


Abstract
Time-dependent inhibition (TDI) of cytochrome P450 (P450) enzymes, especially CYP3A4, is an important attribute of drugs in evaluating the potential for pharmacokinetic drug-drug interactions. The analysis of TDI data for P450 enzymes can be challenging, yet it is important to be able to reliably evaluate whether a drug is a TDI or not, and if so, how best to derive the inactivation kinetic parameters KI and kinact. In the present investigation a two-step statistical evaluation was developed to evaluate CYP3A4 TDI data. In the first step, a two-sided two-sample z-test is used to compare the kobs values measured in the absence and presence of the test compound to answer the question of whether the test compound is a TDI or not. In the second step, kobs values are plotted versus both [I] and ln[I] to determine whether a significant correlation exists, which can then inform the investigator of whether the inactivation kinetic parameters, KI and kinact, can be reliably estimated. Use of this two-step statistical evaluation is illustrated with the examination of five drugs of varying capabilities to inactivate CYP3A4: ketoconazole, erythromycin, raloxifene, rosiglitazone, and pioglitazone. The use of a set statistical algorithm offers a more robust and objective approach to the analysis of P450 TDI data than frequently employed empirically derived or heuristic approaches.
Introduction
Time-dependent inhibition (TDI) represents a more severe form of inhibition for cytochrome P450 (P450) enzymes relative to reversible inhibition because inactivated enzymes have to be regenerated to restore their function by de novo protein synthesis (Kalgutkar et al., 2007; Obach et al., 2007; Venkatakrishnan and Obach, 2007). TDI inactivates P450 enzymes that are critical for metabolizing many drugs and it often causes prolonged effects, even after the inactivator(s) has been removed from the body. TDI can lead to clinically relevant drug-drug interactions (DDI), the effects of which can result in potential toxicity or loss of efficacy concerns (Zhou et al., 2007). For example, the failure of several late-stage clinical candidates was attributed to TDI (Zimmerlin et al., 2011), drugs have been withdrawn from the market because of TDI (e.g., mibefradil), and severe restrictions have been imposed on clinical usage for concomitant medications due to TDI effects (Wienkers and Heath, 2005). For these reasons, regulatory agencies (Food and Drug Administration, European Medicines Agency) have issued strict guidelines pertaining to the conduct of TDI studies, and these data are required for regulatory submission of a new drug application (U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research, 2012).
Currently, measuring TDI is one of the most challenging enzymology experiments to conduct in the area of drug metabolism. The assay includes complex experimental steps with multiple concentrations and time points, preincubation, secondary incubation, multiple transfer steps, and intricate timing. Automation, using robotic systems, can help alleviate some of the complexity of the assay (Zimmerlin et al., 2011). The analysis of TDI experimental data is also quite involved because of the need to fit and interpret multiple kinetic parameters. To address these TDI challenges, Pharmaceutical Research and Manufacturers of America (Washington DC) provided guidance on best practices for conducting in vitro TDI studies (Grimm et al., 2009). This perspective provided a clear roadmap on how to evaluate TDI in drug discovery. However, two questions remained unresolved: 1) when is TDI detectable for a drug; and 2) how to best estimate KI and kinact? In this study, we apply a statistical approach to objectively evaluate whether a compound is a TDI [current standard practices are somewhat arbitrary in our opinion (Grimm et al., 2009; Zimmerlin et al., 2011; U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research, 2012)] and if we can determine KI and kinact with confidence using CYP3A4 TDI experimental data.
Materials and Methods
Materials.
Pooled human liver microsomes from 50 male and female donors were purchased from BD Biosciences (Woburn, MA). Solvents, β-NADPH, monobasic and dibasic potassium phosphate, and magnesium chloride were purchased from Sigma-Aldrich (St. Louis, MO). Probe substrate, internal standard, and inhibitors were obtained from the following sources: midazolam (Cerilliant Corporation, Round Rock, TX), rosiglitazone and pioglitazone (Sequoia Research Products, Pangbourne, UK), erythromycin and raloxifene (Sigma-Aldrich), and ketoconazole (Enzo Life Sciences, Farmingdale, NY);[2H4]1′-hydroxymidazolam was prepared at Pfizer (Groton, CT).
Time-Dependent Inhibition Assay.
Phosphate buffer (100 mM, pH 7.4) was prepared from 100 mM monobasic and dibasic potassium phosphate solutions. MgCl2 stock solution was prepared in water (200 mM). NADPH solution (13 mM) was prepared in potassium phosphate buffer immediately before use. Inhibitor stock solutions were prepared at 100× the final preincubation concentration. Inhibitor concentration ranges were selected to surround the expected KI. Concentrations were approximately evenly spaced after applying a log transformation.
