
Abstract
Background: Physiologically based pharmacokinetic (PBPK) modelling can assist in the development of drug therapies and regimens suitable for challenging patient populations such as very young children. This study describes a strategy employing PBPK models to investigate the intravenous use of the neuraminidase inhibitor oseltamivir in infants and neonates with influenza.
Methods: Models of marmoset monkeys and humans were constructed for oseltamivir and its active metabolite oseltamivir carboxylate (OC). These models incorporated physicochemical properties and in vitro metabolism data into mechanistic representations of pharmacokinetic processes. Modelled processes included absorption, whole-body distribution, renal clearance, metabolic conversion of the pro-drug, permeability-limited hepatic disposition of OC and age dependencies for all of these processes. Models were refined after comparison of simulations in monkeys with plasma and liver concentrations measured in adult and newborn marmosets after intravenous and oral dosing. Then simulations with a human model were compared with clinical data taken from intravenous and oral studies in healthy adults and oral studies in infants and neonates. Finally, exposures after intravenous dosing in neonates were predicted.
Results: Good simulations in adult marmosets could be obtained after model optimizations for pro-drug conversion, hepatic disposition of OC and renal clearance. After adjustment for age dependencies, including reductions in liver enzyme expression and renal function, the model simulations matched the trend for increased exposures in newborn marmosets compared with those in adults. For adult humans, simulated and observed data after both intravenous and oral dosing showed good agreement and although the data are currently limited, simulations in 1-year-olds and neonates are in reasonable agreement with published results for oral doses. Simulated intravenous infusion plasma profiles in neonates deliver therapeutic concentrations of OC that closely mimic the oral profiles, with 3-fold higher exposures of oseltamivir than those observed with the same oral dose.
Conclusions: This work exemplifies the utility of PBPK models in predicting pharmacokinetics in the very young. Simulations showed agreement with a wide range of observational data, indicating that the processes determining the age-dependent pharmacokinetics of oseltamivir are well described.
Similar content being viewed by others
Background
Oseltamivir is a neuraminidase inhibitor indicated for the treatment and prophylaxis of influenza A and B infections. The phosphate pro-drug is given orally, after which it is readily absorbed from the gastrointestinal tract and extensively converted to the active metabolite oseltamivir carboxylate (OC).[1] There is an unmet need for intravenous neuraminidase inhibitor regimens suitable for use in patients with influenza who cannot tolerate, swallow or absorb orally administered medication. Such patients include the critically ill and the very young. In recognition of this need and in light of current public health concerns,[2] preclinical development work is being undertaken to explore and support the intravenous use of oseltamivir.
Part of this effort involves physiologically based pharmacokinetic (PBPK) modelling. Such models assist the development of therapies and regimens suitable for challenging patient populations by integrating wide-ranging physiological and biochemical factors and capturing the complex relationships between them. PBPK models can be used to extrapolate the drug pharmacokinetics to new situations and populations,[3] and have been recognized as an important means of bridging adult and paediatric studies and for predicting doses and drug exposures in children and infants.[4]
The present report describes, for the first time, mechanistic PBPK models that simulate the pharmacokinetics of oseltamivir and its active metabolite OC after oral and intravenous dosing. The models integrate measured physicochemical properties and in vitro data and simulate drug concentration-time profiles in plasma and tissues. In addition to the human model, a model for marmoset monkeys has been created and model assumptions have been verified by comparison of simulations with pharmacokinetic data obtained in a pharmacokinetic study in adult and newborn marmosets.
Although population models characterizing the pharmacokinetics of oseltamivir and OC in humans have been reported previously,[5] the present work represents the first attempt to use PBPK models to describe the pharmacokinetics of oseltamivir and its metabolite in both animals and humans, with an emphasis on potential use in the very young. Issues addressed include (i) the suitability of marmosets to represent age-dependent changes in human pharmacokinetics; (ii) the sensitivity of oseltamivir and OC concentrations in vivo to age-related changes in metabolism and renal function; and (iii) the suitability of data obtained via the oral route to represent exposures after intravenous infusion.
Methods
Modelling Strategy
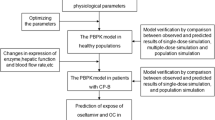
A strategy for prediction of human pharmacokinetics, using PBPK models verified by comparison of simulations with observed data in pre-clinical species, has been developed by our group.[6] This strategy has been successfully applied to predict the pharmacokinetics of many recent Roche phase I drug candidates, and an extension to include extrapolation to juveniles has been developed in the work described herein (figure 1). In this strategy, the first step is the simulation of pharmacokinetics in the adult animal species. Comparison with observed in vivo data may lead to refinement of the model if any significant mismatches are seen. When a reasonable simulation of pharmacokinetics in the adult animal is obtained, prediction of human pharmacokinetics will proceed using a model informed by the refinements that were needed in the animal model. For prediction of juvenile pharmacokinetics in humans, the same principle is followed. First, a juvenile animal model is developed, which accounts for age-dependent changes that are known to affect the pharmacokinetics of the drug, and the model is verified by comparison with data obtained in a juvenile animal study. Then the prediction in juvenile humans is made using a PBPK model that accounts for the age dependencies in humans and is informed by the refinements that were developed for adult humans and juvenile animals.

