


Abstract
Human UDP-glucuronosyltransferase (UGT) 1A1 is a critical enzyme responsible for detoxification and metabolism of endogenous and exogenous lipophilic compounds such as bilirubin. The present study shows how cyclin-dependent kinase (CDK) inhibitor roscovitine stimulated the expression of UGT1A1 in HepG2 cells. Pregnane X receptor (PXR)-mediated transactivation of UGT1A1 reporter gene was more prominently enhanced by roscovitine, compared with the basal-, constitutive androstane receptor (CAR)-, and aryl hydrocarbon receptor-mediated activities. We determined the regulatory mechanism of UGT1A1 expression through PXR's stimulation by roscovitine. Although phosphomimetic mutations at Thr290 and Thr408 retained the PXR protein in cytoplasm and attenuated the induction of UGT1A1 expression by both roscovitine and rifampicin, a mutation at Ser350 specifically reduced the activity of PXR induced by roscovitine. Immunoprecipitation analysis revealed that the T290D but not T408D mutant protein remained in cytoplasm by forming a complex with heat shock protein 90 and cytoplasmic CAR retention protein, whereas treatment with proteasome inhibitor MG-132 accumulated the T408D mutant protein in cytoplasm. Transfection with anti-CDK2 small interfering RNA (siRNA) but not anti-CDK1 or CDK5 siRNA led to enhanced expression of UGT1A1. S350D yellow fluorescent protein-PXR fusion protein could translocate from cytoplasm to nucleus similar to the wild-type protein but was detected as an acetylated protein, whose binding with retinoid X receptor (RXR) and histone deacetylase was impaired. Cotransfection with coactivator steroid receptor coactivator (SRC) 2 but not SRC-1 partly recovered its PXR activity. These results indicate that roscovitine stimulated the expression of UGT1A1 by inhibiting CDK2, which phosphorylated PXR at Ser350 to suppress binding with RXR and coactivator and maintain the acetylation of PXR protein.
Introduction
The constitutive androstane receptor [CAR; nuclear receptor subfamily 1 (NR1), group I, member 3] and pregnane X receptor (PXR; NR1, group I, member 2) were originally characterized as nuclear hormone receptors that interact with a subset of retinoic acid response elements and have been recognized as xenobiotic-sensing nuclear receptors that transcriptionally regulate the expression of genes of phase I, II, and III metabolic enzymes and transporters involved in the metabolism and elimination of endogenous and exogenous substances such as bilirubin, steroid hormones, and xenobiotics (Timsit and Negishi, 2007). UDP-glucuronosyltransferase (UGT) 1A1 plays a critical role in the detoxification of potentially neurotoxic bilirubin by conjugating it with glucuronic acid for excretion in bile (Ostrow and Murphy, 1970) and conjugates drugs and other xenobiotics (Radominska-Pandya et al., 1999; Tukey and Strassburg, 2000). Reduced bilirubin glucuronosyltransferase (UGT1A1) activity is associated with the development of unconjugated hyperbilirubinemia (Crigler-Najjar syndrome and Gilbert's syndrome) (Mackenzie et al., 1997) and increased side effects of drug treatment such as the predisposition of patients to toxicity initiated by 7-ethyl-10-hydroxycamptothecin (SN-38), an active metabolite of the anticancer drug irinotecan (Gagné et al., 2002; Tukey et al., 2002). Understanding the molecular mechanisms of the induction of human UGT1A1 may provide information for the prevention and treatment of unconjugated hyperbilirubinemia and the side effects of drugs. We identified a phenobarbital-responsive enhancer module (PBREM) at −3499/−3210 from the transcription start site of UGT1A1, gtPBREM (Sugatani et al., 2001). gtPBREM is a composite 290-base pair element consisting of CAR, PXR, aryl hydrocarbon receptor (AhR), glucocorticoid receptor, peroxisome proliferator-activated receptor α, and nuclear factor erythroid-derived 2-related factor 2 response elements (Sugatani et al., 2001, 2004, 2005a; Yueh et al., 2003; Senekeo-Effenberger et al., 2007; Yueh and Tukey, 2007), and we demonstrated that the gtNR1 (−3382/−3367) within gtPBREM plays a central role in the expression of UGT1A1 mediated by both CAR and PXR (Sugatani et al., 2005b).
We previously demonstrated the following: 1) hepatocyte growth factor (HGF) increased mRNA and protein levels of UGT1A1 and CYP2B6 as well as the endogenous cyclin-dependent kinase (CDK) 2/4 inhibitors p16, p21, and p27 in HepG2 cells; 2) the CDK inhibitor roscovitine also enhanced the expression of UGT1A1, CYP2B6, and CYP3A4; and 3) transfection of anti-CDK2 small interfering RNA (siRNA) led to elevated levels of UGT1A1, CYP2B6, and CYP3A4 in HepG2 cells (Sugatani et al., 2010). Although HGF is a potent mitogen for hepatocytes, it is an antimitogenic factor for some tumor cell lines including HepG2; HGF induces p16 and p21 expression and decreases CDK2 activity, leading to inhibition of cell growth (Shima et al., 1998; Han et al., 2005). In addition, we found that CDK2 activity was decreased in HGF-treated HepG2 cells, and there was a clear dissociation among the activation of CDK2 and the expression of UGT1A1, CYP2B6, and CYP3A4. Thus, we concluded that the expression of UGT1A1 and CYP2B6 is negatively regulated through a CDK2 signaling pathway linked to cell-cycle progression in HepG2 cells. Lin et al. (2008) demonstrated that roscovitine activates PXR-mediated CYP3A4 gene expression through inhibition of CDKs in a ligand-independent manner and that CDK2 negatively regulates the activity of PXR in HepG2 cells. However, it remains to be fully elucidated how the expression of UGT1A1 is regulated through PXR or other nuclear receptors by roscovitine.
Thus, in this study, we investigated the nuclear receptor involved in the roscovitine-stimulated expression of UGT1A1 in HepG2 cells and found that PXR contributes to the expression, using cells expressing CAR, PXR, and AhR. It remains to be clarified whether the regulatory mechanism of UGT1A1 gene expression through PXR stimulation by roscovitine is different from that by rifampicin in a ligand-dependent manner. PXR activity has been shown to be modulated by translocation from the cytoplasm to nucleus, as well as the formation of heterodimers with retinoid X receptor (RXR) and interaction with cofactors in the nucleus (Squires et al., 2004; Timsit and Negishi, 2007). CAR and PXR form a complex with cytoplasmic CAR retention protein (CCRP; designated Dnajc7 in the National Center for Biotechnology Information database) and heat shock protein (Hsp) 90 to maintain the cytoplasmic localization (Kobayashi et al., 2003; Squires et al., 2004). Phosphorylation of PXR at specific amino acid residues and other posttranslational modifications modulate the cytoplasmic localization and the nuclear activation that affect the PXR activity (Lichti-Kaiser et al., 2009a; Pondugula et al., 2009; Staudinger et al., 2011). Here, we identified critical residues involved in the enhanced expression of UGT1A1 by roscovitine compared with those by rifampicin and investigated the role of the posttranslational modification in PXR activation leading to the induction of UGT1A1 expression.
Materials and Methods
Materials.
Roscovitine and N-(benzyloxycarbonyl)leucinylleucinylleucinal (MG-132) were purchased from Calbiochem (Darmstadt, Germany). Rifampicin and benzo[a]pyrene were obtained from Sigma-Aldrich (St. Louis, MO). Actinomycin D was obtained from P-L Biochemicals (Milwaukee, WI), ATP was obtained from Roche Diagnostics (Mannheim, Germany), and [γ-32P]ATP was obtained from PerkinElmer Life and Analytical Sciences (Waltham, MA). All other chemicals and solvents were of analytical grade and were obtained from commercial sources.
Plasmids.