Inactivation kinetic experiments have been described previously (Ghanbari et al., 2006; Obach et al., 2007; Polasek and Miners, 2007; Zimmerlin et al., 2011). Experiments were conducted in two steps, an inactivation preincubation, followed by a secondary incubation to assess remaining CYP3A4 activity, both conducted at 37°C. In the inactivation preincubation, human liver microsomes (0.3 mg/ml), MgCl2 (3.3 mM), and NADPH (1.3 mM) in potassium phosphate buffer (100 mM, pH 7.4) were prewarmed at 37°C for 5 min. Preincubation was commenced with the addition of inhibitor or vehicle (2 μl). The final preincubation solvent concentration was 0.9% (v/v) acetonitrile and 0.1% (v/v) DMSO (ketoconazole, pioglitazone, raloxifene, erythromycin), or 1% (v/v) DMSO (rosiglitazone) (Chauret et al., 1998). At each time point (0, 2, 4, 7, 15, and 30 min), an aliquot of the preincubation mixture (10 μl) was transferred to a prewarmed secondary incubation mixture, consisting of midazolam (23 μM, approximately 10-fold Km), MgCl2 (3.3 mM), and NADPH (1.3 mM) in potassium phosphate buffer (100 mM, pH 7.4) at a final volume of 200 μl (20-fold dilution). After 6 min, the secondary incubation was terminated by transferring 100 μl of the solution from the second incubation to 200 μl of acetonitrile containing internal standard ([2H4]1′-hydroxymidazolam, 0.1 μg/ml). Incubations were conducted in duplicate. Samples were then vortexed and centrifuged for 10 min at 2000g. Supernatant was transferred, mixed with an equal volume of water containing formic acid (0.2%), and analyzed via liquid chromatography/tandem mass spectrometry (LC-MS/MS). The assay was conducted at six nonzero concentrations and six time points for estimating KI and kinact.
LC-MS/MS Quantification.
A Sciex API4000 QTRAP LC-MS/MS (Foster City, CA) triple quadrupole mass spectrometer fitted with a turbo ion-spray interface operated in positive ion mode was used to monitor for 1′-hydroxymidazolam and [2H4]1′-hydroxymidazolam. A Shimadzu (Columbia, MD) LC-20AD HPLC system with a CTC LEAP autosampler (LEAP Technology, Carrboro, NC) was programmed to inject 10 μl of sample on a Halo 2.7 μm C18 3.0 × 30 mm column (Advanced Materials Technology, Wilmington, DE). Analytes were eluted using a binary gradient mixture consisting of 0.1% (v/v) formic acid in water (solvent A) and 0.1% (v/v) formic acid in acetonitrile (solvent B), at a flow rate of 1.2 ml/min and monitored using the multiple reaction monitoring mode for the mass-to-charge (m/z) transitions 342.0 → 203.3 (1′-hydroxymidazolam) and 346.0 → 207.3 ([2H4]1′-hydroxymidazolam). Average difference between all replicates reported was 4.1%.
Parameter Estimates.
Resulting analyte-to-internal standard peak area ratios from MS were normalized to averaged analyte-to-internal standard peak area ratios observed at 0 min in the solvent controls (hereafter referred to as activity); individual replicate activity was then used to estimate kobs. The linear regressions, Spearman's correlation coefficient calculations, and various slope tests were performed using Excel (Microsoft Corporation, Redmond, WA). The two- and three-parameter nonlinear fits were obtained using JMP 8.0.1 (SAS Institute Inc., Cary, NC).
Statistical Analysis.
Estimating the kinact and KI kinetic parameters for an inactivation experiment is a two-step process (Shou and Dai, 2008; Obach et al., 2010). Statistical tests are applied here in an augmented procedure to test for TDI. First, for each compound concentration j the inactivation rate, kobs,j, is estimated using a linear regression of the natural logarithm of activity, using k individual observations, versus time, that is, ln(Activity)k = β1Timek + β0 + εk, and εk is the experimental variance N(0,σ12). By definition, for each concentration j we estimate kobs,j using the estimate for −β1. A set of kobs,j rates is generated across the concentrations tested using the various individual regression β1 slope estimates. If appropriate, nonlinear regression is then used to estimate a three-parameter Michaelis-Menten equation, that is,
 where εj is the combined experimental and modeling variance N(0,σ22). Two two-parameter models were also tested: one in which all kobs data were uncorrected for activity decreases observed at [I] = 0 μM, and the resulting kobs versus [I] relationship was forced through the origin (i.e., “2pU”-uncorrected), and a second wherein kobs[0 μM] is set to zero and kobs values are normalized against kobs[0 μM] at each time point (“2pC”-corrected). For each linear regression three useful estimates are produced: root mean squared error (RMSE), an estimate for σ1, slope, and intercept. Using standard normal theory assumptions one can form hypothesis tests for both β0 and β1. Of particular interest to a test for TDI is the set of hypotheses H0: −β1[I] + β1[0 μM] = kobs[I]− kobs[0 μM] = 0 versus H1: −β1[I] + β1[0 μM] = kobs[I] − kobs[0 μM] ≠ 0 for each nonzero concentration. Despite the traditional use of a t test here, we evaluate these contrasts using a large sample normal theory test statistic, that is,