Physiologically based pharmacokinetic modelling strategy employed to predict exposure in human neonates and infants. A stepwise approach is followed with verification against in vivo data at each step. Simulations in juveniles are based on a model incorporating age dependencies in physiology and incorporating data from relevant in vitro systems. Verification in juvenile animals allows for model refinement before prediction in children. PK = pharmacokinetics.
Physicochemical and In Vitro Input Data
The key measured input data for oseltamivir and OC are listed in table I.

Physicochemical and in vitro data used in the physiologically based pharmacokinetic models for marmosets and humans
For scaling of hepatic metabolic conversion in marmosets with age, intrinsic clearance (CLint) in S9 sub-cellular fractions from 1-day-old marmosets was measured and showed 13-fold lower activity than in adults (table I). For humans, a similar trend for lower activity of the human carboxylesterase responsible for oseltamivir cleavage, HCE1, has been reported.[7,8]
Physiologically Based Pharmacokinetic (PBPK) Model Structures
PBPK models for disposition and absorption were created in GastroPlus™ version 6.1 (Simulations Plus, Inc., Lancaster, CA, USA). Brief model descriptions are provided below, while full model details are provided in Supplemental Digital Content 1 (available online at http://links.adisonline.com/CPZ/A18).
Absorption
Absorption was predicted with the advanced compartmental and transit (ACAT) model.[9] Physiological values for intestinal volumes, transit times, lengths and pH in humans and marmosets were taken from published data.[10–12] For age dependency, gut size was scaled down with body weight and intestinal transit times in children were incorporated,[13] while intestinal pH, permeability and all other factors were assumed to be the same in children and adults. In the newborn marmoset model, transit times, regional lengths and radii were scaled down on the basis of the relationship between human adults and neonates.
Whole-Body Disposition
Separate whole-body models were constructed for the disposition of oseltamivir and OC, and these were linked through the hepatic conversion, allowing simulation of both oseltamivir and OC concentrations after a dose of oseltamivir. Age-dependent human disposition models were generated using the Population Estimates for Age-Related Physiology (PEAR) module in GastroPlus™, on the basis of body weight, height, body mass index and bioelectrical impedance data from the National Health and Nutrition Examination Survey.[14] The mean data used to estimate height, body weight and tissue/organ perfusion were based on those published by Price et al.[15] and Haddad.[16] For this study, the simulated adult was an 86 kg, 30-year-old man and the infant was a 10 kg 1-year-old. A model for a 4 kg neonate was extrapolated using the PEAR module.
The disposition model for the marmoset was based on in-house and published data.[12,17,18] Adult marmoset body weight was 400 g. Tissue perfusions and volumes for newborn marmosets were based on the ratios determined from human neonates and adults. This scaling resulted in a body weight of 30 g for the newborn marmoset, in good agreement with measured values, which ranged from 24 g to 34 g.
Scaling of In Vitro Metabolism and Permeability Data
Scaling of in vitro metabolism to the in vivo situation was based on liver size and on measurements of the amount of S9 protein in 1 g of liver. The GastroPlus™ absorption model requires scaling of in vitro permeability to in vivo permeability, and this was achieved on the basis of correlations to measured human jejunal permeability for reference drugs.[19]
Tissue Distribution
For the lipophilic pro-drug oseltamivir, well stirred perfusion-limited compartments were used to model distribution into tissues. For OC, although tissue permeability is low and likely to limit the distribution rate, the tissue concentration data needed to parameterize models for such behaviour were not available for most tissues, and well stirred models were therefore used with partition coefficient (Kp) values predicted using published tissue composition-based equations.[20–22]
Summing the contributions of all tissues in the body predicts the steady-state volume of distribution (Vss) [equation 1]:
where Vp is the plasma volume; Ve is the erythrocyte volume; Rep is the erythrocyte : plasma concentration ratio; VT is the tissue volume; KpT is the tissue/plasma partition coefficient; and ERT is the extraction ratio in the tissue.
Altered extracellular space in neonates[23] was accounted for by increasing the Vss of OC by 70%.
