


Abstract
Allometric scaling may be used in drug development to predict the pharmacokinetics of xenobiotics in humans from animal data. Although allometry may be successful for compounds that are excreted unchanged or that are oxidatively metabolized (with corrections for metabolic capacity), it has been more challenging for compounds excreted primarily as conjugates in bile. (S)-10,11-Dihydro-3-[3-(pyridin-2-ylamino)-1-propyloxy]-5H-dibenzo[a,d]cycloheptene-10-acetic acid (SB-265123) is a novel αvβ3 (“vitronectin receptor”) antagonist. In this study, the in vivo pharmacokinetics and in vitro plasma protein binding of SB-265123 were examined in four species: mice, rats, dogs, and monkeys. In monkeys and dogs, SB-265123 exhibited moderate clearance, whereas low clearance (<20% hepatic blood flow) was observed in the rat, and high clearance (>70% hepatic blood flow) was seen in the mouse. The concentration-time profiles indicated the possibility of enterohepatic recirculation; subsequent studies in bile duct-cannulated rats demonstrated extensive biliary excretion of an acyl-glucuronide of SB-265123. In allometric scaling to predict the disposition of SB-265123 in humans, various standard correction factors were applied, including protein binding, maximum lifespan potential, and brain weight; each failed to produce adequate interspecies scaling of clearance (r2< 0.72). Consequently, a novel correction factor incorporating bile flow and microsomal UDP-glucuronosyltransferase activity in each species was applied, demonstrating substantial improvement in the correlation of the allometric plot (r2= 0.96). This study demonstrates a novel allometric correction that may be applicable to compounds that undergo conjugation and biliary excretion.
The process by which new drug candidates are characterized in preclinical models has been subjected to much scrutiny in an effort to optimize selection processes and thereby minimize time and cost of drug development (DiMasi, 1994; Kuhlmann, 1997). Obviously, an accurate prediction of the clinical pharmacokinetic behavior of a new drug based on the available preclinical data would be a very useful tool in the selection of drug candidates to carry forward into development. Numerous methods have been used, with varying degrees of success, to attempt such predictions (Ings, 1990). Perhaps the most common and well understood technique for interspecies extrapolation of pharmacokinetic data is allometric scaling (Ritschel et al., 1992), in which quantitative relationships are established between drug disposition and physiological parameters such as body weight or organ perfusion rate. Successful allometry depends on a number of factors, including data quality, number of species included in the extrapolation, and appropriate identification of the combination of physiological parameters and scaling factors that best relate to the disposition of a given molecule. Because of its potential complexity, allometry is most frequently conducted on a compound-by-compound basis, although there have been recent attempts to perform simultaneous allometry for a series of structurally diverse compounds (Bachmann et al., 1996;Mahmood and Balian, 1996a; Obach et al., 1997; Riviere et al., 1997).
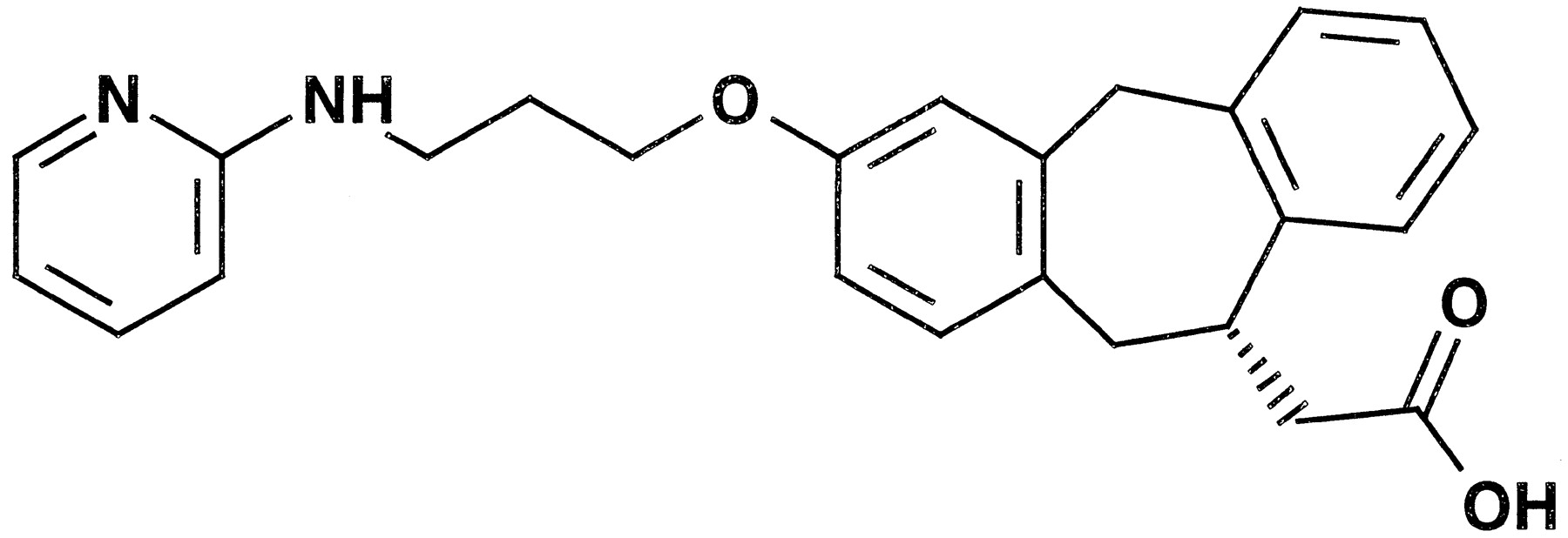
αvβ3 (the “vitronectin receptor”) is a member of the integrin superfamily of adhesion molecules expressed on the surface of a wide variety of cell types, including vascular smooth muscle cells, endothelial cells, various tumor cells, and osteoclasts (Horton, 1997). The vitronectin receptor mediates several important biological processes involving these cell types, including the adhesion of osteoclasts to the bone matrix, angiogenesis, and smooth muscle cell migration; antagonism of this receptor may provide effective therapy for disease processes regulated by these cell types. The identification of potent nonpeptide vitronectin receptor antagonists has been described previously (Keenan et al., 1997, 1998); however, these molecules have generally possessed poor pharmacokinetic properties and low oral bioavailability. Recently, Miller and colleagues (1999)have described (S)-10,11-dihydro-3-[3-(pyridin-2-ylamino)-1-propyloxy]-5H-dibenzo[a,d]cycloheptene-10-acetic acid (SB-265123) (Fig. 1), a potent nonpeptide antagonist of the vitronectin receptor. Preliminary data indicated the possibility of favorable pharmacokinetic properties for SB-265123, including good oral bioavailability.