where εj is the combined experimental and modeling variance N(0,σ22). Two two-parameter models were also tested: one in which all kobs data were uncorrected for activity decreases observed at [I] = 0 μM, and the resulting kobs versus [I] relationship was forced through the origin (i.e., “2pU”-uncorrected), and a second wherein kobs[0 μM] is set to zero and kobs values are normalized against kobs[0 μM] at each time point (“2pC”-corrected). For each linear regression three useful estimates are produced: root mean squared error (RMSE), an estimate for σ1, slope, and intercept. Using standard normal theory assumptions one can form hypothesis tests for both β0 and β1. Of particular interest to a test for TDI is the set of hypotheses H0: −β1[I] + β1[0 μM] = kobs[I]− kobs[0 μM] = 0 versus H1: −β1[I] + β1[0 μM] = kobs[I] − kobs[0 μM] ≠ 0 for each nonzero concentration. Despite the traditional use of a t test here, we evaluate these contrasts using a large sample normal theory test statistic, that is,
 where the S.E. is based on the individual RMSE estimates. Each test is conducted using a nominal level, for example, α = 0.05. If any of these hypothesis tests yields a significant finding we conclude that the compound exhibits TDI. Although not formally discussed here, similar tests for β0 may reflect specific compound behavior or be used to better understand the experimental protocol.
where the S.E. is based on the individual RMSE estimates. Each test is conducted using a nominal level, for example, α = 0.05. If any of these hypothesis tests yields a significant finding we conclude that the compound exhibits TDI. Although not formally discussed here, similar tests for β0 may reflect specific compound behavior or be used to better understand the experimental protocol.
The previous step implements a statistical test for TDI. Because weak inhibitors or suboptimal designs (e.g., number or spacing of concentrations tested) affect the subsequent nonlinear model, we propose a statistical test before estimating the kinetic equation. The two tests proposed here are analogous to a structured comparison of means for a gradient treatment with general spacing (Lentner and Bishop, 1993) and allow us to qualify the kinetic estimates. Qualifying estimate quality may be needed because correlated (or unstable) kinetic estimates are common for the Michaelis-Menten model (Raaijmakers, 1987). In practice, preincubation compound concentrations are often diluted in a logarithmic-like manner, for example, two-fold dilutions. Comparable to bioassay or dose-response models (DeLean et al., 1978), we first consider a relationship between the observed kobs rates and ln[I]. To help mitigate the inferential complexities for a small-sample nonlinear model, we test a linear regression of kobs,j = γ1ln[I]j + γ0 + εj before fitting the three-parameter Michaelis-Menten equation. Here, we are specifically interested in a test of H0: γ1 = 0 versus H1: γ1 > 0. If we reject this one-sided t test at a predetermined level α we have likely improved our ability to estimate the kinetic parameters via nonlinear regression. Failing to reject this parametric test may be because of an inadequate kobs−ln[I] linear approximation for a TDI compound, that is, γ1 was biased toward 0 for some inhibitors investigated. To further control for possible false negatives we apply a second test, the rank-based Spearman's correlation coefficient rs, using all of the untransformed ([I]j, kobs,j) pairs as an additional inferential gate before calculating the kinetic parameters. rs is well-suited for detecting a monotonically increasing relationship between kobs and [I], an intrinsic feature of a TDI compound. A standard test of H0: rs = 0 versus H1: rs > 0 is then performed. Rejecting H0 suggests that meaningful kinetic parameters are likely to be estimable; failing to reject H0 suggests additional scientific and statistical considerations.
Standard least-squares nonlinear regression is used to estimate kobs[0 μM], kinact and KI. The initial starting value for each parameter was the following: kobs[0 μM] = 0, kinact = max(kobs,j), and KI = median[I]j. We suggest caution in the use and interpretation of inferential procedures for the various Michaelis-Menten kinetic parameters. It is well-understood that fit summaries, for example, R2, may suggest a satisfactory nonlinear model, and yet the model's estimated kinetic parameters are very imprecise (Seber and Wild, 2003). Although approximate S.E.s can be used to generate large-sample confidence intervals for kobs[0 μM], kinact, and KI, we recommend and report here profile likelihood confidence intervals, which are more robust and possess better coverage properties for small sample estimates, for characterizing the uncertainty in the kinetic estimates. We advocate the supplemental use of regression-based tests for TDI compounds here due to the prevalence of small sample TDI experiments and a reduction in the number of parameters to test. A single intermediate test for γ1 or rs may be preferred to a formal test for, at a minimum, kinact and KI.