Reporter plasmids containing the UGT1A1 −3570/−3180 fragment linked to the −181/−1 fragment-pGL4-tk-firefly luciferase have been described previously (Sugatani et al., 2008). The expression vector pcDNA3.1-human steroid receptor coactivator (SRC)-2 has also been described previously (Sugatani et al., 2005a). The human CAR, human PXR, and human SRC-1 expression vectors were generously provided by Dr. Masahiko Negishi (National Institute of Environmental Health Sciences, Research Triangle Park, NC). The human AhR and human AhR nuclear translocator (ARNT) expression vectors were from Dr. Yoshiaki Fujii-Kuriyama (University of Tokyo, Tokyo, Japan). Mutations of PXR were performed with a QuickChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer's instructions (Sugatani et al., 2005a). The following primers were used: 5′-CTATCACTTCAATGTCATGGCATGTGAAGGATGCAAGGG-3′, 5′-CTGGCTATCACTTCAATGTCATGGATTGTGAAGGATGCAAGGGCTTTTT-3′, 5′-CAGGGGTGCTTAGCGCTGGCTGCGAGTTGC-3′, 5′-CCAGGGGTGCTTAGCGATGGCTGCGAGTTGCC-3′, 5′-CTCTGCAGGCCCCAGCGAGGGAAGAAG-3′, 5′-CAGAGTCTCTGCAGGCCCCAGATAGGGAAGAAGCTGCC-3′, 5′-TCCGGAAAGATCTGTGCGCTTTGAAGGTCTCTCTG-3′, 5′-GGTCCGGAAAGATCTGTGCGATTTGAAGGTCTCTCTGCAG-3′, 5′-ACCCCCAGCCGACGCTGGCGGGAAAGAG-3′, 5′-CAAACCCCCAGCCGACGATGGCGGGAAAGAGATC-3′, 5′-CGAGGACCAGATCGCCCTGCTGAAGGG-3′, 5′-ATCGAGGACCAGATCGACCTGCTGAAGGGGGC-3′, 5′-GTCAACTGAGATTCAACGCAGTGTTCAACGCGGAG-3′, 5′-CTGTGTCAACTGAGATTCAACGATGTGTTCAACGCGGAGACTGGA-3′, 5′-GTGTGGCCGGCTGGCCTACTGCTTGGA-3′, 5′-GGAGTGTGGCCGGCTGGACTACTGCTTGGAAGAC-3′, 5′-CATCTCCCTCTTCGCCCCAGACCGCCC-3′, 5′-CCATCTCCCTCTTCGACCCAGACCGCCCAG-3′, 5′-CAATGCTCAGCACGCCCAGCGGCTGCT-3′, 5′-ATCAATGCTCAGCACGACCAGCGGCTGCTGCG-3′, and their complements for human PXR T57A, T57D, S180A, S180D, S192A, S192D, S208A, S208D, S231A, S231D, S274A, S274D, T290A, T290D, S305A, S305D, S350A, S350D, T408A, and T408D. Vectors for the expression of 1) pCR3-human CAR-ΔC8, 2) pCR3-human PXR-ΔC9, 3) glutathione transferase (GST)-human PXR, and 4) yellow fluorescent protein (YFP)-human PXR were constructed by inserting the corresponding cDNAs, which were amplified by polymerase chain reaction (PCR) with the following primers: 1) 5′-gtttccggatccATGGCCAGTAGGGAAGATGAGCT-2′ and 5′-gtttaactcgagTCACATCATGGCAGACAGG-3′; 2) 5′-gtttccggatccATGGAGGTGAGACCCAAAGAAAGC-3′ and 5′-gtttaactcgagTCACATGAGGGGCGTAGCAAAGGGGTG-3′; 3) 5′-ttcaagggatccATGGAGGTGAGACCCAAAGAA-3′ and 5′-gtcttcctcgagTCAGCTACCTGTGATGCCGAA-3′; and 4) 5′-ttcaagctcgagCTATGGAGGTGAGACCCAAAGAA-3′ and 5′-gtcttcggatccTCAGCTACCTGTGATGCCGAA-3′, respectively, into the BamHI and XhoI sites of pCR3 and pGEX-6P-3 (GE Healthcare Japan, Tokyo, Japan) and XhoI/BamHI sites of enhanced YFP (pEYFP)-C1 (Clontech, Mountain View, CA) digested with the same enzymes, respectively. Bases in lowercase letters were added to allow digestion of the oligonucleotides with the restriction enzymes. CAR-ΔC8 and PXR-ΔC9 were generated by the deletion of eight and nine amino acids from the C terminus, respectively. All expression vectors were sequenced by dye-terminator automated sequencing.
Cell Culture, Transfection, and Luciferase Assays.
Human liver-derived cells (HepG2 cells line; RIKEN BioResource Center, Tsukuba-shi, Ibaraki Japan) and SW480 human colon cancer cells (American Type Culture Collection, Manassas, VA) were seeded onto 24-well plates at 1 × 105 cells/ml in Dulbecco's modified Eagle's medium and RPMI 1640 medium supplemented with 10% fetal calf serum (HyClone; Thermo Fisher Scientific, Waltham, MA), respectively. Twenty-four hours later, HepG2 cells were transfected with UGT1A1-luciferase reporter constructs including the distal (−3570/−3180) and proximal (−181/−1) regions (Sugatani et al., 2008) (0.2 μg) and expression vectors [pCR3-CAR (0.2 μg), pCR3-CAR (0.2 μg), pCR3-PXR (0.2 μg), or pCR3-AhR (0.2 μg) and pCR3-ARNT (0.2 μg), and pRL-SV40 (0.2 μg)] using a calcium phosphate coprecipitation method (CellPhect Transfection Kit; GE Healthcare Japan). The medium (1 ml/well) was replaced after 12 h with the same medium. The cells were subsequently treated for 24 h with roscovitine (1 × 10−7 to 1 × 10−5 M) or rifampicin (5 × 10−6 M) plus roscovitine (1 × 10−7 to 1 × 10−5 M) as 400 times concentrated stocks in dimethyl sulfoxide (DMSO); controls received an equivalent volume of DMSO. The total amount of DNA transfected was held constant in each transfection by using the corresponding empty vector. Transfected cells were washed once in phosphate-buffered saline and harvested in 1 times passive lysis buffer, and luciferase activity was measured simultaneously with the Dual-Luciferase reporter assay system according to the manufacturer's instructions (Promega, Madison, WI); the firefly luciferase values were normalized to the Renilla values for each sample.
Quantitative Real-Time PCR.
HepG2 cells (2 × 105) seeded onto six-well plates and cultured for 24 h were transfected with expression vectors [pCR3 (0.2 μg), pCR3-CAR (0.2 μg), pCR3-CAR-ΔC8 (0.2 μg), pCR3-PXR (0.2 μg), pCR3-PXR-ΔC9 (0.2 μg), or pCR3-PXR mutant (0.2 μg)] or [pCR3 (0.2 μg), pCR3-PXR (0.2 μg), or pCR3-PXR mutant (0.2 μg) and pcDNA3.1 (0.2 μg), pcDNA3.1-SRC-1 (0.2 μg), or pcDNA3.1-SRC-2 (0.2 μg)] using TransIT-LT1 (Mirus Bio, Madison, WI) according to the manufacturer's instructions. At 24 h after transfection, cells were given fresh medium, further transfected with the expression vectors, and then cultured with roscovitine (5 μM) or vehicle for an additional 24 h unless otherwise stated; the transfection efficiency was increased by the repeat transfection compared with the single transfection [the level of PXR mRNA was increased 2.29 ± 0.02-fold, and the levels of UGT1A1 mRNA induced by roscovitine and rifampicin were increased 2.96 ± 0.15- and 3.44 ± 0.14-fold by the repeat transfection with pCR3-PXR expression vector, respectively (n = 4)]. Total RNA was extracted using TRIzol reagent from Invitrogen (Carlsbad, CA), and cDNAs were synthesized with a PrimeScript RT reagent kit (Takara Bio, Otsu, Japan) according to the instructions. cDNA synthesized from 100 ng of total RNA was subjected to quantitative real-time PCR with an ABI PRISM 7000 Sequence Detector (Applied Biosystems, Foster City, CA) using Premix Ex Taq reagent or SYBR Premix Ex Taq reagent (Takara Bio) for CDK1 (GenBank accession number NM_001786), CDK2 (GenBank accession number NM_001798), CDK5 (GenBank accession number NM_004935), UGT1A1 (GenBank accession number NM_000463), CYP3A4 (GenBank accession number NM_017460), and β-actin (GenBank accession number NM_001101) as described previously (Sugatani et al., 2010). The thermal cycle conditions were as follows: hold for 10 s at 95°C, followed by two-step PCR for 40 cycles of 95°C for 5 s followed by 60°C for 30 s. β-Actin was used to normalize gene expression in all samples. Fold-induction values were calculated by subtracting the mean difference of gene and β-actin cycle threshold Ct numbers for each treatment group from the mean difference of gene and β-actin Ct numbers for the vehicle group and raising the difference to the power of 2 (2−▵▵Ct).
Western Blot Analysis.
HepG2 cells (2 × 106) seeded into 75-cm2 flasks and cultured for 24 h were transfected with expression vectors [pCR3 (10 μg), pCR3-CAR (10 μg), pCR3-PXR (10 μg), pEYFP (10 μg), pEYFP-PXR (10 μg), or pEYFP-PXR mutant (10 μg)] using TransIT-LT1 (Mirus Bio) according to the manufacturer's instructions. At 24 h after transfection, cells were given fresh medium, further transfected with the expression vectors, and then cultured with roscovitine (5 μM) or vehicle for an additional 24 h unless stated otherwise. Treated and untreated cells were washed three times with ice-cold phosphate-buffered saline and lysed with a freshly prepared lysis buffer containing 50 mM Tris-HCl (pH 7.4), 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1 mM Na3VO4, 1 mM NaF, 1 mM benzamidine, 1 μg/ml aprotinin, and 1 μg/ml leupeptin at 4°C. The lysates were centrifuged at 110,000g for 10 min. Nuclear extracts and cytoplasmic fractions were prepared using a nuclear extract kit (Active Motif, Inc., Carlsbad, CA). The cytoplasmic fractions including microsomal proteins were used to investigate the expression of UGT1A1 and CYP3A4. The protein concentrations were determined by the Bradford assay (Bio-Rad Laboratories, Hercules, CA). Western blotting was performed as described previously (Osabe et al., 2008). Nuclear extracts, cytoplasmic fractions, or cell lysates (50 μg) were resolved on 12.5% SDS-polyacrylamide gels, and the proteins were transferred electrophoretically to polyvinylidene difluoride membranes (Millipore Corporation, Billerica, MA). Membranes were blocked at 4°C overnight in Blocking One (Nacalai Tesque, Kyoto, Japan) and probed for 1 h with primary antibodies including anti-UGT1A1 and anti-CYP3A4 from BD Gentest (Woburn, MA), anti-PXR, anti-RXR, and anti-histone H1 from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); anti-Hsp90, anti-acetylated lysine, and anti-histone deacetylases (HDACs) 1, 2, 3, 4, 5, and 7 from Cell Signaling Technology (Danvers, MA); anti-green fluorescent protein (GFP) from Medical Biological Laboratories (Nagoya, Japan); anti-CCRP from Abnova (Taipei, Taiwan); and anti-α-tubulin from Calbiochem. Antigen-antibody complexes were detected using the appropriate secondary antibody conjugated to horseradish peroxidase [horseradish peroxidase-conjugated anti-rabbit, anti-goat, or anti-mouse immunoglobulin (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA)] and visualized with an enhanced chemiluminescence system (GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Small Interfering RNA-Mediated Protein Knockdown.