Chemical structure of SB-265123.
In the present investigation, the preclinical pharmacokinetic characteristics of SB-265123 were investigated, and various allometric scaling techniques for this compound were explored across four species. The objectives of this study were to define the pharmacokinetic behavior of SB-265123 in the mouse, rat, dog, and monkey, and to identify a preferred allometric scaling technique to describe the disposition of SB-265123.
Experimental Procedures
Materials.
SB-265123 was synthesized by the Department of Medicinal Chemistry at SmithKline Beecham Pharmaceuticals (King of Prussia, PA). All drug material was determined to be at least 95% pure by thin layer chromatography, mass spectrometry (MS)2, and/or HPLC. All other reagents and materials were purchased from standard vendors and were of the highest available purity.
Animals.
Male (FVB/N)FBR mice (Taconic Farms, Inc., Germantown, NY) weighing 22 to 32 g, male Sprague-Dawley rats (Charles River Breeding Laboratories, Inc., Raleigh, NC) weighing 290 to 350 g, male purebred beagle dogs (Marshall Research Animals, Inc., North Rose, NY) weighing 9.2 to 10.8 kg, and male Cynomolgus monkeys (Charles River Primate Labs, Houston, TX) weighing 6.3 to 6.5 kg were used for the pharmacokinetic studies. All animals were housed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals in individual cages in unidirectional airflow rooms with controlled temperature (22 ± 2°C) and relative humidity (50 ± 10%) and 12-h light/dark cycles (6:00 AM–6:00 PM). Filtered tap water was available ad libitum and was periodically analyzed by Philadelphia Suburban Water Company. Animals were fed a standard animal diet (Purina Mills, St. Louis, MO); food was available ad libitum except for certain overnight periods before dosing, as described below. Whenever overnight fasting was used, food was provided after the 240-min blood sample was obtained on the following study day. All animal use was conducted according to protocols reviewed and approved by the Institutional Animal Care and Use Committee before the study, and all surgical procedures were conducted with aseptic techniques in special-purpose operating suites.
In Vivo Pharmacokinetic Studies.
All dosages were administered as solutions in either isotonic saline (i.v. doses) or analytical grade water (oral doses), and contained up to 3% dimethyl sulfoxide and/or 5% polyethylene glycol-300 (v/v) to aid in dissolution of the test compound. Previous experience with this class of compounds has indicated that use of these dose vehicles does not alter the pharmacokinetic behavior of these molecules. Verification of the concentration of test compound in each dose solution was performed by HPLC/MS/MS. In all studies, the same animals received the i.v. and oral dosage at least two (mice and rats) or seven (dogs and monkeys) days apart, except as noted below. A complete blood count screen was performed on each dog and monkey before each study day to obtain baseline values and ensure hematological recovery. Blood samples were collected from each study animal at various times during and after drug administration, as noted in the figures; plasma (50 μl for rats, dogs, and monkeys; 20 μl for mice) was isolated by centrifugation and stored at −70°C until analysis.
Mice.
Four mice received two surgically implanted jugular vein catheters each for blood sampling and drug administration. The surgery was performed under ketamine/xylazine (80:50 mg/kg i.m.) anesthesia at least 3 days before the 1st day of the experiment; each catheter was filled with heparinized saline when not in use to maintain patency. The catheters were positioned such that blood collection from one catheter was not influenced by drug administration from the second catheter. On each study day, the mice were placed in plastic restrainers for up to 2 h before being returned to their cages. On study day 1, the mice received SB-265123 (2.8 mg/kg) as a 30-min i.v. infusion (4.0 ml/kg). On study day 2, after an overnight fast, the mice received SB-265123 (2.7 mg/kg) as an oral gavage (16 ml/kg).
Rats.
Six rats received surgically implanted femoral vein catheters for drug administration. The surgery was performed under ketamine/xylazine (87:13 mg/kg, i.m.) anesthesia 4 to 5 days before the 1st day of the experiment; the catheters were filled with heparinized saline when not in use to maintain patency. On each study day, the rats were held in restrainers to facilitate drug administration and blood sampling (from a lateral tail vein) for up to 2 h before being returned to their cages. On study day 1, the rats received SB-265123 (0.8 mg/kg) as a 30-min i.v. infusion (4.0 ml/kg). On study day 2, after an overnight fast, the rats received SB-265123 (0.75 mg/kg) as an oral gavage (4 ml/kg).
Bile Duct-Cannulated Rats.
Two rats received surgically implanted femoral artery catheters in addition to femoral vein catheters for blood sampling and drug administration, as described above. Also, the bile duct of each rat was cannulated in a loop fashion, such that normal bile flow through the catheter into the duodenum was maintained before and after the conduct of each day of the study (Tomlinson et al., 1981). On study day 1, the rats received SB-265123 (0.8 mg/kg) as a 30-min i.v. infusion (4.0 ml/kg). On study day 2, after an overnight fast, the rats received SB-265123 (0.75 mg/kg) as an oral gavage (4 ml/kg). Before each dose administration, the bile duct catheter loop was disconnected, and control bile was collected for 15 min. On each study day, bile was collected continuously over dry ice at 2-h intervals through 12-h postdose, and also for the 12- to 24-h interval before the catheter loop was reconnected.
Dogs.