As a final aide to qualify kinetic estimate quality, we propose a heuristic based on the empirical estimate for KI. We suggest that the estimate for KI be more than three times the smallest experimental concentration and less than roughly one-third the largest concentration for a TDI compound. Estimates for KI that are outside of this region may suggest a suboptimal experimental design or an unstable nonlinear regression. Refer to Fig. 1 for a summary of the TDI decision tree containing the various statistical tests proposed here.

TDI decision tree.
Results and Discussion
Importance of Statistical Methods for Assessing TDI.
Considerable attention has been given to the TDI of P450 enzymes and its role in clinical DDI. Much of this attention has focused on the use of in vitro inactivation data, specifically KI and kinact, in the prediction of clinical DDI using various static and dynamic models (Mayhew et al., 2000; Wang et al., 2004; Obach et al., 2007; Yang et al., 2008). Despite wide interest in methodologies for accurate TDI measurement and prediction, there has not been an investigation of the statistical rigor that needs to be employed to appropriately interpret in vitro TDI data. One of the most important questions is how to decide whether a test compound is, in fact, a TDI or not. Statistical methods may help provide a nonbiased assessment of TDI and offer a quantitative interpretation of the U.S. Food and Drug Administration draft guidance statement pertaining to TDI “any time-dependent loss of initial product formation rate may indicate time-dependent inhibition” (U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research, 2012).
Statistical tests, despite their inherent limitations as decision instruments, offer some general advantages for testing for TDI. Apart from declaring the level of the test (e.g., α = 0.05), these tests are data-driven and incorporate the observed variability. This lessens the need for ad hoc tests or investigator-specific heuristics. Statistical uncertainty estimates may protect against data over-interpretation, especially for weak TDI compounds. Continuous-valued test statistics are more informative than discrete decisions. Various statistical estimates, for example, the RMSE or S.E. of a given kobs fit, provide an investigator with valuable data. Such information, apart from qualifying the interpretation of a single experiment, can inform the planning and conduct of future TDI experiments. An increased understanding of a putative TDI compound is also possible, for example, a correlation between the RMSE and concentration for a set of kobs. In contrast to simulation- or numerical-estimation procedures, linear models (or an assumed linear approximation) are more workable for evaluating design proposals.
Two-Step Statistical Analysis of TDI Data.
In this study we use a two-step process in the statistical analysis of experimental TDI data (Fig. 1) to determine 1) whether a compound exhibits TDI, and 2) whether KI and kinact can be estimated. Statistical analysis is applied to TDI data generated by using an experimental design of 6 nonzero test concentrations at 6 time points of preincubation. Five drugs (ketoconazole, pioglitazone, rosiglitazone, raloxifene, and erythromycin) with varying degrees of TDI for CYP3A4 are used to illustrate the statistical approach.
Step I: Is a Compound a TDI?
In the first step, the natural logarithm of activity is plotted at each time point for each concentration (Fig. 2A). Linear regressions are used to estimate the inactivation rate constants (kobs) at each of the concentrations. This plot makes it easier to identify potential data edits needed to improve kobs accuracy. For example, to better estimate kobs for the potent inactivator raloxifene, several later time points were removed at multiple inhibitor concentration levels. Each kobs is assessed in reference to the vehicle control (typically DMSO, acetonitrile, or methanol) using a two-sided two-sample z-test (see under Materials and Methods). If an estimated p value is less than 0.05, this result suggests that the compound is a TDI at that concentration. Other choices for the level of the test, for example, α equal to 0.01, are possible and may safeguard the reproducibility of the observed finding. A qualitative assessment of the RMSE estimates may be warranted because this quantity is critical in estimating the various kobs S.E.s. If a compound does not demonstrate a significant difference in kobs between the vehicle and compound at one or more concentrations (p ≥ 0.05), this suggests, within the confines of the experimental approach used, that the test compound is not a TDI. Hence, if at least one significant kobs difference is observed, then the conclusion should be made that TDI has occurred. Using this rationale, the statistical tests for TDI of five drugs are summarized in Table 1. These data suggest that all of the compounds are TDI at all concentrations (p < 0.05), with the exception of pioglitazone at low concentrations (3, 6, and 10 μM) and ketoconazole at all concentrations (potent, reversible inhibition was observed for ketoconazole).