Hepatic Disposition
Because of the importance of hepatic disposition in the pharmacokinetics of OC, a well stirred model (such as was applied for all other tissues) was not appropriate for the liver, and so a multi-compartment permeability-limited liver model was used. This is described by equation 2:
where Cb is the concentration in blood entering tissue; Q is the liver blood flow; Kp is the partition coefficient for the extracellular space; Rbp is the blood : plasma concentration ratio; Ce is the drug concentration in the extracellular space; subscript u denotes the unbound concentration; CT is the drug concentration in the intracellular space; Vv, Ve and VT are the volumes of the vascular, extracellular and intracellular compartments, respectively; PSTC is the permeability and surface area product for liver cells; and CLint,u is unbound intrinsic clearance.
Kp was calculated as (equation 3):
where fu,p is the fraction unbound in plasma and fu,T is the fraction unbound in tissue. fu,T was estimated from fu,p, assuming that the drug binds solely to albumin in the plasma and extracellular spaces.[6]
PSTC represents a permeability limitation and is the product of the drug-specific cellular membrane permeability and the surface area available for transfer between the systemic circulation and the hepatocytes. PSTC was optimized to fit the plasma profile of OC in adult marmosets and was then scaled across ages and species, using an allometric relationship based on liver weight (LW) [e.g. equation 4 was used to scale from marmoset to human]:
Assuming that the drug-specific permeability is constant, this relationship would represent how the extracellular space to cellular surface area changes with liver size. The limiting range for this exponent can be estimated on the basis of geometric considerations. If it is assumed that all liver cells are bathed in plasma and that the number of cells in the liver is directly proportional to the liver weight, then this exponent should be 1.0. Alternatively, a lower limit can be estimated from the change in the liver surface area with liver volume which, assuming a spherical geometry, would be 0.67.[24] In this modelling, an exponent between these two limits, 0.8, was used for scaling of PSTC both between and within species.
Renal Clearance
Published data on age-dependent changes in glomerular filtration and in renal clearance of para-aminohippuric acid (a substance that is filtered and actively secreted by the renal tubules) show a similar trend for maturation of renal function in human infants.[23] On the basis of these data, a 10-fold reduction in renal clearance in newborns versus adults was assumed for both humans and monkeys.
Pharmacokinetic Data: Marmosets
The animal data used in this work were based on a single-dose study in adult and newborn marmosets. Aqueous solutions of oseltamivir were administered by oral gavage at nominal doses of 2 and 10 mg/kg and by slow intravenous bolus at 5 mg/kg. Four adults and four newborns 1–3 days old were used for each dose. Blood samples were taken at 0.5, 1, 2, 5 and 8 hours post-dose (oral), and at 0.083, 0.25, 1, 5 and 8 hours post-dose (intravenous).
Plasma samples were assayed by a validated liquid chromatography-mass spectrometry method with a lower limit of detection of 0.1 ng/mL for both oseltamivir and OC.
Plasma concentration-time profiles were analysed by using WinNonlin Professional version 5.2 (Pharsight Corporation, Palo Alto, CA, USA). For the reporting of pharmacokinetic parameters, the maximum (peak) plasma concentration (Cmax) was determined directly from the plasma concentration-time profiles, while the area under the plasma concentration-time curve (AUC) was calculated non-compartmentally, using the linear trapezoidal method. For the AUC from time zero to infinity (AUC∞), the apparent terminal elimination rate was obtained by log-linear regression of the terminal phase of the plasma concentration-time curve with extrapolation from the observed concentration at the last time point.
Pharmacokinetic Data: Humans
Pharmacokinetic data in adult humans were based on intravenous doses of OC 150 mg and oseltamivir 15 mg given over 1 hour in six healthy male subjects.[5] Oral data were obtained after a single dose of oseltamivir 100 mg in six healthy male subjects.[5]
For human infants and neonates, few data are available, although preliminary results are emerging from two ongoing trials.[25] CASG114 is a study where nominal doses of 3 mg/kg were given every 12 hours orally for 5 days to infants aged less than 2 years with confirmed influenza infection. CASG119 is a study in neonates (many of whom were preterm) of postnatal age 1.5–17.5 weeks who received 12-hourly oral doses of 1.7 mg/kg for 5 days. These babies had been exposed to pandemic influenza A (H1N1) and were receiving oseltamivir prophylactically.[25]
Results
Simulations in adult marmosets were based on measured physicochemical and in vitro inputs, as described in the Methods section. Tissue distribution in monkeys was predicted using published equations based on human tissue composition. For oseltamivir, a lipophilic basic molecule, the predicted tissue partition coefficients are mostly greater than 1 and result in a predicted Vss of 1.9 L/kg. OC is a hydrophilic acid, which is mostly ionized at physiological pH and has poor penetration across lipid membranes. The predicted tissue partition coefficients are all less than 1 and result in a Vss of 0.5 L/kg. Both of these predicted volumes were within the range of the observed values in monkeys.