On each study day, a cephalic vein catheter was placed in each dog for blood sampling; on study day 1, a catheter also was placed in the saphenous vein of each dog for drug administration. On each study day, the dogs were restrained in slings to facilitate drug administration and blood sampling for up to 2 h before being returned to their cages. On study day 1, the dogs received SB-265123 (1 mg/kg) as a 60-min i.v. infusion (4.0 ml/kg). On study day 2, after an overnight fast, the dogs received SB-265123 (1 mg/kg) as an oral gavage (4 ml/kg).
Monkeys.
Each monkey had a previously implanted chronic vascular access port in a femoral vein that was used for blood sampling; on study day 1, a catheter also was placed in a saphenous vein for drug administration. On each study day, the monkeys were placed in restraining chairs for the first 4 h of the study to facilitate drug administration and blood sampling. On study day 1, after an overnight fast, the monkeys received SB-265123 (2 mg/kg) as a 60-min i.v. infusion (4.0 ml/kg). On study day 2, after an overnight fast, the monkeys received SB-265123 (4 mg/kg) as an oral gavage (8 ml/kg).
In Vitro Plasma Protein Binding.
Plasma protein binding of SB-265123 was investigated in each of the four preclinical species, as well as in humans. Approximately 20 ml of whole blood (10 ml for mouse) was collected by cardiac puncture from mice and rats, and by peripheral venipuncture from dogs, monkeys, and one male human volunteer. Plasma was obtained by centrifugation. Incubation mixtures were prepared in plasma of each species at final concentrations of 1000 and 10,000 ng/ml; similar solutions were prepared in Krebs-Ringer bicarbonate (pH 7.4) to test for nonspecific binding of test compound to the filtration device. For each incubation mixture, 4 ml (2 ml for mouse) of plasma or buffer was prewarmed for 5 min in a 37°C water bath; 20 μl (10 μl for mouse) of a 0.2 or 2.0 mg/ml stock solution of SB-265123 in 10% aqueous dimethyl sulfoxide was added to the prewarmed plasma or buffer to achieve the desired final concentrations. After addition of the stock solutions, each incubation mixture was vortexed gently to ensure uniform mixing, and a single 50-μl (20-μl for mouse) aliquot was collected to verify initial concentrations. Mixtures were incubated in a 37°C water bath for at least 15 min. After incubation, triplicate 1-ml (0.5-ml for mouse) aliquots were collected from each incubation mixture and placed into Centrifree micropartition devices (Amicon, Inc., Beverly, MA; MW cutoff = 30,000). The tubes were centrifuged at ∼200g for 30 min to generate an ultrafiltrate ≤10% of the volume of the original sample. One 50-μl (20-μl for mouse) aliquot was collected from the ultrafiltrate (protein-free plasma) of each tube (three samples per mixture). One 50-μl (20-μl for mouse) aliquot was collected from the plasma in the top portion of one tube from each incubation mixture to determine recovery of test compound (one sample per mixture). Each sample was quick-frozen on dry ice and stored at −70°C until analysis.
Analytical Procedure.
Quantitative analysis of plasma, bile, and buffer samples for SB-265123 was performed with a HPLC/dual mass spectometry (MS/MS) method. SB-265123 was isolated from 50 μl of rat, dog, or monkey plasma or rat bile, or from 20 μl of mouse plasma, by protein precipitation and quantified with a Sciex API 365 instrument with a turbo-ionspray interface. Samples containing SB-265123 were injected onto a Genesis C18 (3 × 50 mm, 3 μm packing) column under isocratic conditions [60:40, 10 mM ammonium formate (pH 3.0)/acetonitrile] at a flow rate of 300 μl/min. Positive-ion multiple reaction monitoring was used for the MS/MS detection of SB-265123. The lower limit of quantitation was 10.0 ng/ml. Additionally, qualitative and semiquantitative analysis was performed on bile samples collected from the bile duct-cannulated rats to determine the presence of metabolites of SB-265123 in bile. HPLC/MS/MS analysis of SB-265123 in rat bile for the identification of metabolites was performed by diluting the neat bile 1:1 in 10 mM ammonium formate buffer (pH 3.0) and injecting 10 μl onto a BDS Hypersil C8 (2.1 × 50 mm, 3 μm packing) column. A Hitachi L7100 ternary pump delivered a gradient from 5 to 85% acetonitrile over 15 min at 0.2 ml/min, with 10 mM ammonium formate (pH 3.0). A Finnigan LCQ ion trap mass spectrometer was used with full scan data-dependent MSn, in positive ion electrospray mode. The metabolite analysis was semiquantitative due to the unavailability of analytical standards of the identified metabolites.
Pharmacokinetic Analysis.
Concentration-time profiles after both i.v. and oral administration of SB-265123 were obtained for each animal. Several different compartmental models were fit to each individual data set with the nonlinear least-squares regression analysis program WinNonlin Professional Version 1.0 (Scientific Consultants, Inc., Apex, NC). The most appropriate model to describe the data was chosen based on Akaike’s Information Criterion (Yamaoka et al., 1978) and lack of bias in residual error. For each data set, the model that best described the observed data was a two-compartment model with first order elimination from the central compartment (Gabrielsson and Weiner, 1997). WinNonlin was used to generate the best-fit critical pharmacokinetic parameters for each animal, including the elimination rate constant (k10, min−1), the intercompartmental transfer rate constants (k12 and k21, min−1), and, in the case of oral administration, the absorption rate constant (ka, min−1). Other pharmacokinetic parameters also were estimated with WinNonlin, including maximum plasma concentration (Cmax, nanograms per milliliter), the distribution and elimination half-lives (T½α andT½β, min), systemic plasma clearance (CL, kilograms · milliliter/minute), the mean residence time (min), and steady-state volume of distribution (Vdss, milliliters per kilogram). Oral bioavailability also was estimated for each oral dosage by dividing the dose-normalized area under the concentration-time curve resulting from oral administration by the dose-normalized area under the concentration-time curve resulting from i.v. administration. Individual pharmacokinetic parameters were generated for each study animal; the data for each pharmacokinetic parameter were averaged and reported as means ± S.D. For analysis of plasma protein binding in each species, the concentration of SB-265123 in the plasma ultrafiltrate (corrected for nonspecific binding by dividing by the buffer free fraction) was divided by the concentration in the plasma reservoir to yield the unbound fraction (fu) in the sample; values were multiplied by and then subtracted from 100 to arrive at the percentage of SB-265123 bound to plasma proteins (fb). Values are reported as means ± S.D. for three ultrafiltrate determinations.