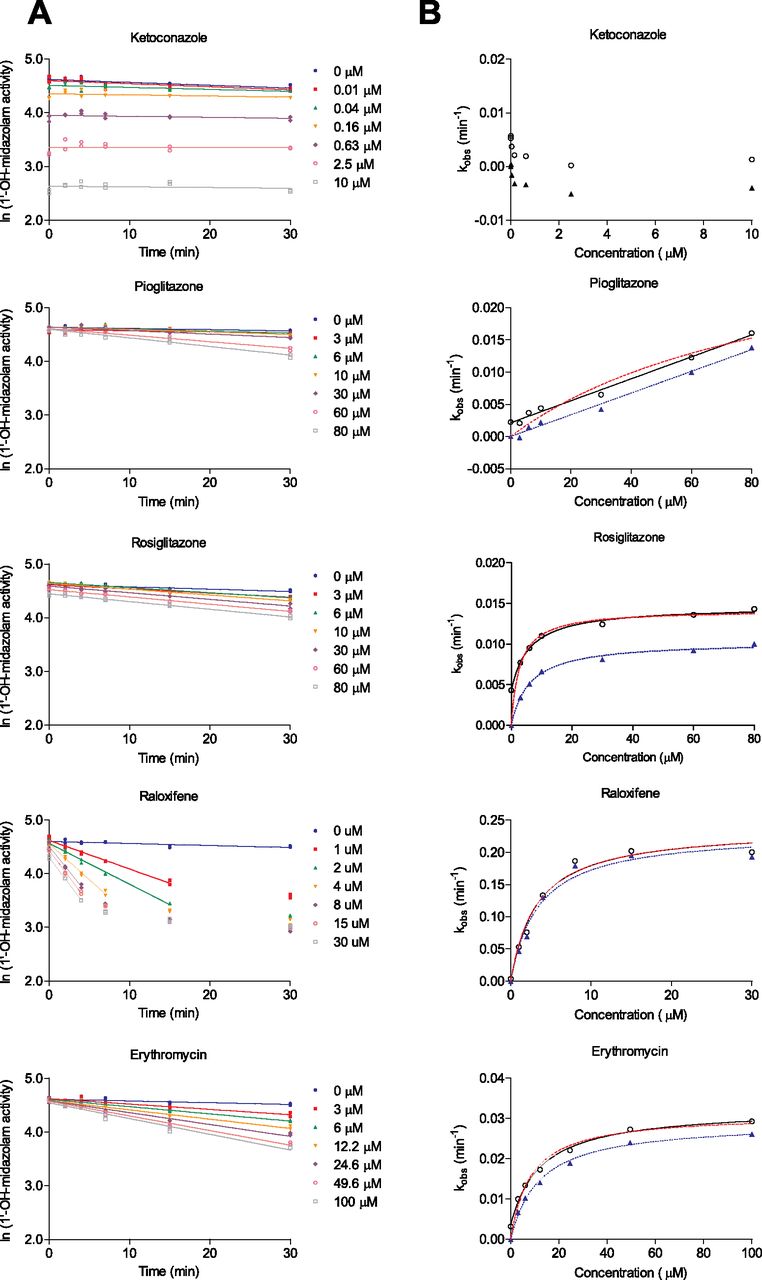
Inactivation of CYP3A4-mediated midazolam hydroxylation by ketoconazole, pioglitazone, rosiglitazone, erythromycin, and raloxifene. Inactivation plots to determine kobs (A) and nonlinear regression to determine KI and kinact (B). Incubations were done in duplicate, and individual activity values were plotted and analyzed. In B, solid lines and hollow circles represent the 3p fit; red dashed lines represent the 2pU fit to the hollow circles; blue dotted lines and filled triangles represent the 2pC fit. Experimental observations for raloxifene that were excluded from the kobs estimates are also indicated.
Summary of kobs and the statistical tests for kobs[I] vs. kobs[0 μM]
A compound is declared to be TDI when at least one p < 0.05.
Compounds with low empirical kobs values (e.g., 0.004–0.01 min−1) can be declared as TDI, albeit very weak, should kobs test significantly different from vehicle. Based on an empirical median S.E. estimate of 0.0012 min−1 (Table 1), the least significant difference that could be detected for a kobs[I] versus kobs[0 μM] comparison for our laboratory is 0.0033 min−1 at a p = 0.05 level. The clinical relevance of weak inactivators, for example, their potential for DDI in the clinic, and our ability to accurately predict DDI in humans is beyond the scope of this article. In a recent report it was suggested that kobs values less than 0.02 min−1 at 10 μM serve as a boundary between compounds that show a DDI for CYP3A and those that do not (Zimmerlin et al., 2011). Establishing a boundary on the limit of detection for kobs is currently subjective. If an artificially high limit is set the potential impact of weak inactivators will be overlooked. In our view, it is more important to determine whether kobs is significant based on statistical analysis. The estimated kinetic parameters can be used to model and predict in vivo DDI. It has been previously reported that even a weak TDI can cause significant DDI in the clinic (Obach et al., 2007). Because TDI is a safety endpoint we advocate a conservative approach toward evaluating and declaring TDI.
Step II: Can KI and kinact Be Determined?