Absorption of oseltamivir in monkeys was predicted using the human jejunal permeability scaled from in vitro studies. This was 1.5 × 10−4 cm/s, a relatively high permeability similar to that seen with metoprolol.
After comparison of initial simulations with observed data, further model refinements were made as follows:
-
(i)
Renal clearance based on glomerular filtration of the unbound drug produced simulations that underestimated the amounts of oseltamivir and OC measured in urine. Therefore, renal clearances were increased to match the observed urinary excretion. This gave values of unbound renal clearance more than 6-fold higher than the glomerular filtration rate (GFR) in the marmosets, indicating significant active secretion.
-
(ii)
Metabolic conversion of oseltamivir to OC in the liver, scaled directly from in vitro data, led to an overestimate of the oseltamivir half-life. The scaled CLint was increased by a 4-fold factor.
-
(iii)
A perfusion-limited liver model for OC led to a profile that underestimated the observed half-life of OC. A permeability-limited liver model was introduced and the PSTC parameter was optimized to match the observed longer half-life.
After making these model adjustments, simulated and observed pharmacokinetic results for adult marmosets were in good agreement, as shown in table II and figure 2.

Observed and simulated exposures to oseltamivir and its active metabolite oseltamivir carboxylate (OC) after intravenous (IV) or oral (PO) dosing in adult and newborn marmosets

Simulated plasma concentration-time profiles (using the physiologically based pharmacokinetic model) and observed plasma concentrations of oseltamivir and oseltamivir carboxylate (OC) in (a) adult marmosets (n = 4) after intravenous dosing; (b) juvenile marmosets (n = 4) after intravenous dosing; (c) adult marmosets (n = 4) after oral dosing; and (d) juvenile marmosets (n = 4) after oral dosing.
Simulations for newborn marmosets allowed for reduced renal function in newborns and included hepatic metabolic conversion scaled from in vitro data that were relevant for newborns with the additional 4-fold factor as needed in adults. Simulated profiles (figure 2) and estimated AUCs (table II) matched well the observed trend for increased oseltamivir exposures in newborns compared with adults after the same intravenous or oral doses. However, the observed exposures of OC in newborns were higher than the simulated values, particularly at the highest oral dose. A more than dose-proportional increase in exposures was observed between the two oral doses. A larger underestimation of the Cmax of oseltamivir in newborn marmosets than in adult marmosets was also found, and a model sensitivity analysis showed that the simulations of the Cmax and the time to reach the Cmax (tmax) of oseltamivir and OC could both be improved by increasing intestinal permeability in the juvenile model.
As further verification of the hepatic disposition model, the simulated concentrations of OC in the livers of adult and newborn marmosets at the 8-hour time point were compared with measured values. The liver/plasma partition coefficients simulated with the model were all within 2-fold of the observed values and captured well the trends seen in the measurements, where values for OC were significantly higher in adults (observed: 36–48; simulated: 64) than in newborns (observed: 4–7; simulated: 7).
Simulations in adult humans were made with the PBPK model, including metabolic conversion scaled from in vitro data (table I) and the 4-fold correction factor that had been found necessary in monkeys. The liver permeability parameter, PSTC, was scaled from monkeys to humans on the basis of liver weight to the power of 0.8, and renal clearances of oseltamivir and OC were adjusted to match the reported urinary excretion in humans.
Simulated plasma profiles for 150 mg OC given by 1-hour intravenous infusion were compared with published data[5] and were in reasonable agreement, with a simulated Cmax of 3700 ng/mL (observed range 4000–5700 ng/mL) and a simulated AUC of 9400 ng · h/mL (observed range 7300–11 400 ng · h/mL). Simulated profiles for a 15 mg intravenous infusion and a 100 mg oral dose of oseltamivir are compared with observed data in figure 3 and table III. These simulations are close to the observed data, indicating that the model appropriately describes the processes determining the human pharmacokinetics of oseltamivir and OC in adults.

Simulated plasma concentration-time profiles (using the physiologically based pharmacokinetic model) and observed plasma concentrations of oseltamivir and oseltamivir carboxylate (OC) in adult human male subjects (a) after a 15 mg intravenous infusion of oseltamivir and (b) after a 100 mg oral dose of oseltamivir.

Observed and simulated exposures to oseltamivir and its active carboxylate metabolite (OC) after intravenous (IV) or oral (PO) dosing of oseltamivir in six healthy adult human males
Model sensitivity analyses showed that OC release from hepatocytes is rate limiting for plasma concentrations after either intravenous or oral dosing. Thus, the Cmax of OC was sensitive to changes in PSTC and insensitive to changes in hepatic CLint. For an oral dose, plasma concentrations of oseltamivir were sensitive to hepatic first-pass metabolism, with both the Cmax and AUC of oseltamivir affected by small changes in hepatic CLint.