Allometric Scaling.
For interspecies scaling, the values of CL and Vdss obtained for each animal with compartmental analysis were plotted against individual body weight (kilograms) on a log-log scale (Ritschel et al., 1992). Linear least-squares regression analysis was performed on these plots to fit these relationships to the equations
Physiological parameters used in the allometric scaling of SB-265123
Statistical Analysis.
An unpaired Student’s t test was used to assess the significance of differences between the pharmacokinetic parameters determined in control and in bile duct-cannulated rats. In instances where parameters possessed unequal variances, statistical comparison was made with the Mann-Whitney nonparametric U test. One-way ANOVA was used to assess the significance of differences in pharmacokinetic parameters for SB-265123 across species. In all cases, a probability level of p < .05 was predetermined as the criterion of significance.
Results
Figure 2A displays representative concentration-time profiles for SB-265123 in the rat, as well as a representation of the model fit to the observed data, after both i.v. and oral administration. In general, the two-compartment model with first order elimination from the central compartment provided a good fit to the observed data. The pharmacokinetic parameters generated from this model are displayed in Table 2. SB-265123 exhibited low plasma clearance in the rat (<20% hepatic blood flow), with an elimination half-life of 2.5 to 3 h, and a Vdss somewhat greater than total body water. After oral administration, SB-265123 was well absorbed in the rat, with excellent oral bioavailability. Similarly, Fig. 2B displays the concentration-time profiles for SB-265123 in the dog, as well as a representation of the model fit to the observed data, after both i.v. and oral administration. As with the rat, the selected two-compartment model provided a good fit to the observed data; the calculated pharmacokinetic parameters for SB-265123 are displayed in Table 2. In contrast to the rat, the compound exhibited moderate clearance in the dog (∼50% hepatic blood flow). After oral administration, SB-265123 was rapidly absorbed in the dog, and possessed good bioavailability.

Representative concentration-time profiles for SB-265123 after i.v. (○, solid lines) or oral (▵, dashed lines) administration to the rat (A) or dog (B).
Lines indicate best fit of a two-compartment model with first order elimination from the central compartment to the observed data.
Pharmacokinetic parameters defining systemic disposition of SB-265123 in various species after i.v. and oral administration
Examination of the concentration-time profiles and pharmacokinetic parameters for SB-265123 in the rat revealed that the compound displayed an apparent oral bioavailability >100%. One explanation for this phenomenon could be enterohepatic recirculation, either via excretion of unchanged compound or a conjugated derivative into bile. The possible existence of enterohepatic recirculation of SB-265123 was supported by the apparent increase in plasma concentrations at later time points in the rat (Fig. 2A), dog (Fig. 2B), and monkey (data not shown). To examine this possibility, an experiment was conducted in bile duct-cannulated rats. The concentration-time profiles for representative bile duct-exteriorized rats are compared with those for noncannulated rats in Fig. 3; the pharmacokinetic parameters associated with bile duct cannulation in the individual rats are presented in Table 2. In general, bile duct cannulation appeared to produce an increase in overall elimination; systemic clearance increased, and mean residence time and terminal half-life decreased substantially. Also, the AUC after both i.v. and oral administration decreased dramatically in the bile duct-cannulated animals, and the apparent recirculatory phenomenon evident from the concentration-time profiles in intact animals (Fig. 2) disappeared from the profiles for the cannulated rats (Fig. 3). However, apparent oral bioavailability in the bile duct-cannulated rats was still in excess of 100%, suggesting that enterohepatic recirculation is not the sole explanation for this phenomenon. A more detailed investigation of the oral bioavailability of SB-265123 in the rat is being conducted and will be presented in a separate publication.