siRNAs targeting human CDK1 (stealth RNAi), human CDK2 (validated stealth RNAi), and human CDK5 (stealth RNAi) were obtained from Invitrogen. SW480 cells cultured for 24 h were transfected with siRNA duplexes (100 nM) using TransIT-siQUEST (Mirus Bio) according to the manufacturer's instructions. At 24 h after transfection, cells were given fresh medium and further transfected with the siRNA duplexes for an additional 24 h unless stated otherwise.
In Vitro Kinase Assay.
GST-PXR and GST-PXR S350D fusion proteins were expressed in Escherichia coli BL21 [DE3(pLys)] and purified using GSH-Sepharose (GE Healthcare) according to the manufacturer's protocols. CDK2 kinase assays were performed as described previously (Sugatani et al., 2010). Cell lysates prepared from SW480 cells at 2 h after release from the thymidine block (Osabe et al., 2008) were cleared by centrifugation, and equal amounts of protein in the cell extracts (50 μg) were immunoprecipitated with 1 μg of anti-CDK2 antibody for 3 h at 4°C. Precipitated immune complexes were washed three times with the cell lysis buffer and twice with a kinase buffer [Tris-HCl (50 mM, pH 7.5) containing 10 mM MgCl2, 1 mM dithiothreitol, 20 mM EGTA, 10 mM β-glycerophosphate, 0.1 mM sodium orthovanadate, and 1 mM NaF]. Kinase reactions were performed in 10 μl of the kinase buffer containing 25 μg of GST-fused protein, 100 μM ATP, and 0.37 MBq of [γ-32P]ATP at 30°C for 30 min. Reactions were stopped by addition of 10 μl of two times Laemmli sample buffer [125 mM Tris-HCl (pH 6.8), 4% SDS, 20% glycerol, 0.002% bromphenol blue, and 10% 2-mecaptoethanol], and samples were resolved by SDS-polyacrylamide gel electrophoresis on a 5 to 20% gradient gel (ATTO Corporation, Tokyo, Japan) after heat denaturation. Phosphorylation of the substrate was visualized by autoradiography.
Immunocytochemistry.
To detect the localization of PXR and PXR mutants, HepG2 cells (6 × 103/well) seeded on three-well glass coverslips (Teflon printed glass slides; Erie Scientific Company, Portsmouth, NH) were transfected with expression vectors [pEYFP (0.2 μg), pEYFP-PXR (0.2 μg), or pEYFP-PXR mutant (0.2 μg)] using TransIT-LT1 (Mirus Bio) according to the manufacturer's instructions. At 24 h after transfection, cells were given fresh medium, further transfected with the expression vectors, and then were cultured with roscovitine (5 μM) or vehicle for an additional 24 h. Cells were fixed with methanol and nuclein was stained with 3 μM 4,6-diamidino-2-phenylindole-2-HC (DAPI; Dojin Laboratories, Kumamoto, Japan). Subsequently, cells were washed and mounted with Antifade reagent (Bio-Rad Laboratories). Fluorescence was visualized using a LSM510 confocal microscope (Carl Zeiss GmbH, Jena, Germany). For quantitation, the subcellular localization of YFP-PXR, YFP-PXR T290D, YFP-PXR S350D, and YFP-PXR T408D was scored in at least 100 cells.
Coimmunoprecipitation.
HepG2 cells (2 × 106) seeded into 75-cm2 flasks and cultured for 24 h were transfected with expression vectors [pEYFP (10 μg), pEYFP-PXR (10 μg), or pEYFP-PXR mutant (10 μg)] using TransIT-LT1 (Mirus Bio) according to the manufacturer's instructions. At 24 h after transfection, cells were given fresh medium, further transfected with the expression vectors, and cultured with roscovitine (5 μM) or vehicle for an additional 24 h. The nuclear extract (280 μg) or cytoplasmic fraction (460 μg) was precleared with protein G-agarose beads and rabbit IgG (Santa Cruz Biotechnology) for 30 min at 4°C. YFP-tagged proteins were immunoprecipitated using the anti-GFP antibody or anti-CCRP antibody combined with the protein G-agarose beads. The agarose beads were washed three times with Dulbecco's phosphate buffer and subjected to SDS-polyacrylamide gel electrophoresis on a 5 to 20% gradient gel (ATTO Corporation). The proteins were transferred to an Immobilon-P Transfer Membrane (Millipore Corporation), and the blots were probed with antibodies as indicated.
Statistics.
Values are expressed as the mean ± S.E. All data were analyzed using a one-way analysis of variance. The statistical significance of differences between groups was analyzed using the analysis of variance or an unpaired t test. The level for statistically significant differences was set at p < 0.05.
Results
Enhancement of PXR-Mediated Transactivation of UGT1A1 Gene by the CDK Inhibitor Roscovitine.
Previously (Sugatani et al., 2010), we found that roscovitine markedly stimulated the expression of UGT1A1 mRNA and protein, but only slightly stimulated that of CYP1A1, CYP2B6, and CYP3A4 mRNA and protein, in HepG2 and SW480 cells. To investigate the transactivation mechanism for UGT1A1 expression induced by roscovitine, we investigated the transcriptional activities of a distal (−3570/−3180) enhancer motif linked to a proximal (−185/−1) promoter motif placed in front of the reporter luciferase gene in HepG2 cells in the presence of exogenously expressed CAR, PXR, or AhR/ARNT. PXR-mediated transactivation of the reporter gene in the absence of rifampicin was enhanced by roscovitine in a dose-dependent manner and peaked at 5 μM roscovitine (Fig. 1). The extent of PXR-mediated transactivation by 5 μM roscovitine was almost the same as the extent of that by 5 μM rifampicin. The PXR-mediated transcriptional activity of the reporter gene in the absence of rifampicin with 5 μM roscovitine (3.3 ± 0.1-fold the control) was more prominently enhanced, compared with the basal activity (1.9 ± 0.1-fold the control), CAR-mediated activity (1.4 ± 0.1-fold the control), PXR-mediated activity in the presence of 5 μM rifampicin (1.1 ± 0.0-fold the control), and AhR/ARNT-mediated activity in the absence and presence of benzo[a]pyrene (1.1 ± 0.0- and 1.4 ± 0.0-fold the control, respectively) (Fig. 1).

Effect of roscovitine on the transcriptional activities of the UGT1A1 distal (−3570/−3180) enhancer motif and proximal (−181/−1) promoter motif mediated by CAR, PXR, or AhR/ARNT, assessed by expression of a reporter gene encoding firefly luciferase. HepG2 cells were cotransfected with expression plasmids for CAR (B), PXR (C and D), AhR/ARNT (E and F), or the control vector pCR3 (A), as well as the pGL4-thymidine kinase-firefly luciferase vector reporter gene (open bar) or the UGT1A1 reporter plasmid (closed bar). The transfected cells were treated with vehicle (DMSO) (C and E), 5 μM rifampicin (RIF) (D), 1 μM benzo[a]pyrene (B[a]P) (F), and then roscovitine at the indicated concentration for 24 h and were harvested and assayed for luciferase activity. Luciferase activity was measured using the dual-luciferase reporter assay system. Transcriptional activity of the UGT1A1 reporter gene in the absence of roscovitine was calculated as one. Data presented are the average for three independent experiments ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001 roscovitine-treated UGT1A1 reporter gene group versus vehicle-treated UGT1A1 reporter gene group.