Simulations for a 1-year-old infant were performed with 2-fold lower CLint than that in adults, based on in vitro measurements in infant livers,[7,8] and as renal function is reported to be close to maturity by this age,[23] renal clearances of oseltamivir and OC were scaled from adult values on the basis of body surface area. Pharmacokinetic data pertaining to human infants remain scarce; however, it was recently reported that an oseltamivir dose of 3 mg/kg, when given to infants aged <2 years, resulted in an AUC from 0 to 12 hours (AUC12) of 4300 ± 1900 ng · h/mL for OC.[25] The simulated steady-state AUC12 is 5700 ng · h/mL, which is within the reported range.
For simulations in newborns, 10-fold lower metabolic activity than adults was incorporated and, because of the immature renal function, urinary clearances of oseltamivir and OC were scaled to one-tenth of the adult values when based on body surface area.
As reported,[25] a dose of 1.73 mg/kg oseltamivir, when given to premature neonates in study CASG119, resulted in a steady-state AUC12 of 9250 ng · h/mL for OC. The simulated steady-state AUC12 values for this dose were 23 000 ng · h/mL for OC and 620 ng · h/mL for oseltamivir.
Investigations using liver samples from children have shown a large interindividual variability in the expression of hCE1, which is responsible for metabolism of oseltamivir.[7,8] The possible impact of such variability on plasma concentrations in neonates was explored by running simulations where hepatic CLint was varied. OC concentrations in neonates remained relatively insensitive to variability in hepatic metabolism of oseltamivir while, as seen with the adult model, oseltamivir exposures were affected by variation in hepatic first pass. Such sensitivity was obviously not present for the intravenous route.
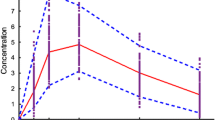
Finally, to compare the exposures via the intravenous and oral routes in neonates, simulations were performed for a 2 mg/kg twice-daily intravenous infusion (figure 4). These delivered exposures of OC the same as those achieved with a 2 mg/kg twice-daily oral dose. In these simulations, the oseltamivir Cmax and AUC were both 3-fold higher after infusion than when given orally, because of the lack of hepatic first pass for the intravenous route.

Simulated plasma concentration-time profiles (using the physiologically based pharmacokinetic model) of oseltamivir and oseltamivir carboxylate (OC) in neonates after administration of 2 mg/kg twice daily for 5 days, given either as 2-hour intravenous (IV) infusions or as oral doses (PO).
Discussion
A strategy for the application of PBPK models to predict pharmacokinetics in neonates and infants has been developed and applied to oseltamivir. The PBPK model integrated drug-specific knowledge, including physicochemical properties, solubility and permeability, blood cell and plasma protein binding, and in vitro data on the metabolism of oseltamivir by HCE1. Simulations of plasma and tissue concentrations after intravenous and oral dosing were made for both oseltamivir and its active metabolite by means of whole-body models linked through hepatic metabolic conversion.
The strategy employed for prediction of human pharmacokinetics involved model verification in the marmoset monkey, a species chosen because its pharmacokinetic behaviour is similar to that of humans, with the liver being the major site of metabolism. A PBPK marmoset model was constructed, and initial simulations with this model showed that a number of refinements were needed.
First, the assumption that renal clearance is determined by filtration led to underestimation of the amounts eliminated in urine for both oseltamivir and OC. Unbound renal clearances in marmosets are more than 6-fold higher than the GFR, indicating that significant secretion of both molecules occurs. Furthermore, it has been reported that OC is a substrate of transporters expressed on the basolateral membrane of kidney epithelia, organic anion transporter (OAT)-1 and OAT3[26,27] and also of multidrug resistance-associated protein (MRP)-4,[26] a transporter expressed on the apical membrane. Although these three transporters have all been reported to have high expression levels in the kidneys of both monkeys and humans,[28] methods for scaling of in vitro data to the in vivo situation have not been established. In the absence of a mechanistic model for active renal secretion, an empirical approach was taken and renal clearances were adjusted to match the urinary excretion.
A second adjustment of the model was needed when direct physiological scaling of in vitro data measured in S9 fractions led to a conversion rate of oseltamivir to OC that was too slow to match the in vivo profile of oseltamivir. It was necessary to increase this conversion rate by an additional 4-fold factor to match the observed plasma half-life. The reason that this correction factor is needed is unclear but might involve an underestimation of the amount of S9 protein in the liver.