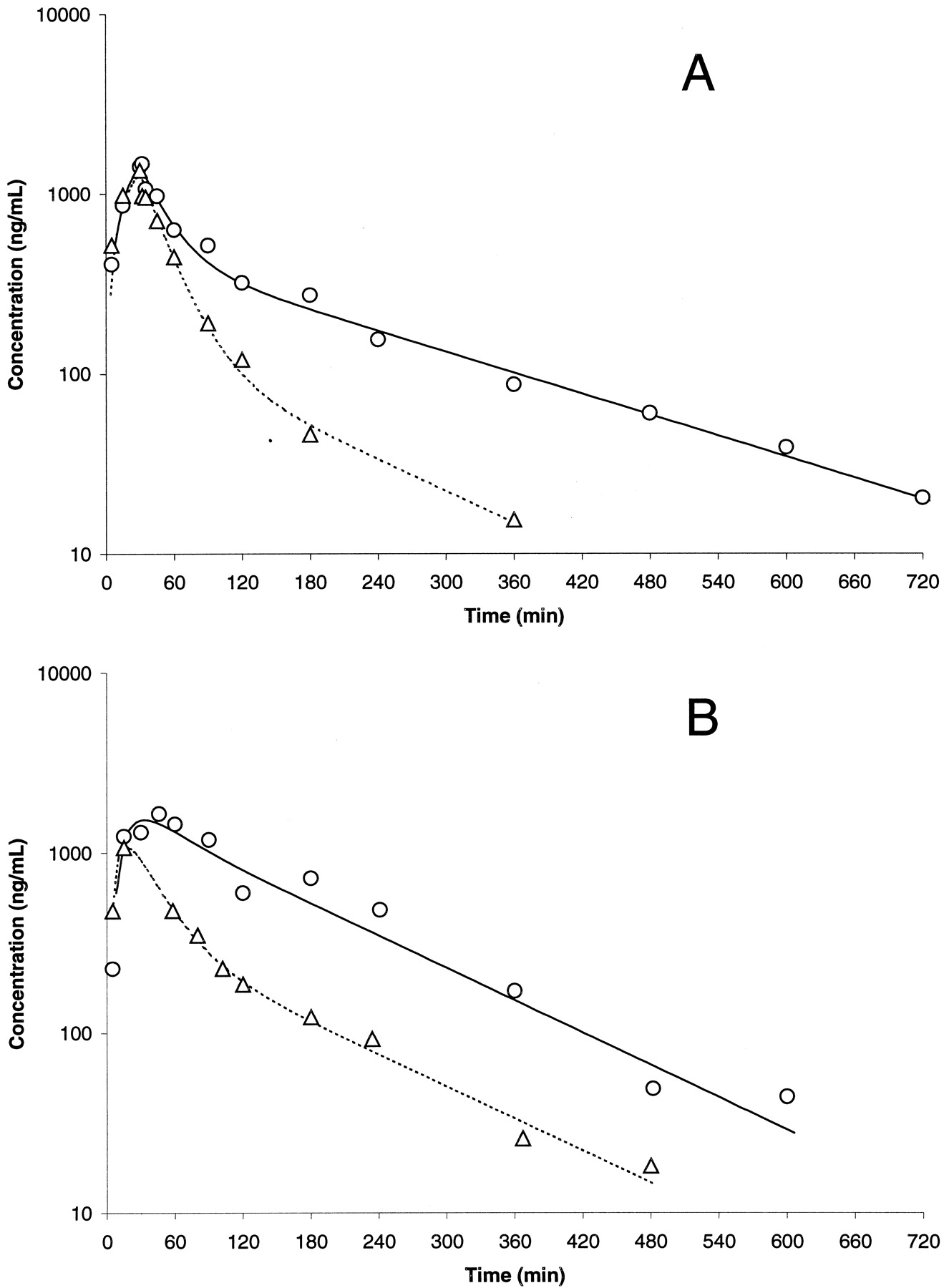
Representative concentration-time profiles in control (○, solid lines) and bile duct-cannulated (▵, dashed lines) rat for SB-265123 after i.v. (A) or oral (B) administration.
Lines indicate best fit of a two-compartment model with first order elimination from the central compartment to the observed data.
In addition to the plasma samples collected from these animals, SB-265123 and its major metabolites were identified in bile from the bile-exteriorized animals after both i.v. and oral administration by HPLC/MS/MS (Table 3). SB-265123 was excreted into bile after both i.v. and oral administration; an acyl-glucuronide of SB-265123 was the only metabolite observed in bile. Although a precise analytical response factor for the glucuronide was not known because of the lack of a true analytical standard, it was apparent that substantial amounts of this biotransformation product were excreted in bile. With an approximate (and likely underestimated) response factor for the glucuronide, at least 20% of the administered dose was excreted in the bile as either parent or conjugated material after i.v. administration, and at least 15 to 35% after oral administration
Biliary excretion of SB-265123 and the acyl-glucuronide of SB-265123 in bile duct-cannulated rats
The pharmacokinetic parameters obtained after administration of SB-265123 to the mouse and the monkey are displayed in Table 2. In the mouse, plasma clearance of SB-265123 was high (>70% hepatic blood flow), with an associated terminal half-life of <30 min and a distributional volume about three times body water. After oral administration, the molecule was well absorbed and exhibited good bioavailability. In the monkey, the disposition of SB-265123 was somewhat different, with a low-to-moderate clearance (∼30% hepatic blood flow) and a long, but variable terminal half-life. The oral bioavailability of SB-265123 in the monkey was ∼24%. Taking all the pharmacokinetic data for each of the four species examined, a statistical analysis was performed by one-way ANOVA to compare the pharmacokinetic parameters of SB-265123 in each species. Significant differences were noted across species for all parameters except half-life and the fitted oral time to peak plasma concentrations.
In addition to the in vivo pharmacokinetic studies, the in vitro plasma protein binding of SB-265123 also was examined. As detailed in Table4, plasma protein binding was relatively high in all species examined. The unbound fraction was slightly, although significantly, greater at the 10,000 ng/ml concentration compared with the value obtained at 1000 ng/ml in all species except monkey. Furthermore, significant differences were noted across species at both concentrations tested, but not for the mean value of both concentrations. Post hoc comparisons with a Bonferroni correction with results from the one-way ANOVA across species with each concentration revealed significant differences between mouse and human at the 1000 ng/ml incubation, and between all species except rat and dog at the 10,000 ng/ml concentration. Because the in vivo pharmacokinetic profiles spanned a range that incorporated both values tested, the mean protein binding value was used for all allometric scaling corrections.