Moreover, to evaluate the role of PXR in the induction of UGT1A1 expression by roscovitine, the basal, CAR-, or PXR-expressing plasmid was transfected into HepG2 cells. The mRNA level of UGT1A1 in the cells expressing exogenous PXR was markedly enhanced by roscovitine (52.2 ± 7.2-fold), compared with levels in the control cells and the cells expressing CAR (8.7 ± 1.8- and 2.7 ± 0.2-fold, respectively), and that of CYP3A4 in the cells expressing PXR was also enhanced by roscovitine (3.6 ± 0.6-fold) but not in the cells expressing CAR (Fig. 2, A and B). The induction of UGT1A1 and CYP3A4 mRNA expression by PXR stimulated by roscovitine was abolished by the deletion of eight and nine amino acids from the C terminus of CAR (CAR-ΔC8) and PXR (PXR-ΔC9), respectively. Furthermore, the protein levels of UGT1A1 and CYP3A4 were enhanced by roscovitine in the cells that expressed PXR. The levels of UGT1A1 and CYP3A4 were also enhanced in the cells expressing CAR, but not by roscovitine (Fig. 2C).

Effect of roscovitine on expression of UGT1A1 and CYP3A4 in HepG2 cells expressing CAR or PXR. HepG2 cells cultured for 24 h were transfected with expression plasmids for CAR, CAR-ΔC8, PXR, and PXR-ΔC9 or the control vector pCR3. At 24 h after transfection, the medium was replaced, and the cells were further transfected and then cultured with roscovitine (5 μM, closed bar) or vehicle (DMSO, open bar) for an additional 24 h. In A and B, the mRNA levels in cells transfected with pCR3 and treated with vehicle were taken as one. Data presented are the average ± S.E. for three experiments. ***, p < 0.001 versus vehicle-treated cells; ###, p < 0.001 versus CAR or PXR-expressing cells. In C, protein levels were determined by Western blotting.
Effects of Phosphomimetic and Phosphodeficient Mutations of PXR on UGT1A1 Expression Stimulated by Roscovitine and Rifampicin.
To determine whether the regulatory mechanism of UGT1A1 expression through PXR stimulation by roscovitine is different from that by the PXR ligand rifampicin, we were prompted to investigate effects of phosphomimetic mutations of PXR on UGT1A1 and CYP3A4 expression stimulated by roscovitine and rifampicin. Thr57, Ser180, Ser192, Ser208, Ser230, Ser274, Thr290, Ser305, Ser350, and Thr408 were selected for mutagenesis, on the basis of in silico consensus kinase site predictions and the reports by Lichti-Kaiser et al. (2009a) and Pondugula et al. (2009). As shown in Figs. 3 and 4, phosphomimetic mutations at Thr57, Thr290, Ser350, and Thr408 attenuated the induction of UGT1A1 and CYP3A4 mRNA expression by roscovitine. Although it was not easy to determine the residues associated with the induction of CYP3A4 mRNA by roscovitine and rifampicin, it resulted from UGT1A1 mRNA being more stable than CYP3A4 mRNA (Fig. 5). Thus, we determined the residues associated with PXR transactivation capacity for UGT1A1 expression by roscovitine and rifampicin. Phosphomimetic mutations at Thr57, Ser274, Thr290, Ser305, Ser350, and Thr408 reduced the induction of UGT1A1 mRNA expression by rifampicin to below 0.5-fold that by the wild-type PXR (Figs. 3 and 4). Both the phosphomimetic mutation and the phosphodeficient mutation at Thr57 attenuated PXR activation by rifampicin and roscovitine (Fig. 4). The S350D mutation attenuated the activation by roscovitine, whereas the phosphodeficient mutation increased it (Fig. 4).

Transactivation capacity of UGT1A1 (A) and CYP3A4 (B) by phosphomimetic mutant PXR proteins. HepG2 cells cultured for 24 h were transfected with expression vectors encoding wild-type or mutant PXR or the control vector pCR3. At 24 h after transfection, the medium was replaced, and the cells were further transfected and then cultured with roscovitine (5 μM, closed bar), rifampicin (5 μM, hatched bar), or vehicle (DMSO, open bar) for an additional 24 h. The mRNA levels are expressed by taking the control value obtained from the pCR3-transfected and vehicle-treated cells as one. Data presented are the average ± S.E. of 4 to 11. *, p < 0.05; **, p < 0.01; ***, p < 0.01 versus rifampicin-stimulated wild-type PXR; #, p < 0.05; ##, p < 0.01; ###, p < 0.01 versus roscovitine-stimulated wild-type PXR.

Relative transactivation capacity of UGT1A1 by phosphodeficient (A and C) and phosphomimetic (B and D) mutant PXR. HepG2 cells cultured for 24 h were transfected with expression vectors encoding wild-type or mutant PXR or the control vector pCR3. At 24 h after transfection, the medium was replaced, and the cells were further transfected and then cultured with roscovitine (5 μM) (A and B), rifampicin (5 μM) (C and D), or vehicle (DMSO) for an additional 24 h. The UGT1A1 mRNA levels are expressed by taking the value obtained from the wild-type PXR expression vector-transfected and roscovitine-treated cells as one. Data presented are the average ± S.E. of 4 to 11. *, p < 0.05; **, p < 0.01; ***, p < 0.01 versus rifampicin-stimulated wild-type PXR; #, p < 0.05; ##, p < 0.01; ###, p < 0.01 versus roscovitine-stimulated wild-type PXR.
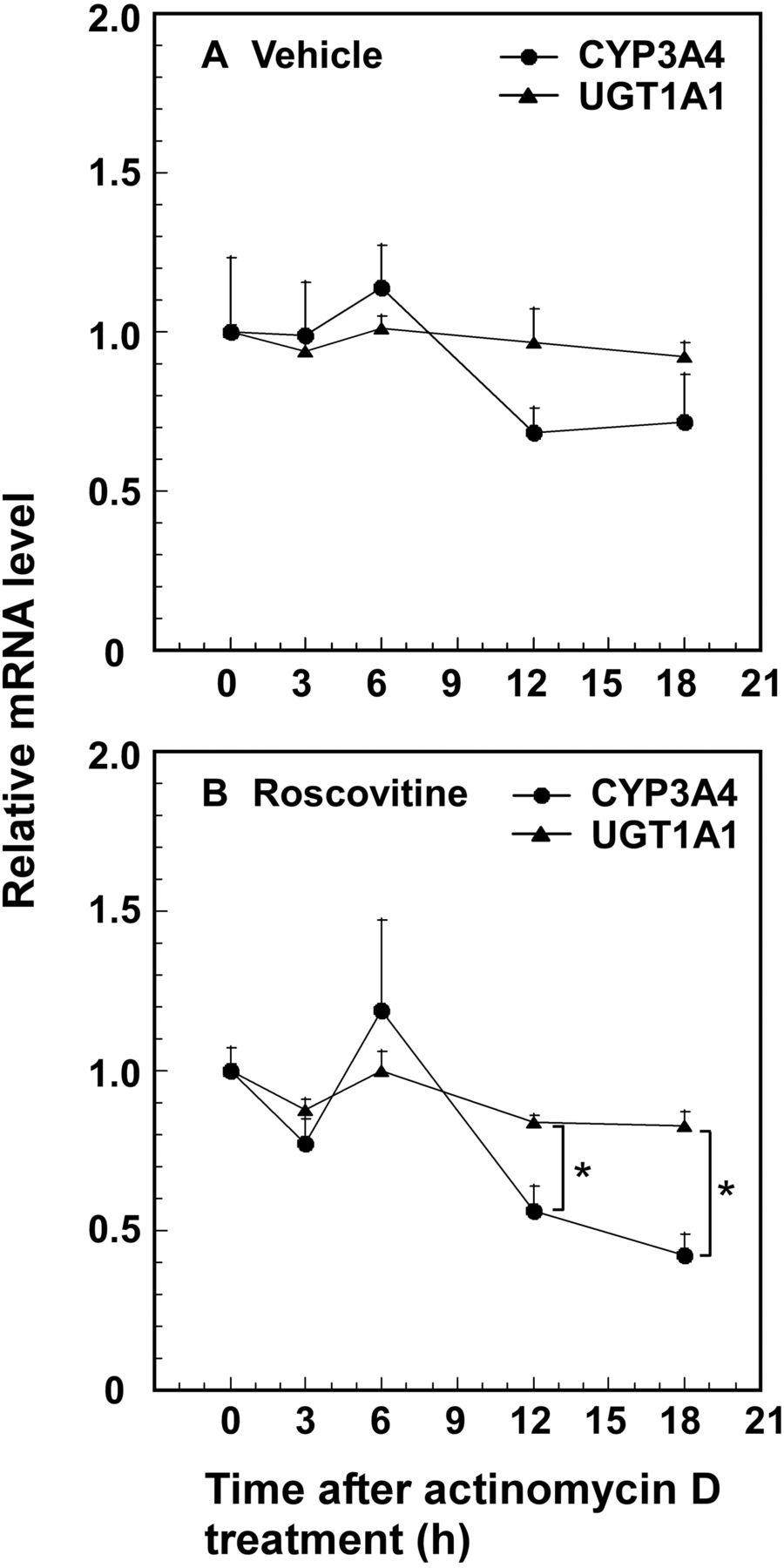
Stability of CYP3A4 and UGT1A1 mRNAs. HepG2 cells cultured for 48 h were treated with vehicle (DMSO) (A) or roscovitine (5 μM) (B) for an additional 24 h and then actinomycin D (2 μg) was added to block translation. CYP3A4 and UGT1A1 mRNA levels remaining after addition of actinomycin D were determined. The mRNA levels at time 0 (CYP3A4, 2.90 ± 0.21-fold induction; UGT1A1, 6.31 ± 0.16-fold induction) are expressed as one. Data presented are the average ± S.E. for three experiments. *, p < 0.05, CYP3A4 mRNA levels versus UGT1A1 mRNA levels.