A third refinement was introduced because a perfusion-limited liver model was not able to deliver a reasonable simulation of the plasma profile of OC. The poor in vitro permeability of OC suggested that escape from the liver would be rate limiting, and the liver disposition model was modified accordingly. As a priori prediction of the model parameter PSTC (a combination of the permeation of OC across hepatocyte cell membranes and the available surface for exchange between blood and hepatocytes) was not possible, its value was optimized to match the observed OC plasma concentrations.
These model refinements were made on the basis of the in vivo data in adult marmosets and subsequently needed to be scaled across species to adult humans and with age to neonates. Mechanistic scaling of active renal secretion between monkeys and humans again proved to be problematic because of lack of quantitative methods for transporter scaling. Unbound renal clearances were higher than the GFR in both species, indicating that significant secretion of both molecules must occur. However, although the use of the ratio of GFRs between rats and humans has been proposed as a method to scale renal clearance between species in such cases,[29] this approach did not work well to scale for oseltamivir or OC between monkeys and humans. The renal clearances in adult humans were therefore adjusted to match the measured urinary excretion, which gave unbound renal clearance 2-fold higher than the GFR of oseltamivir and 4-fold higher for OC. Verification of the scaling of renal clearance with age in marmosets was limited by lack of data on the ontogeny of renal processes in monkeys. On the basis of the development of renal function reported for human neonates,[23] 10-fold reduced renal function was assumed to apply also to marmosets. Although simulations showed that this gave the correct trend for increased plasma concentrations in neonates, the actual renal clearance appears to be lower than predicted and also saturable (this remains uncertain, as urine could not be collected in neonates). However, given the uncertainties of the monkey model, it was considered appropriate to retain the 10-fold reduction in humans without further refinement.
The approach for scaling of hepatic metabolism developed in adult monkeys worked well for simulations in adult humans. This method was also successful when the reduced CLint measured in vitro for newborn marmosets was incorporated and produced reasonable simulations. Also the use of an allometric relationship based on liver size proved appropriate to scale the optimized liver permeability parameter from adult monkeys to adult humans; furthermore, the modified liver model and scaled parameter gave reasonable estimations of the liver to plasma partition coefficient in both adult and newborn marmosets. The differences in liver partitioning with age were explainable as being due to the balance between OC metabolic production and permeability-limited release from the liver. Thus, in newborns, the reduced metabolic conversion from oseltamivir resulted in an OC liver partition coefficient that was roughly 10-fold lower than that seen in adults.
Modelling the absorption of oseltamivir in marmosets did not present major problems. High solubility and good permeability of oseltamivir led to simulated complete absorption of the doses in both adults and newborns. Nonetheless, simulations of oral profiles in newborns were improved (both the Cmax and tmax) if it was assumed that permeability was higher than in adults. One reason for this could be the reported higher paracellular permeability due to wider tight junctions in neonates.[30]
Overall, simulations in adult humans were found to agree well with observed data, indicating that the models capture the major processes driving the pharmacokinetics of oseltamivir and OC. Furthermore, early data after oral dosing in human infants are also in reasonable agreement with simulated profiles and although the observed AUC of OC in neonates was slightly more than 2-fold higher than predicted, it is noted that most of the babies in this study were pre-term, and this may have played a role. In spite of the greatly reduced HCE1 activity in neonates, therapeutic concentrations of OC are still achieved and the PBPK modelling shows that this is as expected when accounting for both the reduced HCE1 expression and reduced renal function. Moreover, the similarities between simulated intravenous and oral profiles in neonates support the relevance of oral studies for the intravenous dosing. Further data in infants after oral and intravenous dosing will be needed to confirm these preliminary conclusions.
Non-clinical oral safety studies in juvenile animals have shown that oseltamivir is devoid of specific behavioural and other central nervous system effects and has a high no-observed-effect level in these animals, with maximal exposures ≥200 times the efficacious concentrations of both oseltamivir and OC.[31] This PBPK modelling supports the relevance of these oral studies for intravenous dosing.
Paediatric doses of medication have traditionally been scaled from adult doses, using functions related to body size, but such simple allometric measures are questionable when complex processes in absorption and disposition need to be accounted for, and they often fail to predict exposure accurately, particularly in the very young.[32] Developing children show rapid changes in rates of organ maturation, blood flow, body composition and ontogeny of drug elimination and transport. Mechanistic modelling, as carried out in the present study, represents a more rational approach, as it integrates a wide variety of biological factors to predict drug absorption, distribution, metabolism and elimination. Examples of paediatric PBPK models have been published for drugs[33–36] and for chemical compounds to assess inadvertent exposure to unborn and breastfed infants[37] and children.[15,38] These PBPK models have allowed for the prediction of differences in pharmacokinetics between children and adults for several drugs. For example, Bjorkman[33] predicted drug disposition of theophylline and midazolam in infants and children; Edginton et al.[34] described scaling of pharmacokinetics from adults to children for paracetamol (acetaminophen), alfentanil, morphine and levofloxacin; and Johnson et al.[36] predicted clearance and its variability for 11 drugs in children and infants.