The second step tests whether reliable KI and kinact values can be determined for a TDI compound. A statistical test for this is partially motivated via a plot of kobs versus ln[I] (Fig. 3) or [I] (Fig. 2B). Ketoconazole is not included in Fig. 3 because it is not a TDI; this compound is furthermore excluded from potential subsequent nonlinear modeling efforts. A significant positive correlation (p < 0.05) suggests that reliable kinetic parameter estimates may be achievable. One-sided tests, based on both a linear regression (ln[I]) and a nonparametric correlation estimator ([I]), are used. The suitability of the kobs versus ln[I] linear approximation may help gauge design quality and inactivator potency. The nonparametric test, although not as powerful as its parametric counterpart for small samples, provides a robust intermediate test for a monotonic increasing association. If both correlations are not significant, KI and kinact may not be reliably determined even though the compound is a TDI. If either test is significant, KI and kinact are calculated using a three-parameter nonlinear regression model (Fig. 2B).

Plots of estimated inactivation rates (kobs) versus the natural log of inactivator concentration (ln[I]) for four compounds. A linear fit (no nonzero concentration points were excluded) is used to evaluate the relationship between inactivation rate and concentration. Results using Spearman's correlation, a supplementary test, are listed in Table 2.
Reliable estimates for KI and kinact will, at a minimum, depend on the relationship between the estimate of KI and the range of [I] used in the experiment. Because this is true for any enzyme kinetic or binding experiment, we added an additional provision to guard against unreliable kinetic estimates (e.g., 3 [I]min < KI < 1/3 [I]max for a TDI compound likely to generate reliable kinetic estimates). A poor positioning of the KI estimate relative to the range of [I] may affect the S.E. for KI, reflect an intrinsically ill-conditioned regression, or a suboptimal experimental design. For example, the KI and kinact estimates for pioglitazone are not reported due to KI > 1/3 [I]max (Table 2). However, it should be noted that the KI for rosiglitazone was only two-fold greater than the lowest tested concentration, but nevertheless offered a quality fit for the kobs versus [I] relationship. This is evident in the small confidence interval in the parameter estimates (Table 2), and illustrates the notion that even when the KI approaches the maximum or minimum concentration tested good estimates of parameters are attainable.
Summary of pioglitazone, rosiglitazone, raloxifene, and erythromycin kinetic parameter estimates for two- (corrected and uncorrected kobs) and three-parameter models, Spearman's rank correlation coefficient (rs), and a test for a positive linear regression of kobs versus ln[I]
Ninety-five percent profile-likelihood confidence intervals for each kinetic estimate are listed in parentheses. The rank correlation and linear regression tests do not depend on the nonlinear model fit and are reported on the same row as the three-parameter model estimates. kobs[0 μM] is not estimated in the 2p model.
For the remaining four compounds examined in this article significant associations, for both the linear regression and Spearman's rank coefficient (rs) tests (Table 2), suggest that the kinetic parameters can be calculated via the Michaelis-Menten equation. In Fig. 3, we see a V shape in a plot of kobs versus ln[I] for the weak inactivator pioglitazone, an inverted V shape for the potent inactivator raloxifene, and a near linear relationship for the intermediate inactivators rosiglitazone and erythromycin. Such plots can serve as an ad hoc diagnostic for inactivator potency or concentration spacing and magnitude. We did not attempt to improve the quality of the linear approximation by removing selected kobs values; for some compounds and designs such an effort may be contraindicated. Applying the final provision to pioglitazone's estimate for KI suggested that the kinetic estimates for this weak inactivator are unreliable (KI > 1/3 [I]max; see Table 2). Because there are other experimental designs for TDI, such as the IC50 shift assay or simpler experiments that use one concentration and one preincubation time (Grimm et al., 2009), it is recommended that statistically based decision points be derived for these experimental designs as well.
Two- Versus Three-Parameter Michaelis-Menten Models.
TDI kinetic parameters are commonly estimated using either a two-parameter Michaelis-Menten model (kinact and KI; here, we either set kobs[0 μM] at time 0 to zero and normalize all of the kobs values against kobs[0 μM] for each time point, or did not perform this adjustment to any of the data; other “normalization” strategies are possible) or a three-parameter model (kinact, KI, and kobs[0 μM]). Depending on the number of observations, experimental design and strength of the nonlinear model, including kobs[0 μM] in the fitting of the kinetic parameters can be important to generate reliable estimates for kinact and KI. Assuming kobs[0 μM] equals zero in a two-parameter fit can bias estimates for kinact and KI for some inactivators and is influenced by strategies to “normalize” or “correct” the set of kobs relative to the vehicle kobs. Two- and three-parameter Michaelis-Menten kinetic estimates for the four inactivators are listed in Table 2. KI estimates for rosiglitazone and erythromycin, using uncorrected kobs estimates in a two-parameter model, differed noticeably from the three-parameter estimates for KI. Apart from rosiglitazone, the kinact estimates were comparable across the two- and three-parameter models. A graphical comparison of the models (Fig. 2B) was inconclusive in its ability to identify the most suitable model form. At a minimum, this makes apparent the challenges associated with generating precise kinetic estimates based on small samples. As evident in Table 2, assuming a two-parameter model also affects the width of the confidence intervals due to a more parsimonious model parameterization. Historical estimates for kobs[0 μM] from our laboratory ranges from 0.0002 to 0.0081 min−1 (mean ± S.D. = 0.0036 ± 0.0016, n = 60). This measureable uncertainty is explicitly included in a three-parameter model, whereas a two-parameter model may discount (or disguise) this fact via intermediate adjustments to the observed data or the estimates for kobs.