Ser350 Is Involved in the Down-Regulation of UGT1A1 Expression and Phosphorylation of PXR by CDK2.
In a previous study (Sugatani et al., 2010), transfection with the anti-CDK2 siRNA in HepG2 and SW480 cells led to stimulated expression of UGT1A1, CYP2B6, and CYP3A4 mRNAs and proteins, with the extent of the stimulation by anti-CDK2 siRNA greater in SW480 cells than in HepG2 cells. Because roscovitine is a CDK1, CDK2, and CDK5 inhibitor (Meijer et al., 1997), anti-CDK1 siRNA, anti-CDK2 siRNA, or anti-CDK5 siRNA was introduced into SW480 cells. Transfection with the anti-CDK2 siRNA was followed by an increase in the UGT1A1 mRNA level, but transfections with anti-CDK1 siRNA and anti-CDK5 siRNA were not (Fig. 6A). Because the residue at Ser350 is predicted to be the only one phosphorylated by CDK in the PXR protein, we analyzed the extent of phosphorylation of a GST-PXR or GST-S350D PXR fusion protein after incubation with the cell lysate. As shown in Fig. 6B, the extent of phosphorylation was decreased in S350D-mutated PXR compared with wild-type PXR, and CDK2 activity was required for down-regulation of UGT1A1 expression by roscovitine.
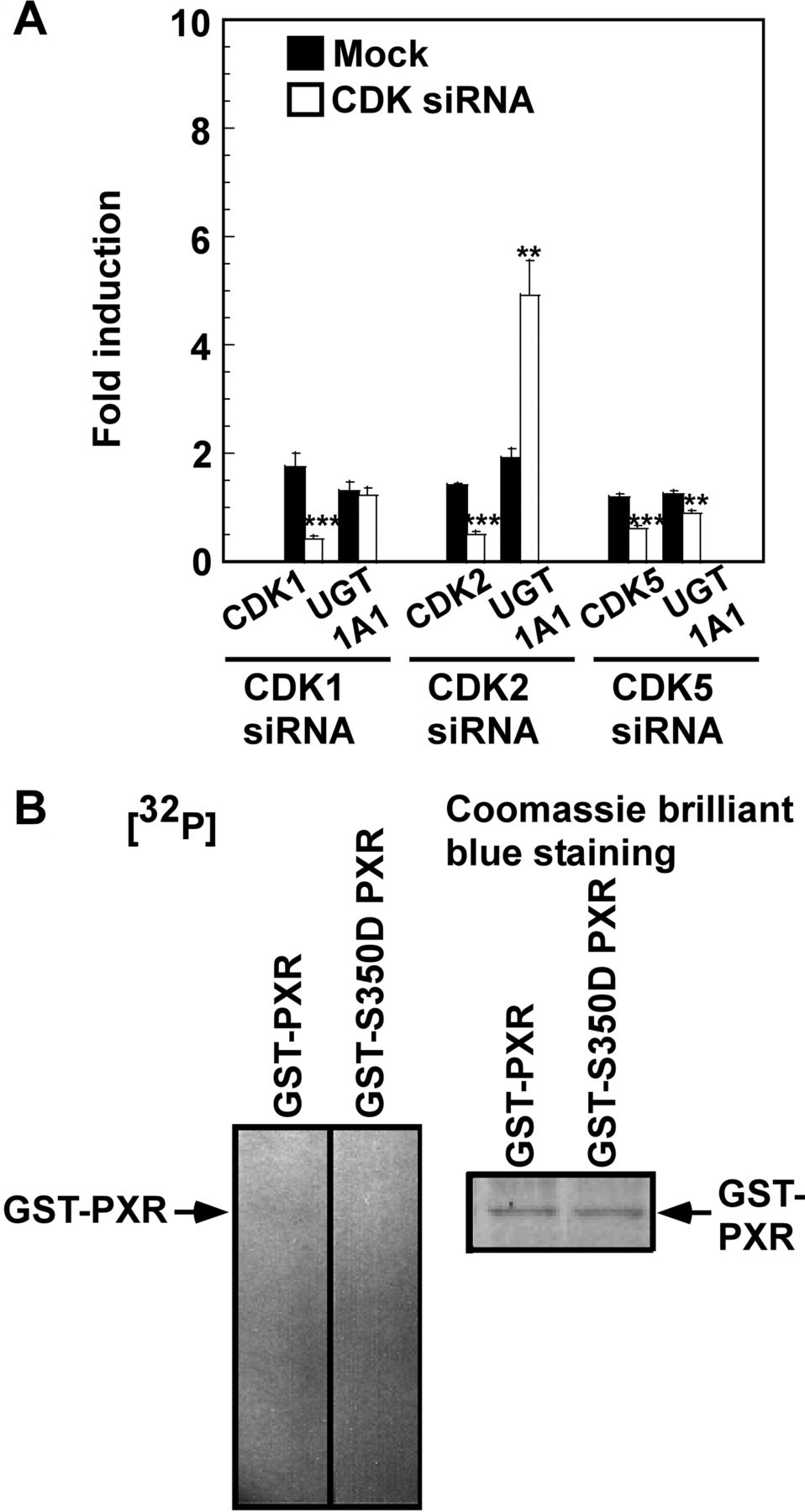
Effect of anti-CDK1, anti-CDK2, and anti-CDK5 siRNA transfection on expression of UGT1A1 mRNA and phosphorylation of PXR at Ser350 by CDK2. In A, SW480 cells cultured for 24 h were transfected with anti-CDK siRNA (100 nM) or mock-transfected. At 24 h after transfection, the medium was replaced, and the cells were further transfected with anti-CDK siRNA (100 nM) or mock-transfected and then were cultured for an additional 24 h. The mRNA levels in cells cultured for 48 h (control group) were taken as one. Data presented are the average ± S.E. for three experiments. **, p < 0.01; ***, p < 0.001 versus the mock-transfected group. In B, whole-cell lysate was prepared from SW480 cells at 2 h after release from the thymidine block and immunoprecipitated with anti-CDK2 antibody. Immune complex kinase assays were performed using GST-fused forms of wild-type and S350D mutant PXR as substrates. Experiments were performed twice with similar results, and representative data are shown.
Effects of Phosphomimetic Mutations at Thr290, Ser350, and Thr408 on the Subcellular Localization of the PXR Proteins.