Regulatory authorities are increasingly focusing on the use of modelled scenarios to complement and rationalize clinical studies in children,[39] and PBPK modelling is one approach to optimize study design and minimize exposure of real patients.[4] By integrating available pre-clinical in vitro and in vivo data, clinical data and a detailed understanding of developmental pharmacology PBPK modelling can enhance the learning phase of the ‘learning-confirming’ cycle of clinical trials and optimize the collection of the data needed for efficacy and safety in special patient populations.[40]
Conclusions
The PBPK models discussed in this article have shown good agreement with observed data in adult and newborn animals and humans. This modelling enhances our understanding of the pharmacokinetic behaviour of oseltamivir in the very young and supports the use of oral safety studies as a precursor to the development of intravenous regimens for this patient group.
References
Davies BE. Pharmacokinetics of oseltamivir: an oral antiviral for the treatment and prophylaxis of influenza in diverse populations. J Antimicrob Chemother 2010; 65 Suppl. 2: ii5–10
Funk DJ, Siddiqui F, Wiebe K, et al. Practical lessons from the first outbreaks: clinical presentation, obstacles, and management strategies for severe pandemic (pH1N1) 2009 influenza pneumonitis. Crit Care Med 2010; 38(4): e30–7
Rowland M, Balant L, Peck C. Physiologically based pharmacokinetics in drug development and regulatory science: a workshop report (Georgetown University, Washington, DC, May 29–30, 2002). AAPS PharmSci 2004; 6(1): E6
Manolis E, Pons G. Proposals for model-based paediatric medicinal development within the current European Union regulatory framework. Br J Clin Pharmacol 2009 Oct; 68(4): 493–501
Rayner CR, Chanu P, Gieschke R, et al. Population pharmacokinetics of oseltamivir when coadministered with probenecid. J Clin Pharmacol 2008; 48(8): 935–47
Jones H, Parrott N, Jorga K, et al. A novel strategy for physiologically based predictions of human pharmacokinetics. Clin Pharmacokinet 2006; 45(5): 511–42
Yang D, Pearce RE, Wang X, et al. Human carboxylesterases HCE1 and HCE2: ontogenic expression, inter-individual variability and differential hydrolysis of oseltamivir, aspirin, deltamethrin and permethrin. Biochem Pharmacol 2009; 7(7): 238–47
Shi D, Yang D, Prinssen EP, et al. Surge in expression of carboxylesterase-1 during the post-neonatal stage enables a rapid gain of the capacity to activate the anti-influenza pro-drug oseltamivir. J Infect Dis 2011; 203(7): 937–42
Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev 2001; 50 Suppl. 1: S41–67
Dressman JB, Amidon GL, Reppas C, et al. Dissolution testing as a prognostic tool for oral drug absorption: immediate release dosage forms. Pharm Res 1998; 15(1): 11–22
de Zwart LL, Rompelberg CJM, Sips AJAM, et al. Anatomical and physiological differences between various species used in studies on the pharmacokinetics and toxicology of xenobiotics: a review of literature [report no. 623860010]. Bilthoven: Rijksinstitute voor Volksgezondheit en Milieu, 1999 [online]. Available from URL: http://www.rivm.nl/bibliotheek/rapporten/623860010.pdf [Accessed 2011 Jun 14]
Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res 1993; 10(7): 1093–5
Bodé S, Dreyer T, Greisen G. Gastric emptying and small intestinal transit time in preterm infants: a scintigraphic method. J Pediatric Gastroenterol Nutr 2004; 39(4): 378–82
National Center for Health Statistics (NCHS), Centers for Disease Control and Prevention (CDC). National Health and Nutrition Examination Survey data. Hyattsville, MD: CDC, 2010
Price PS, Conolly RB, Chaisson CF, et al. Modeling interindividual variation in physiological factors used in PBPK models of humans. Crit Rev Toxicol 2003; 33(5): 469–503
Haddad S. Characterization of age-related changes in body weight and organ weights from birth to adolescence in humans. J Toxicol Environ Health 2001; 64(6): 453–64
Brown RP, Delp MD, Lindstedt SL, et al. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health 1997; 13(4): 407–84
Sweeney RE, Langenberg JP, Maxwell DM. A physiologically based pharmacokinetic (PB/PK) model for multiple exposure routes of soman in multiple species. Arch Toxicol 2006; 80(11): 719–31
Parrott N, Lave T. Applications of physiologically based absorption models in drug discovery and development. Mol Pharmaceutics 2008; 5(5): 760–75