The correlation estimates between the various kinetic parameters using both corrected and uncorrected two- and three-parameter Michaelis-Menten fits are summarized in Table 3. Here, a two-parameter model more strongly affects the correlation between kinact and KI as compared with a three-parameter model. This illustrates the coupling of design-modeling complexities for producing estimates for kobs[0 μM], KI, and kinact using a limited number of observations/concentration range. Large correlation differences between the corrected and uncorrected two-parameter kinetic estimates are not evident. For pioglitazone, the evident lack of nonlinearity in kobs (Fig. 2B) suggests that both the two- and three-parameter models are over-parameterized. The near-perfect correlation between KI and kinact reflects this result. Despite observable curvature in kobs for the remaining three compounds (Fig. 2B), the kinetic correlations for the two-parameter model are high. Transitioning to a three-parameter model reduced the correlation between KI and kinact while making evident the correlation of these parameters with kobs[0 μM]. The three-parameter correlation structure reflects the observable experimental interdependencies between the three kinetic estimates and, although not formally presented here, can be used to diagnose the individual kinetic estimates or the design. Seber and Wild (2003) discuss common problems pertinent to nonlinear regression, for example, inherently ill-conditioned models or very imprecise/highly correlated estimates. Our experimental data indicates that the stochastic behavior of kobs[0 μM] has a nonzero mean, and we capture this uncertainty in our statistical model definition. It is clear that kobs[0 μM] possesses a nonzero value, and that attempts to force the kobs versus [I] relationship through the origin without correcting for this factor is inappropriate and yields estimates of KI that are incorrect (Table 2). Although extreme differences between the kinetic estimates under the two model parameterizations are not grossly evident in Table 2, other methods to adjust for nonzero kobs[0 μM] or more variable experimental data can result in large kinetic model discrepancies. For estimating TDI kinetic parameters, we advocate the use of the three-parameter form of the Michaelis-Menten equation.
Nontrivial correlation estimates for the Michaelis-Menten kinetic parameters using both two- and three-parameter nonlinear models
Others Methods Considerations.
Various kobs comparisons, for example, kobs[60 μM] versus kobs[30 μM] or kobs[0 μM], could be tested using a combined regression-ANOVA statistical model (Myers, 1990). We did not apply such a model for several reasons: specifying/fitting a suitable statistical model combines aspects of the experimental protocol with more complex estimation algorithms, (unplanned) tests of various kobs combinations increases the need for more careful Type I error control (an incorrect rejection of the null hypothesis), and the slope-based inactivation rate estimate at one or more concentrations may be poor in the log-linear approximation for strong TDI compounds [i.e., more pronounced activity attenuation can occur at long time points (Obach et al., 2010)]. Here, we removed several experimental observations to improve the kobs estimates for raloxifene. Removing select observations to create a more defensible model creates imbalances in these data, affects estimate S.E.s (only the lowest raloxifene concentration S.E.s resembled those obtained for the other compounds), and affects degrees of freedom calculations. Concerns over how best to count the “right” number of degrees of freedom, in the presence of a potentially unbalanced complex experimental structure, combined with a conservative testing approach, motivated our use of a z-test in place of a t test. The large sample-based test statistic calculations proposed here, although open to statistical criticism, are straightforward and easily calculated in widely available software. A closed-form z-test easily supports the possibility of plug-in S.E. estimators should severe under- or overestimates for one or more RMSEs be observed. Aliquot extraction at various time intervals suggests that the distinction between experimental and sampling units may need careful consideration or induce a nontrivial correlation structure between observations. More complex statistical models or experimental designs may be employed here. Bayesian methods may also be suitable for experimentalists who routinely conduct TDI experiments.
We are not recommending that multiplicity corrections, for example, Dunnett's test, be performed across the j-1 kobs[I] − kobs[0 μM] comparisons. In practice, only a small number of compound concentrations are evaluated across a limited (empirically determined) potency range. We consider an (in)ability to differentiate or test pairs of kobs estimates using unadjusted p values more relevant to the TDI decision process than controlling for an overall family-wise Type I error or false discovery rate. As suggested earlier, a careful consideration of the precise α level used for testing can mitigate false alarm concerns. The interplay between aspects of testing and the magnitudes of the observed experimental variances across one or more experiments is nontrivial. Furthermore, for TDI compounds the kobs values must be monotonic (kobs,j > kobs,i for some concentrations [I]i and [I]j) in relation to increasing concentration. The tests for the regression-based slope γ1 and rank correlation-based rs proposed here, although not providing rigorous Type I control for the overall test for TDI, should reflect this pattern in addition to providing a post hoc measure of protection against subsequent attempts to fit an ill-conditioned kinetic equation. Potential TDI false positives pose less of a concern relative to allowing a false negative TDI compound to reach the later stages of drug development due to TDI's relation to a safety endpoint. At present, we have adopted a conservative stance and expect that an evolving standard for declaring TDI may emerge in the literature over time.