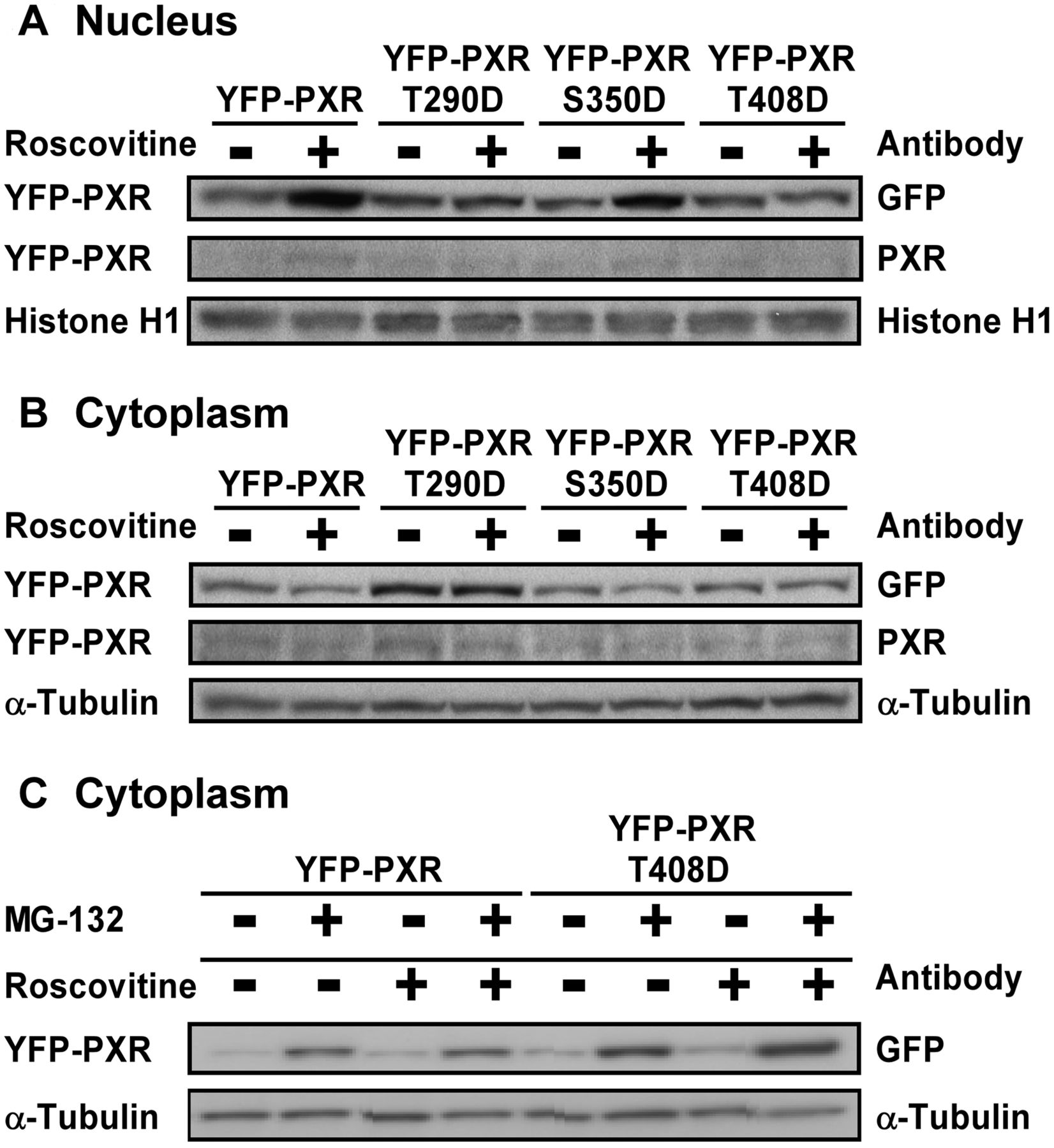
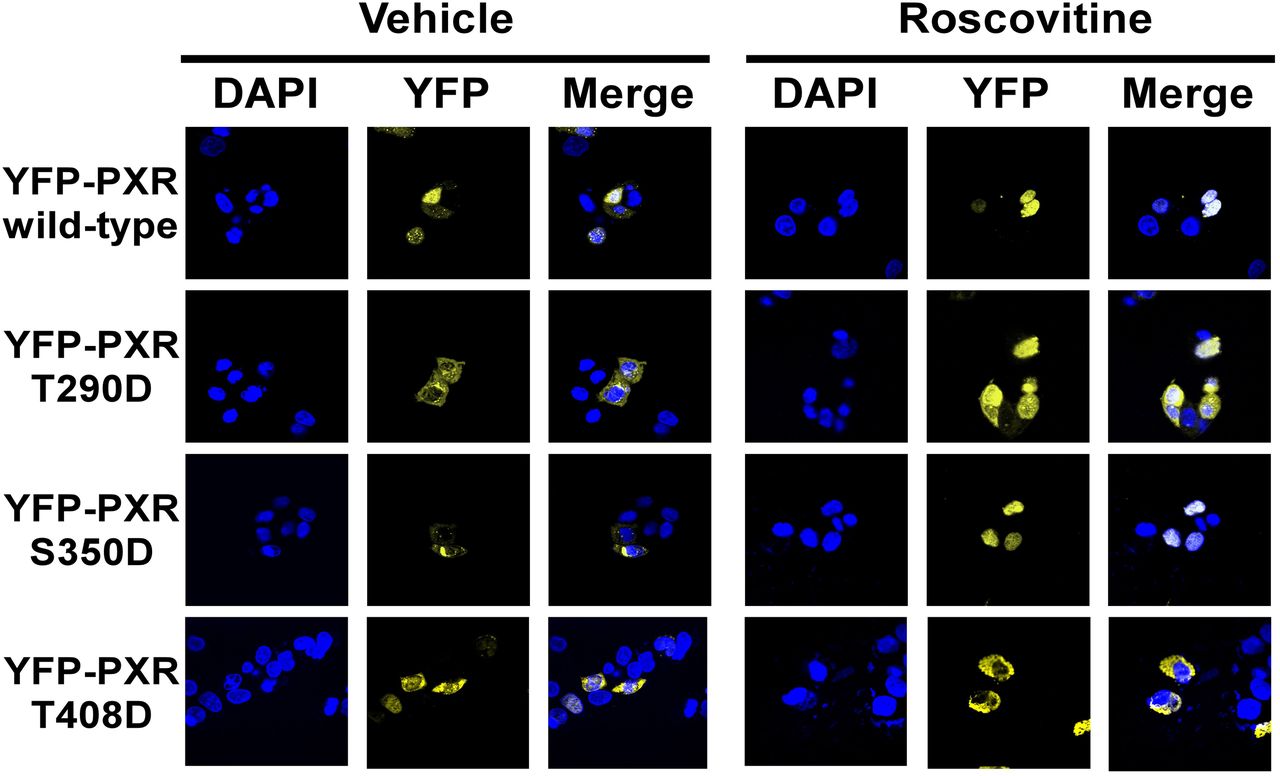
We next investigated whether a phosphomimetic mutation alters the nuclear translocation of PXR in HepG2 cells treated with roscovitine and contributes to impaired transcriptional activity. The S350D mutant YFP-PXR fusion protein was decreased in the cytoplasm and accumulated in the nucleus after roscovitine treatment, similar to the wild-type protein (Fig. 7). The T290D and T408D mutant YFP-PXR fusion proteins were retained in the cytoplasm, and the translocation of both mutant proteins to the nucleus by roscovitine was suppressed (Fig. 7A). The level of the T290D and T408D mutants in the cytoplasm was higher than that of the wild-type protein, whereas the expression of the T408D mutant was lower than that of the T290D mutant (Fig. 7B). However, the levels of the T408D mutant in the cytoplasm were markedly elevated after treatment with proteasome inhibitor MG-132, in the presence and absence of roscovitine, compared with those of the wild-type protein (Fig. 7C). Immunocytochemistry revealed that the S350D mutant YFP-PXR protein, but not T290D and T408D mutant proteins, was localized to the nucleus in the cells stimulated by roscovitine similar to the wild-type protein (Fig. 8). In contrast, the T290D and T408D mutant YFP-PXR proteins revealed a punctate distribution in the cytoplasm and nucleus of the cells treated with and without roscovitine. These observations indicate that phosphorylation at Thr290 and Thr408 abrogated the roscovitine-stimulated nuclear translocation capacity of PXR and resulted in its impaired function.

Western blot analysis of expressed PXR constructs fused with YFP in HepG2 cells. HepG2 cells cultured for 24 h were transfected with constructs expressing the wild-type and mutant YFP-PXR fusion proteins. At 24 h after transfection, the medium was replaced, and the cells were further transfected and then cultured with roscovitine (5 μM) or vehicle (DMSO) in the absence (A, B, and C) or presence (C) of MG-132 (5 μM) for an additional 24 h. The nuclear (A) and cytoplasmic (B and C) YFP-PXR proteins were determined by Western blotting with the indicated antibodies.

Subcellular localization of expressed PXR constructs fused with YFP in HepG2 cells. HepG2 cells cultured for 24 h were transfected with constructs expressing the wild-type and mutant YFP-PXR fusion proteins. At 24 h after transfection, the medium was replaced, and the cells were further transfected and then cultured with roscovitine (5 μM) or vehicle (DMSO) for an additional 24 h. The subcellular localization of the YFP-PXR protein was observed by fluorescence microscopy.
SRC-2 Partly Recovers the Impaired Transcriptional Activity of S350D Mutant PXR in HepG2 Cells.
Because transcriptional coactivators promote the function of nuclear receptors (Glass and Rosenfeld, 2000), we investigated effects of SRC-1 and SRC-2 on impaired transcriptional activity of S350D mutant PXR in HepG2 cells. Whereas SRC-1 and SRC-2 did not alter the transcriptional activities of the basal and exogenously expressed wild-type PXR by roscovitine, SRC-2 but not SRC-1 enhanced the impaired activity of the S350D mutant 1.5-fold, indicating that phosphorylation at Ser350 is likely to alter the binding affinity of PXR with a coactivator such as SRC-2 (Fig. 9).