Rodgers T, Leahy D, Rowland M. Tissue distribution of basic drugs: accounting for enantiomeric, compound and regional differences amongst b-blocking drugs in rat. J Pharm Sci 2005; 94(6): 1237–48
Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci 2006; 95(6): 1238–57
Rodgers T, Rowland M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharmaceutical Research 2007; 24(5): 918–33
Kearns GL, Abdel-Rahman SM, Alander SW, et al. Developmental pharmacology: drug disposition, action, and therapy in infants and children. N Engl J Med 2003; 349(12): 1157–67
Kawai R, Mathew D, Tanaka C, et al. Physiologically based pharmacokinetics of cyclosporine A: extension to tissue distribution kinetics in rats and scale-up to human. J Pharmacol Exp Ther 1998; 287: 457–68
Acosta EP, Jester P, Gal P, et al. Oseltamivir dosing for influenza infection in premature neonates. J Infect Dis 2010; 202(4): 563–6
Ose A, Ito M, Kusuhara H, et al. Limited brain distribution of [3R,4R,5S]-4-acetamido-5-amino-3-(1-ethylpropoxy)-1-cyclohexene-1-carboxylate phosphate (Ro 64-0802), a pharmacologically active form of oseltamivir, by active efflux across the blood-brain barrier mediated by organic anion transporter 3 (Oat3/Slc22a8) and multidrug resistance-associated protein 4 (Mrp4/Abcc4). Drug Metab Dispos 2009; 37: 315–21
Hill G, Cihlar T, Oo C, et al. The anti-influenza drug oseltamivir exhibits low potential to induce pharmacokinetic drug interactions via renal secretion: correlation of in vivo and in vitro studies. Drug Metab Dispos 2003; 30(1): 13–9
Bleasby K, Castle JC, Roberts CJ, et al. Expression profiles of 50 xenobiotic transporter genes in humans and pre-clinical species: a resource for investigations into drug disposition. Xenobiotica 2006; 36: 963–88
Lin JH. Applications and limitations of interspecies scaling and in vitro extrapolation in pharmacokinetics. Drug Metab Dispos 1998; 26: 1202–12
Edginton AN, Fotaki N. Oral drug absorption in pediatric populations. In: Dressman JB, Reppas C, editors. Oral drug absorption. New York: Informa Healthcare, 2010: 108–26
Freichel C, Prinssen E, Hoffmann G, et al. Oseltamivir is devoid of specific behavioral and other central nervous system effects in juvenile rats at supratherapeutic oral doses. Int J Virol 2009; 5: 119–30
Johnson TN. The problems in scaling adult drug doses to children. Arch Dis Child 2008; 93(3): 207–11
Bjorkman S. Prediction of drug disposition in infants and children by means of physiologically based pharmacokinetic (PBPK) modelling: theophylline and midazolam as model drugs. Br J Clin Pharmacol 2005; 59(6): 691–704
Edginton AN, Schmitt W, Willmann S. Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin Pharmacokinet 2006; 45(10): 1013–34
Ginsberg G, Hattis D, Russ A, et al. Physiologically based pharmacokinetic (PBPK) modeling of caffeine and theophylline in neonates and adults: implications for assessing children’s risks from environmental agents. J Toxicol Environ Health 2004; 67(4): 297–329
Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet 2006; 45: 931–56
Gentry PR, Covington TR, Clewell HJ. Evaluation of the potential impact of pharmacokinetic differences on tissue dosimetry in offspring during pregnancy and lactation. Regul Toxicol Pharmacol 2003; 38(1): 1–16
Pelekis M, Gephart LA, Lerman SE. Physiological-model-based derivation of the adult and child pharmacokinetic intraspecies uncertainty factors for volatile organic compounds. Regul Toxicol Pharmacol 2001; 33(1): 12–20
Pons G. Expectations from PK-PD modelling and simulation in the evaluation of medicinal products in children [presentation]. European Medicines Agency Workshop on Modelling in Paediatric Medicines; 2008 Apr 14–15; London [online]. Available from URL: http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2009/11/WC500009643.pdf [Accessed 2011 Jun 14]
Hoppu K. Can we get the necessary clinical trials in children and avoid the unnecessary ones? Eur J Clin Pharmacol 2009; 65(8): 747–8
Acknowledgements
All authors were employees of F. Hoffmann-La Roche Ltd. when this work was carried out. They have no other conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Parrott, N., Davies, B., Hoffmann, G. et al. Development of a Physiologically Based Model for Oseltamivir and Simulation of Pharmacokinetics in Neonates and Infants. Clin Pharmacokinet 50, 613–623 (2011). https://doi.org/10.2165/11592640-000000000-00000
Published:
Issue Date:
DOI: https://doi.org/10.2165/11592640-000000000-00000