The use of traditional statistical estimators allows us to exploit well-understood large sample theory. We acknowledge the inherent risks in such an approach, but numerical routines, such as resample/bootstrap procedures, also pose a concern for investigators (Berger, 2000). One- or two-sided tests may be performed here; one's choice influences the interpretation of the results and the power of the test. Here, we elect to use a two-sided hypothesis test for a pair of kobs (H0: kobs[I] − kobs[0 μM] = 0 versus H1: kobs[I] − kobs[0 μM] ≠ 0), but chose a one-sided test for γ1 and rs (e.g., H0: rs = 0 versus H1: rs > 0). The first case may gauge empirical precision, whereas the latter test may be preferred for further TDI screening efforts. Calibrating the procedure against known positive or negative controls can also be performed. Spearman's rank correlation coefficient rs was preferred to other nonparametric estimators, for example, Kendall's τ or Hoeffding's D (Hollander and Wolfe, 1999), due to its comparability with the widely used Pearson product-moment correlation coefficient and its use as a specific test for a positive association between kobs and [I].
We are not recommending a novel approach to estimating kinact or KI; standard least-squares nonlinear regression was performed here. Unlike other linear-based Michaelis-Menten approximations, such as Lineweaver-Burk or Eadie-Hofstee fits, which are known to be problematic (Ritchie and Prvan, 1996), both the nonparametric and slope-based tests are not used to estimate kinetic parameters. Wald-type or profile-likelihood methods can be used to generate confidence intervals and test the model estimates. An ordinary least-squares test of the kobs-ln[I] slope is understood to be imperfect. But, fitting and diagnosing model quality for an (intercept-adjusted) Michaelis-Menten curve based on a small sample is also challenging. Seber and Wild (2003) document the use of transformations to circumvent nonlinear modeling challenges. Reducing the number of parameters to estimate and test can provide benefit as a preliminary screening measure. A linear fit simplifies model diagnostics, for example, to identify outliers or detect abrupt transitions in kobs. The use of Spearman's coefficient rs helped mitigate concerns regarding inadequate linear approximations for some TDI compounds. A large number of samples for analysis may negate the need for the linear test.
Conclusions
In this report, we have proposed a two-step statistical approach to the analysis of CYP3A4 TDI data. This is particularly important in the research of new drugs because the U.S. Food and Drug Administration recently issued a draft guidance stating that “any time-dependent loss of activity” (sic) caused by a P450 inhibitor merits further investigation. The first step, a two-sided two-sample z-test, offers a simple and effective manner of evaluating TDI data and determining whether a new drug is a TDI, without resorting to empirically derived or heuristic cutoff values. The second step shows whether a set of kobs versus [I] or ln[I] data can be used to estimate KI and kinact, ultimately using a three-parameter Michaelis-Menten model. An important attribute to the successful application of this approach requires that the experimental technique be precise and reproducible; if individual data points of the natural logarithm of activity versus preincubation time relationships show a high degree of scatter, TDI could be missed because the z-test p values will exceed 0.05. In our research, kobs values of approximately 0.005 min−1 can be reliably discerned and yield p < 0.05 when tested for known TDI compounds. Although drugs yielding such low rates of inactivation may not result in meaningful in vivo DDI, it is still nevertheless important to identify such compounds in vitro so that appropriate clinical follow-up can be done. Further improvements in physiologically based pharmacokinetic modeling of CYP3A4 TDI data are needed to better use high quality in vitro data in generating reliable predictions of DDI.
Authorship Contributions
Participated in research design: Yates, Eng, Di, and Obach.
Conducted experiments: Eng.
Performed data analysis: Yates, Eng, Di, and Obach.
Wrote or contributed to the writing of the manuscript: Yates, Eng, Di, and Obach.
Acknowledgments
We thank Mark Niosi for generating some of the experimental data and Dr. Larry Tremaine for leadership and support.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
ABBREVIATIONS:
- TDI
- time-dependent inhibition
- DDI
- drug-drug interaction
- DMSO
- dimethyl sulfoxide
- LC-MS/MS
- liquid chromatography/tandem mass spectrometry
- P450
- cytochrome P450
- RMSE
- root mean square error.
- Received June 13, 2012.
- Accepted August 31, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}