Plasma protein binding of SB-265123 in mouse, rat, dog, monkey, and human
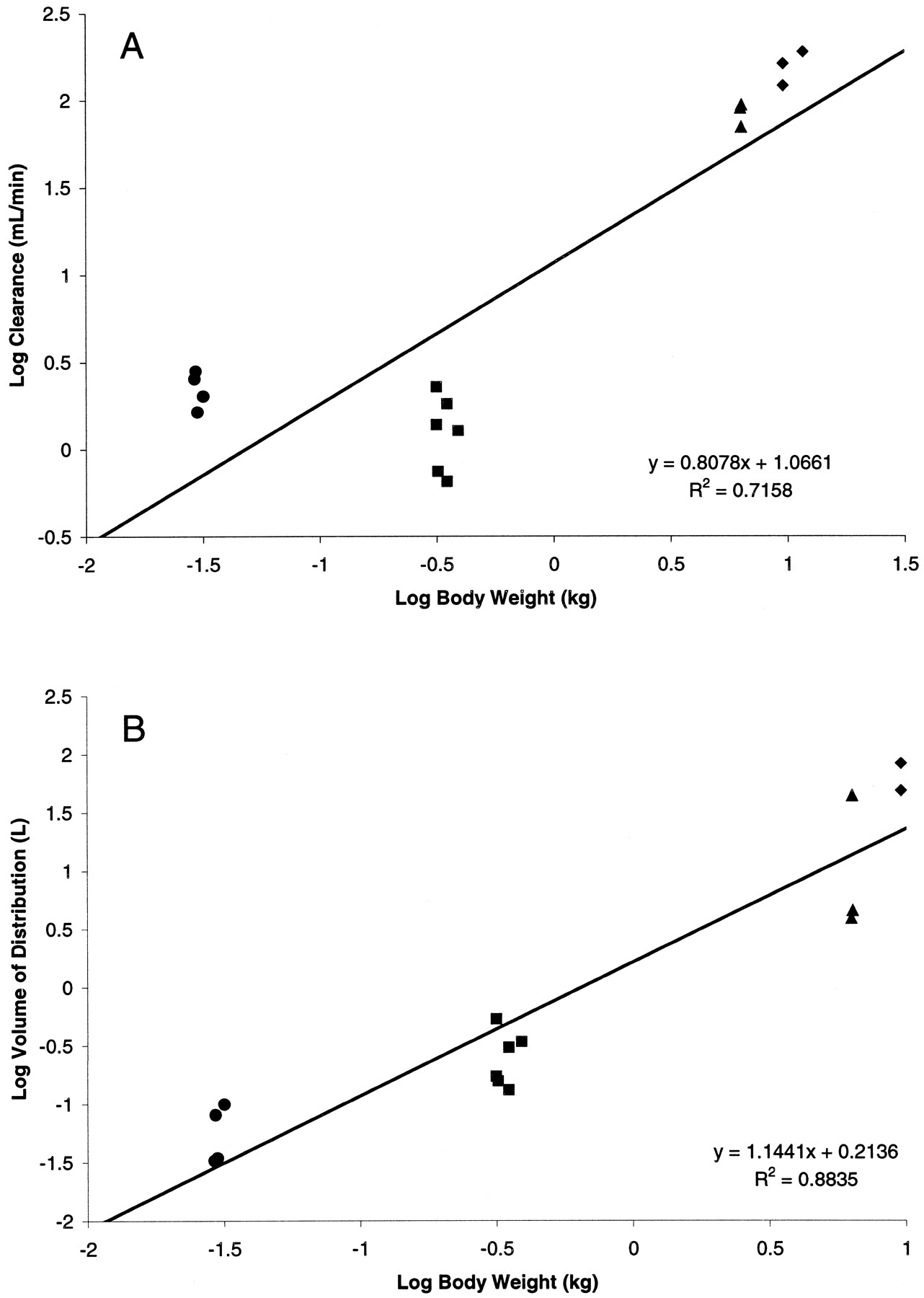
Finally, all the data for each species were incorporated to perform interspecies allometric scaling for SB-265123. Figure4 demonstrates the standard log-log allometric plots for clearance and volume of distribution across species, without the inclusion of any correction factors. Although distributional volume scaled reasonably well across species (r2 = 0.88), plasma clearance did not (r2 = 0.716). Correction of both plasma clearance and distributional volume for interspecies differences in plasma protein binding did not substantially alter the scaling profile; plasma clearance still did not scale well across species, whereas correction for plasma protein binding did not substantially alter the interspecies correlation for distributional volume. In an attempt to improve the scaling of clearance across species, other correction factors reported in the literature were attempted. As detailed in Table 5, although inclusion of a correction factor for maximum life span potential or brain weight somewhat improved the interspecies correlation coefficient for plasma clearance, a good correlation was still not achieved. Given the data from this investigation demonstrating excretion of SB-265123 in the bile as a glucuronide conjugate, a novel correction factor incorporating bile flow and activity of microsomal UDPGT was incorporated. Values for bile flow and hepatic microsomal UDPGT content were derived from the literature (Table 1). Figure5 demonstrates that inclusion of this correction factor substantially improved the interspecies correlation coefficient for plasma clearance (r2 = 0.949).
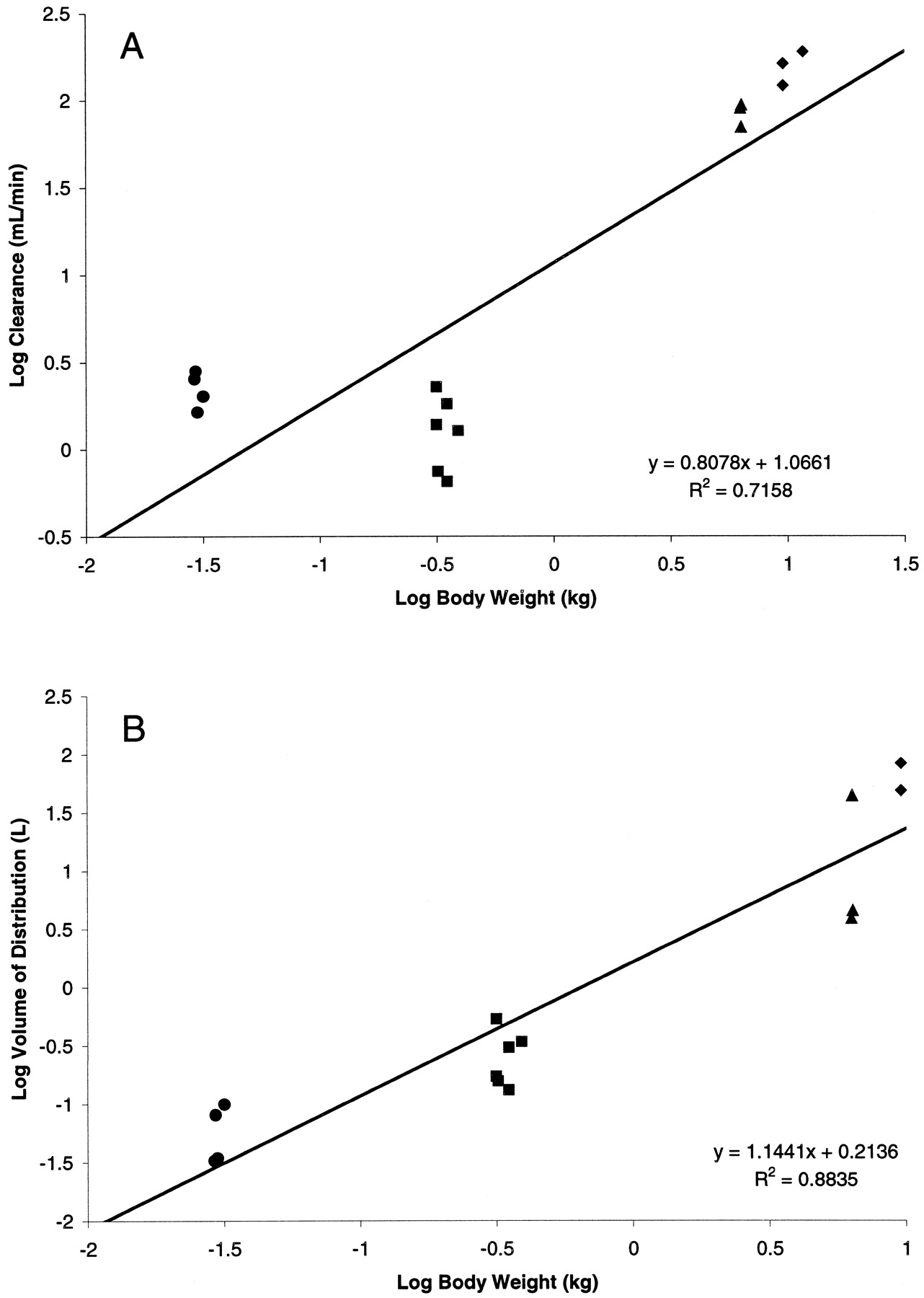
Allometric scaling plot for plasma clearance (A) and distributional volume (B) for SB-265123 in the mouse (●), rat (▪), monkey (▴), and dog (♦).
