,
glutathione S-transferase;
- RARE,
RA response element;
- TRE,
T3 response element.
References
Miller TL, Mayo KE, Growth hormone-releasing hormone (GHRH) receptor mRNA regulation in rat pituitary cells. Program of the 79th Annual Meeting of The Endocrine Society, Minneapolis, MN, 1997 (Abstract P1–79), p 154

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}