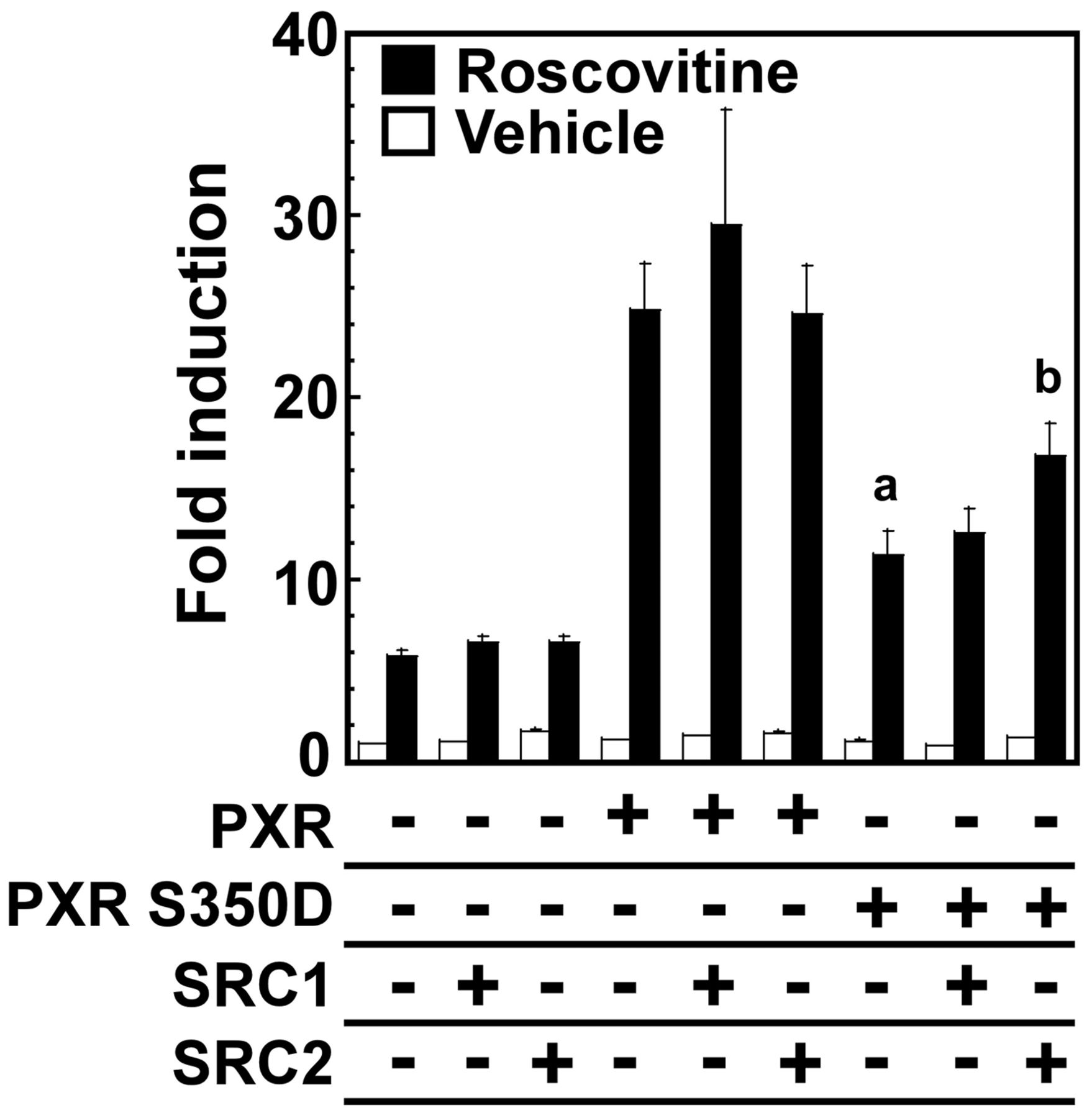
SRC-2 is a critical transcriptional coactivator for S350D mutant PXR in HepG2 cells. HepG2 cells cultured for 24 h were transfected with constructs expressing wild-type or mutant PXR protein and SRC-1 or SRC-2 protein. At 24 h after transfection, the medium was replaced, and the cells were further transfected and then cultured with roscovitine (5 μM) or vehicle (DMSO) for an additional 24 h. The mRNA levels of UGT1A1 are expressed by taking the control value obtained from the control vector pCR3- and pcDNA3.1-transfected and vehicle-treated cells as one. Data presented are the average ± S.E. for three experiments. a, p < 0.001 wild-type PXR versus S350D mutant PXR; b, p < 0.05 S350D mutant PXR versus S350D mutant PXR plus SRC-2.
Role of Hsp90 and CCRP in Cytoplasmic Localization of T290D and T408D Mutant YFP-PXR Fusion Proteins.
Hsp90 and CCRP are involved in maintaining the cytoplasmic localization of PXR (Squires et al., 2004). To test whether Hsp90 or CCRP was involved in the retention of the T290D and T408D mutant YFP-PXR proteins in the cytoplasm, we investigated whether the mutants formed a complex with Hsp90 or CCRP in the cytoplasm. Immunoprecipitation with anti-GFP or anti-CCRP antibody and Western blot analyses demonstrated that the wild-type and T290D and S350D mutant PXR proteins (Fig. 10A) but not YFP protein (data not shown) in the cytoplasm formed a complex with Hsp90 and CCRP. The formation of a T290D mutant PXR-Hsp90-CCRP complex was not disrupted even after roscovitine treatment. In contrast, little T408D mutant-Hsp90-CCRP complex was found in the cells treated with or without roscovitine (Fig. 10A), suggesting that Hsp and CCRP contribute to the retention of the T290D but not T408D mutant in the cytoplasm.

Binding of wild-type or mutant YFP-PXR fusion protein to cytoplasmic Hsp90 and CCRP (A) and to nuclear RXR, HDAC1, and HDAC3 (B), and acetylation of wild-type or mutant protein (B). HepG2 cells cultured for 24 h were transfected with constructs expressing wild-type and mutant YFP-PXR fusion proteins. At 24 h after transfection, the medium was replaced, and the cells were further transfected and then cultured with roscovitine (5 μM) or vehicle (DMSO) for an additional 24 h. Cytoplasmic fractions and nuclear extracts were prepared and used for immunoprecipitation using anti-GFP antibody or anti-CCRP antibody and Western blotting stained with the indicated antibodies.
Phosphomimetic Mutation at Ser350 Alters Deacetylation of PXR and Capacity to Bind with RXR in the Nucleus of Cells Treated with Roscovitine.
Next, we investigated the effect of a phosphomimetic mutation at Ser350 on PXR activation in the nucleus by roscovitine, that is, heterodimerization with RXR and release from acetylation after roscovitine treatment. PXR forms a heterodimer with RXR and helps to activate PXR. Whereas the YFP protein was markedly expressed compared with the YFP-PXR fusion protein and an association of YFP with RXR was found, the extent of binding of the wild-type YFP-PXR fusion protein with RXR was more than that of YFP with RXR and further enhanced by roscovitine, but that of the S350D mutant with RXR was markedly reduced (Fig. 10B). Because the levels of the wild-type and S350D mutant PXR proteins were lower in the nucleus of the control cells than those of the roscovitine-treated cells, acetylated forms were detected in trace amounts (Fig. 10B). However, Western blot analysis using anti-acetylated lysine antibody demonstrated that acetylated S350D mutant YFP-PXR protein but not the wild-type or YFP protein was detected even in the nucleus after roscovitine treatment, although the levels of the wild-type and S350D mutant proteins were similar (Fig. 10B). Furthermore, whereas the level of binding of wild-type YFP-PXR fusion protein with HDAC1 was enhanced after roscovitine treatment, that of the S350D mutant with HDAC1 was reduced (Fig. 10B). In addition, the level of binding of the S350D mutant PXR with HDAC3 was also reduced after roscovitine treatment, compared with the wild-type protein (Fig. 10B). These results suggest that the wild-type YFP-PXR fusion protein but not S350D-mutated protein may be deacetylated by HDAC after roscovitine stimulation.
Discussion
PXR, a member of the nuclear orphan receptor superfamily, was originally characterized as a xenobiotic-activated transcription factor that plays a key role in regulating the activation of genes encoding drug-metabolizing enzymes and drug transporters and subsequently has been found to potentiate a variety of biological effects associated with pharmacological and toxicological consequences (reviewed in Timsit and Negishi, 2007; Staudinger and Lichti, 2008; Ihunnah et al., 2011) and hepatic energy metabolism including gluconeogenesis, β-oxidation of fatty acids, and lipogenesis (reviewed in Konno et al., 2008). Upon the binding of endogenous and exogenous xenobiotics to its ligand-binding domain (LBD), PXR is activated and translocated from the cytoplasm to cell nucleus where it allows interaction with coactivators such as SRC-1 and SRC-2 and activates transcription by binding to the xenobiotic-response element in the promoter region of the target gene. Studies demonstrated that posttranslational modifications of PXR alter its function; Lin et al. (2008) showed that CDK2 directly phosphorylates PXR and negatively affects its activity, and Biswas et al. (2011) found that the acetylation status of PXR regulates its selective function independent of ligand activation. Thus, although posttranslational modifications of PXR are likely to be involved in the regulation of its function, much less is known about the molecular mechanism regulating physiological functions of PXR. Our finding that the CDK inhibitor roscovitine markedly stimulated the expression of UGT1A1 mRNA and protein in HepG2 cells (Sugatani et al., 2010) prompted us to investigate how roscovitine enhances UGT1A1 gene expression. In the reporter gene assay, PXR-mediated transcriptional activity of the reporter gene was more prominently enhanced by roscovitine, compared with the basal, CAR-mediated, and AhR/ARNT-mediated transcriptional activities (Fig. 1). Moreover, expression of UGT1A1 mRNA and protein was markedly enhanced by roscovitine in HepG2 cells expressing PXR (Fig. 2). These observations indicate that PXR contributes to the induction of UGT1A1 expression by roscovitine.