Line represents linear least-squares regression analysis of all the data points; the equation and correlation coefficient for the regression are provided for each plot.
Allometric equation coefficients, correlation coefficients, and predicted human clearance resulting from interspecies scaling of SB-265123, based on the inclusion of various species and correction factors (see text for details of correction)

Allometric scaling plot for plasma clearance of SB-265123 corrected for plasma protein binding, bile flow (Qbile), and UDPGT activity in the mouse (●), rat (▪), monkey (▴), and dog (♦).
Line represents linear least-squares regression analysis of all the data points; the equation and correlation coefficient for the regression are provided for each plot.
The effect of inclusion of each of the above-mentioned correction factors on predicted plasma clearance in humans is depicted in Table 5. Compared with the uncorrected prediction value, inclusion of the novel correction factor increased the predicted plasma clearance in humans of SB-265123 from 5.2 to 10.5 ml/min/kg when data from all the species are included in the scaling exercise. The impact of not including data for either rat or mouse on the scaling to humans also is shown in Table 5. Exclusion of the mouse data would result in a substantial increase of the predicted human clearance of SB-265123 compared with the estimate generated with data from all the species. Without this correction, the predicted clearance value derived without the mouse data is ∼7-fold higher than that obtained when all species are included in the scaling. Inclusion of the correction factor for bile flow and UDPGT activity, however, diminished the consequence of excluding the mouse data, resulting in a 2-fold increase in the predicted human clearance.
Discussion
This study details the pharmacokinetics of SB-265123 across four preclinical species. In general, the compound exhibited favorable pharmacokinetic properties across all species examined with excellent bioavailability in the mouse, rat, dog, and monkey. Previous small-molecule antagonists of αvβ3 have been characterized by very poor oral bioavailability, thus limiting their potential clinical utility (Keenan et al., 1997, 1998). SB-265123, however, appears to have overcome this limitation, and represents an important step in the progression toward an orally available αvβ3 antagonist for the treatment of a variety of diseases.
Because of the potential clinical implications of a tool compound such as SB-265123, various efforts were made to perform interspecies scaling of SB-265123. The volume of distribution of SB-265123 scaled well across species, without the need for any correction factors; inclusion of interspecies differences in plasma protein binding did not substantially impact the allometry for distributional volume. Either with or without inclusion of a protein-binding correction, the allometric exponent for distributional volume was ∼1.1, a value typical for allometric scaling of this parameter (Ings, 1990). These data indicate that for SB-265123, distributional volume scales in approximate proportion to body weight.
For plasma clearance, however, interspecies scaling was less straightforward. Simple allometry, either with or without a correction for plasma protein binding, failed to produce a good interspecies correlation coefficient. It has been noted that although conventional allometry provides reasonably good interspecies scaling for compounds that are rapidly metabolized by phase I reactions or that are excreted unchanged, such allometric relationships typically fail to produce adequate scaling for low to moderate extraction ratio compounds (Lave et al., 1997). Numerous investigations have attempted to use physiological correction factors to improve the interspecies scaling for slowly metabolized compounds. One such technique involves a correction for differences in maximum life span potential across species (Boxenbaum, 1984), whereas another, noted by Mahmood and Balian (1996a), incorporates interspecies differences in brain weight to predict the clearance of slowly metabolized compounds. Both approaches were attempted in the present investigation, and although each did improve the interspecies correlation, the correlation coefficient for clearance remained <0.9. Preliminary in vitro data for SB-265123 (data not shown) indicate that phase I oxidative metabolism of SB-265123 may be a minor contributor to the overall elimination of the compound; this may explain why these metabolic correction factors improved the scaling of the compound. However, as detailed in this study, SB-265123 is excreted in bile in the rat as an acyl-glucuronide conjugate. Furthermore, although direct evidence for such phase II and III elimination of SB-265123 is not present in the other species examined, the plasma concentration-time profiles for the other species indicate the strong likelihood of enterohepatic recirculation of SB-265123, and thus support the assumption that a similar metabolic pathway exists as in the rat. Therefore, it was hypothesized that the additional conjugation pathway contributed to the suboptimal scaling for the compound.