Because roscovitine has been shown to activate PXR with moderate effect but bind to PXR with lower affinity than the PXR ligand tetra-ethyl 2-(3,5-di-tert-butyl-4-hydroxyphenyl)ethenyl-1,1-bisphosphonate (SR12813) (Lin et al., 2008), in the present study, we investigated the mechanism of PXR activation to induce UGT1A1 expression both in a ligand-independent manner by roscovitine and in a PXR ligand rifampicin-inducible manner. We analyzed roles of nine potential phosphorylation sites in the LBD demonstrated using in silico consensus site prediction methods and reporter gene analysis by Lichti-Kaiser et al. (2009a), compared with the functionally significant Thr57 site in the DNA-binding domain (Pondugula et al., 2009). Phosphomimetic mutations at Thr290, Ser350, and Thr408 of PXR strongly attenuated the induction of UGT1A1 expression by roscovitine, as did the phosphomimetic mutation at Thr57, although those at Thr290, Ser305, and Thr408 contributed to the suppression of rifampicin-stimulated UGT1A1 expression (Figs. 3 and 4). As expected, some of the residues involved in PXR activation by roscovitine and rifampicin are different. In both rifampicin- and roscovitine-stimulated cells, the Thr57 phosphomimetic mutant PXR lost all transcriptional activity for the UGT1A1 gene (Figs. 3 and 4). Pondugula et al. (2009) found that Thr57 identified as a potential phosphorylation site for p70 S6K is located in the first zinc-finger motif of the DNA-binding domain, and the mutation T57D impairs the ability to bind to the target CYP3A4 promoter. Thus, Thr57 is considered to play a key role in both ligand-inducible and ligand-independent PXR activation. In contrast, the phosphomimetic mutation at Ser305 more strongly reduced the ligand (rifampicin)-inducible transcriptional activity compared with the ligand-independent activation by roscovitine (Figs. 3 and 4), indicating that the residue at Ser305 in the LBD plays an important role in the ligand-dependent PXR activation. As reported by Lichti-Kaiser et al. (2009a), the impaired activity of the S305D mutant PXR may result from the inhibition of ligand-inducible PXR-RXR heterodimerization and interaction with its cofactors. On the other hand, in our previous study, we demonstrated that the expression of UGT1A1 is negatively regulated through a CDK2 signaling pathway linked to cell-cycle progression in HepG2 cells, but the molecular mechanism remains to be clarified (Sugatani et al., 2010). Phosphomimetic mutation of the residue at Ser350 in only one consensus CDK phosphorylation motif [(S/T)PX(R/K)] more strongly impaired the roscovitine-stimulated transcriptional activity compared with the ligand-inducible activity, whereas the interaction of the S350D mutant PXR with RXR after roscovitine treatment was markedly reduced in coimmunoprecipitation assays (Figs. 3, 4, and 10). In addition, SRC-2 but not SRC-1 enhanced the roscovitine-stimulated activity of the S350D mutant PXR (Fig. 9), although SRC-1 binding has been found to promote the specific interaction between ligands and PXR (Watkins et al., 2003). These observations indicate that phosphorylation at Ser350 is likely to alter the interaction of PXR with its coactivator and influence the formation of a complex with RXR and its coactivator, repressing the cell-based nuclear transactivation of PXR by roscovitine. This study demonstrated that the deletion of CDK2 but not CDK1 or CDK5 using siRNA resulted in enhanced UGT1A1 expression (Fig. 6A), and the wild-type PXR protein but not S350D mutant was phosphorylated by CDK2 (Fig. 6B). These findings that CDK2 phosphorylates the Ser350 residue of the PXR protein, resulting in impaired PXR activity, confirm the speculation by Lin et al. (2008) made on the basis of a reporter gene analysis with mutant PXR protein. Moreover, the present study demonstrated that the S350D mutant but not wild-type PXR was detected as acetylated in the cell nucleus after roscovitine treatment (Fig. 10B). In addition, the level of binding of wild-type PXR with HDAC1 was increased in the cell nucleus after roscovitine treatment, but that of the S350D mutant was decreased (Fig. 10B). These results suggest that the PXR protein would be acetylated in the nucleus of the control cells, resulting in impaired PXR activity, and the deacetylation by HDAC may lead to the PXR activation. It has been shown that PXR is acetylated in vivo and the acetylation status of the PXR protein regulates its selective function independent of ligand activation (Biswas et al., 2011). In addition, HDAC1 is required for carbamazepine-induced CYP3A4 expression (Wu et al., 2012). Although further study of whether HDAC1 is involved in PXR activation by roscovitine as well as by carbamazepine is required, an acetylated S350D mutant PXR may result in impaired activity in the nucleus.
Although site-specific phosphorylation has been shown to occur in the PXR protein to modulate its activity (Lin et al., 2008; Lichti-Kaiser et al., 2009a; Pondugula et al., 2009), the current study has for the first time found the critical role of Thr290 in PXR activation. Phosphomimetic mutation at Thr290 of PXR also suppressed roscovitine-induced translocalization of the YFP-PXR fusion protein from the cytoplasm to nucleus, which may result in the reduced transcriptional activity of PXR in the cells stimulated by roscovitine and rifampicin (Figs. 3, 4, 7, and 8). The formation of a complex of PXR with Hsp90 and CCRP has been found to retain PXR in the cytoplasm (Squires et al., 2004). Western blot and immunoprecipitation analyses demonstrated the presence of T290D mutant YFP-PXR in a complex with Hsp90 and CCRP, and the extent of accumulation in the cytoplasm was not changed after roscovitine treatment (Figs. 7 and 10). Although the ligand-independent mechanism regulating PXR activation remains to be clarified, the present finding demonstrated that phosphorylation at Thr290 is likely to suppress the roscovitine-induced dissociation of PXR from Hsp90 and CCRP, resulting in the accumulation of PXR protein in the cytoplasm. The complex of T408D mutant PXR with Hsp90 and CCRP was rare, but the T408D mutant protein was accumulated in the cytoplasm of the cells treated with MG-132. These observations indicate that PXR phosphorylated at Thr408 may be degraded by proteasome (Figs. 7 and 10), because murine PXR has been reported to be a target of proteasomal degradation (Masuyama et al., 2002). Protein kinase A (PKA) and protein kinase C (PKC) contribute to cell signaling involved in ligand-inducible PXR activation (Ding and Staudinger, 2005; Lichti-Kaiser et al., 2009b). PKC-dependent signaling has been demonstrated to repress the transcriptional activity of PXR by increasing the strength of interaction between PXR and the nuclear receptor corepressor protein (Ding and Staudinger, 2005). Lichti-Kaiser et al. (2009b) demonstrated 1) that PKA signaling has a repressive effect on PXR-mediated gene activation in human hepatocytes and stimulates the interaction of PXR with the nuclear receptor corepressor, and 2) that human PXR protein can serve as an effective substrate for catalytically active PKA in vivo. Further study is needed to determine whether PKA or PKC associated with cell signaling contributes to the phosphorylation of PXR at Thr290 (a predicted PKA/PKC phosphorylation site).
In conclusion, whereas phosphorylation of PXR at Thr290 and Thr408 suppresses the PXR activation both in a ligand-dependent manner by rifampicin and in a ligand-independent manner by roscovitine, roscovitine inhibits phosphorylation of PXR at Ser350 by CDK2 and enhances the translocation of PXR from the cytoplasm to the nucleus, resulting in elevated expression of UGT1A1. The current study provides a clue to examining dynamic regulatory mechanisms through posttranslational modifications such as phosphorylation and acetylation and specific cross-talk with diverse signaling pathways in PXR activation.
Authorship Contributions
Participated in research design: Sugatani.
Conducted experiments: Sugatani, Uchida, and Kurosawa.
Contributed new reagents or analytic tools: Sugatani, Uchida, and Kurosawa.
Performed data analysis: Sugatani, Uchida, Kurosawa, Yamaguchi, Yamazaki, Ikari, and Miwa.
Wrote or contributed to the writing of the manuscript: Sugatani and Miwa.
Acknowledgments
We thank Yoshinori Hattori for excellent technical assistance.
Footnotes
This work was supported by the Global Center of Excellence Program from the Ministry of Education, Culture, Sports, Science and Technology, Japan; and the Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan [Grants 19590070, 19590151, 21590170, 22590068].
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
ABBREVIATIONS:
- CAR
- constitutive androstane receptor
- NR1
- nuclear receptor subfamily 1
- PXR
- pregnane X receptor
- UGT
- UDP-glucuronosyltransferase
- SN-38
- 7-ethyl-10-hydroxycamptothecin
- PBREM
- phenobarbital-responsive enhancer module
- AhR
- aryl hydrocarbon receptor
- HGF
- hepatocyte growth factor
- CDK
- cyclin-dependent kinase
- siRNA
- small interfering RNA
- RXR
- retinoid X receptor
- CCRP
- cytoplasmic CAR retention protein
- Hsp
- heat shock protein
- MG-132
- N-(benzyloxycarbonyl)leucinylleucinylleucinal
- SRC
- steroid receptor coactivator
- ARNT
- aryl hydrocarbon nuclear translocator
- GST
- glutathione transferase
- YFP
- yellow fluorescent protein
- PCR
- polymerase chain reaction
- pEYFP
- enhanced yellow fluorescent protein
- GFP
- green fluorescent protein
- HDAC
- histone deacetylase
- DMSO
- dimethyl sulfoxide
- LBD
- ligand-binding domain
- SR12813
- tetra-ethyl 2-(3,5-di-tert-butyl-4-hydroxyphenyl)ethenyl-1,1-bisphosphonate
- PKA
- protein kinase A
- PKC
- protein kinase C
- Ct
- cycle threshold.
- Received May 13, 2012.
- Accepted July 24, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}