Few examples exist in the literature for interspecies scaling of compounds that undergo phase II metabolism. Izumi et al. (1996)demonstrated that the clearance of troglitazone, an antidiabetic agent that undergoes both phase I and phase II metabolism, demonstrated favorable allometric scaling without the incorporation of a correction factor; however, human i.v. systemic clearance data are not available. Also, Lave et al. (1996) demonstrated improvement of interspecies scaling of tolcapone, a catechol-O-methyltransferase inhibitor primarily metabolized by conjugation, by incorporating in vitro metabolic data in the allometry. Essentially, that study showed an improved scaling for tolcapone clearance when the in vivo plasma clearance was multiplied by the ratio of human intrinsic clearance to the intrinsic clearance of each animal species, as estimated in vitro in isolated hepatocytes. Given these findings, it was hypothesized that the allometric scaling of the present data with SB-265123 would be improved by incorporation of a correction factor taking account of phase II metabolism. Rather than using in vitro metabolism data specific to SB-265123, a more generic approach was attempted that incorporated interspecies differences in UDPGT activity, based on reported literature values (Table 1). As described in Results, this approach improved the allometry for SB-265123 compared with uncorrected extrapolations but was not substantially better than generic phase I corrections for maximum life span potential or brain weight. Another potential influence on compounds excreted in the bile would be the bile flow rate; interspecies differences in bile flow could have an impact on the rate and extent of elimination. Indeed, incorporation of relative bile flow rates did improve the allometry for SB-265123; this was improved further when adjusted for UDPGT activity as monitored with a generic marker substrate in vitro (Fig. 5). Because the present data do not allow a determination of whether phase II or phase III processes are rate-limiting in the biliary excretion of conjugated SB-265123, both correction factors were included in the final analysis. At present, only preclinical data are available for SB-265123; clinical data would be required to validate the allometric prediction. As a partial validation, this novel correction factor was applied to the data of Lave et al. (1996) for tolcapone. The corrected predicted clearance of tolcapone was 5.2 ml/min/kg compared with the actual human value of 1.7 ml/min/kg, and the predicted value with the correction method of Lave and coworkers of 1.2 to 3.2 ml/min/kg. Thus, the correction approach described in the present study would appear to extend to other compounds and may be clinically relevant.
Despite the overall improvement in interspecies scaling with the approach described in this investigation, additional refinement of this correction technique may be required to yield appropriate scaling for specific compounds. Several specific experimental data sets could be gathered for a given compound that could be incorporated into this generic scheme to attempt an enhancement of the interspecies scaling. For example, the specific activity of UDPGT from hepatocytes of different species toward a more specific substrate could be incorporated, rather than the generic UDPGT value used in this example. Also, other model systems, such as isolated perfused liver, liver slices, isolated hepatocytes, or isolated canalicular membrane vesicles, could be used to generate data on the biliary excretion rate, hepatocellular binding and transit time, or canalicular egress rate that might improve the interspecies scaling for a specific compound of interest. Generally, however, these data will not be available early in the drug development process when interspecies extrapolations may play an important role in determining whether or not to advance compounds in development, and they may not be available at all for environmental toxicants for which interspecies scaling data are desired. One generalized modification to the described approach could be the introduction of glutathione-S-transferase orN-acetyltransferase activity for compounds that undergo acetylation or glutathione conjugation, rather than glucuronidation. Such preliminary metabolite data often may be available sufficiently early in the drug development process to enable an accommodation of the appropriate conjugation enzyme in performing such extrapolations to humans.
Inclusion of both rat and mouse data in the present investigation highlights the importance of choosing appropriate preclinical species in performing interspecies extrapolations, or at least of considering the potential impact of the available species when performing allometry. As detailed in Table 5, had the mouse data not been available for SB-265123, the predicted clearance value would have been substantially greater than when both rat and mouse data were considered. Furthermore, the lack of correlation across species would probably not have been noted if either rat or mouse data were not available because a relatively good correlation is observed when either is excluded and only three species are used. The mechanism underlying the relative importance of the mouse and rat data is not clear, but it may be related to the lack of a gall bladder in rats, and thus the physiologic difference in bile flow patterns. Previously, it has been stated that data from three or more species are required for a reliable prediction of clearance with allometric scaling (Mahmood and Balian, 1996b); other investigators have explored the ability of allometry to predict human pharmacokinetics with data from as few as one other species (Bachmann et al., 1996). In contrast, the data from the present investigation suggest that for SB-265123, and perhaps for other compounds that undergo phase II and phase III elimination, the use of more than three species may be important in identifying species that are allometric outliers and in incorporating appropriate correction factors in the scaling process.
In summary, the present investigation describes the preclinical pharmacokinetics of SB-265123 and demonstrates that the compound exhibits good oral bioavailability across species, and thus may be useful as a tool compound for investigating the use of oral αvβ3 antagonists in the treatment of various disease states. Standard techniques for interspecies scaling of SB-265123 failed to produce adequate extrapolation of the clearance of the compound across the preclinical species; therefore, a novel correction factor was used that incorporated corrections for phase II and phase III processes. Although the lack of clinical pharmacokinetic data for SB-265123 do not allow a conclusive determination of whether the correction factor improved the prediction of the pharmacokinetic behavior of SB-265123 in humans, this correction did enable an improved allometric correlation among the preclinical species. This correction may be a useful addition to allometry conducted in drug development or in risk assessment for environmental xenobiotics; further evaluation of this technique will be required to more thoroughly validate its use in interspecies extrapolation and prediction.
Footnotes
-
Send reprint requests to: Keith W. Ward, Ph.D., Drug Metabolism and Pharmacokinetics, SmithKline Beecham Pharmaceuticals R&D, UW 2720, 709 Swedeland Rd., King of Prussia, PA 19406. E-mail:Keith_W_Ward{at}sbphrd.com
-
↵1 This work presented in part at the 1999 Annual Society of Toxicology Meeting, March 14–18, New Orleans, LA.
- Abbreviations used are::
- MS
- mass spectrometry
- MS/MS
- dual mass spectrometry
- CL
- plasma clearance
- Vdss
- steady-state volume of distribution
- fb and fu
- unbound and bound fraction in plasma
- Qbile
- bile flow rate
- UDPGT
- UDP-glucuronosyl transferase
- Received March 16, 1999.
- Accepted July 2, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}