


Abstract
Isoforms of cytochrome P-450 (CYP) involved in the metabolism of gallopamil enantiomers were identified by measuring the disappearance rate of parent drug from an incubation mixture with human liver microsomes and recombinant human CYPs. Mean (± S.D.) intrinsic clearances (CLint) of R(+)- andS(−)-gallopamil in human liver microsomes were 0.320 ± 0.165 and 0.205 ± 0.107 ml/min/mg protein, respectively. These values were highly correlated with the 6β-hydroxylation activity of testosterone, a marker substrate of CYP3A4 (r = 0.977 and 0.900 forR(+)- and S(−)-gallopamil, respectively,p < .001). Ketoconazole and troleandomycin, selective inhibitors of CYP3A4, and polyclonal antibodies raised against CYP3A4/5 markedly reduced the CLint of gallopamil enantiomers in human liver microsomes. Among the 10 recombinant human CYP isoforms, CYP3A4 exhibited the highest CLint of gallopamil enantiomers, and CYP2C8 and CYP2D6 also exhibited appreciable activity. When the contribution of CYP3A4 to the total metabolic clearance of gallopamil enantiomers in human liver microsomes was estimated by relative activity factor, the mean (± S.D.) contributions were 92 ± 18 and 68 ± 19% forR(+)- and S(−)-gallopamil, respectively. These values were comparable to the rates of immunoinhibition by antibodies raised against CYP3A4/5 observed in human liver microsomes. The present study suggests that CYP3A4 is a major isoform involved in the overall metabolic clearance of gallopamil enantiomers in the human liver, and that the present approach based on disappearance rate may be applicable to identify major isoforms of CYP involved in the metabolism of a drug in human liver microsomes.
Gallopamil, a methoxy derivative of verapamil, is a calcium-channel antagonist used for the treatment of coronary artery diseases (e.g., vasospastic or variant angina pectoris). As is the case with verapamil, gallopamil is a chiral compound administered as a racemic mixture of R(+)- and S(−)-enantiomers. It is well established that the pharmacokinetics of racemic verapamil is stereoselective; the total systemic clearance ofS(−)-verapamil is greater than that ofR(+)-enantiomer after i.v. administration, and the preferential first-pass metabolism of S(−)-verapamil occurs after oral administration (Eichelbaum et al., 1984; Vogelgesang et al., 1984). Unlike verapamil, the first-pass metabolism of gallopamil is not stereoselective, despite its similarity in the structure and disposition to verapamil (Gross et al., 1997).
The pharmacokinetics of gallopamil is characterized by a metabolism- and flow-dependent clearance with its distinct first-pass metabolism (Stieren et al., 1983). Major metabolic processes of gallopamil areN-dealkylation and O-demethylation in humans in vivo (Stieren et al., 1983; Weymann et al., 1989) and in rat and human liver microsomes in vitro (Mutlib and Nelson, 1990a,b) (Fig.1). The formation ofN-dealkylated and N-demethylated metabolites of verapamil has been demonstrated to be catalyzed mainly by an isoform of cytochrome P-450 (CYP)1, CYP3A4 (Kroemer et al., 1993). However, the candidate human CYP isoform(s) involved in the metabolism of gallopamil has not been identified.

Metabolic pathways of gallopamil.
The chiral carbon is indicated by the asterisk.
Identification of CYP isoform(s) has generally been performed by measuring the metabolite(s) production rate with a specific pathway of metabolism, although the metabolic process of a new drug is usually not known in the early stage of drug development. In contrast, the disappearance rate of a drug from incubation medium may be equally useful for this purpose, and in certain circumstances this approach appears to be more appropriate than an approach based on use of a specific metabolic pathway (Houston, 1994). However, little information is available whether the disappearance rate is applicable to the identification of CYP isoform(s) involved in the metabolism of new drugs.
The aim of the present study was to identify the major CYP isoform(s) involved in metabolism of R(+)- andS(−)-gallopamil by measuring the disappearance rate of parent compound from an incubation mixture with human liver microsomes and recombinant human CYP isoforms.
Materials and Methods
Chemicals and Reagents.
R(+)- and S(−)-gallopamil HCl, and Lu46973 (internal standard) were obtained from Knoll AG (Ludwigshafen, Germany). Testosterone and troleandomycin (TAO) were purchased from Sigma Chemical Co. (St. Louis, MO). 6β-Hydroxytestosterone and ketoconazole were obtained from Sumika Chemical Analysis Service (Osaka, Japan) and BIOMOL Research Laboratories, Inc. (Plymouth Meeting, PA), respectively. Acetonitrile, methanol, and other reagents of analytical grade were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Microsomal preparations from 10 different recombinant human CYP isoforms (i.e., CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9-Arg, CYP2C9-Cys, CYP2C19, CYP2D6, CYP2E1, and CYP3A4) expressed in the human B lymphoblastoid cell line (AHH-1) were purchased from Gentest Corp. (Woburn, MA). Inhibitory antibody to human CYP3A4/5 was purchased from Amersham International (Tokyo, Japan).
Human Liver Microsomes.
The microsomal fraction was prepared from human livers by differential centrifugation as described previously (Chiba et al., 1993a). Liver samples (HL-31, HL-32, HL-33, HL-34, HL-35, HL-37, HL-38, HL-39, and HL-40) were obtained, as excess material removed during surgery on liver, from nine Japanese patients undergoing partial hepatectomy at the Department of General Surgery, International Medical Center of Japan (Tokyo, Japan). After determination of protein concentration (Lowry et al., 1951), the individual microsomal samples were aliquoted, frozen, and stored at −80°C until used. Use of human liver samples for the study was approved by the Institutional Ethics Committee, International Medical Center of Japan.
Incubation Conditions with Human Liver Microsomes and Recombinant Human CYP Isoforms.
The basic incubation medium contained 0.1 mg/ml human liver microsomes, 15 mM MgCl2, 1.5 mM NADP, 10 mM glucose 6-phosphate, 1.5 I.U./ml glucose 6-phosphate dehydrogenase, 100 mM potassium phosphate buffer (pH 7.4), 0.1 mM EDTA, and 0.5 μMR(+)- and S(−)-gallopamil HCl, in a final volume of 200 μl. The mixtures were incubated at 37°C in a shaking water bath for 0, 5, 10, 15, and 20 min except for the inhibition study in which incubations were carried out for 0, 10, 20, 30, and 40 min. The reaction was terminated by addition of 10 μl of perchloric acid and 50 μl of a methanolic solution of the internal standard (0.2 μg Lu 46973/ml in methanol). After termination of the incubation, the mixtures were centrifuged at 10,000 rpm for 1 min, and the supernatants were injected onto an HPLC apparatus as described below.
Throughout the study, we used 0.5 μM as the substrate concentration to determine the disappearance rates of gallopamil enantiomers. This concentration was determined by a preliminary study that showed the disappearance rates of gallopamil enantiomers at 0.25 μM did not differ from those at 0.5 μM in human liver microsomes, suggesting that metabolism of gallopamil enantiomers is not saturated at these substrate concentrations (i.e., 0.5 μM is estimated to be in the range of gallopamil concentrations in which gallopamil shows the first order disappearance from the incubation mixture). The incubation conditions used for the 10 different recombinant human CYP isoforms obtained from genetically engineered B lymphoblastoid cells were essentially the same as those used for human liver microsomes, except for incubation time (120 min) and the concentration of microsomes used (1 mg/ml).
HPLC Assay.
The determination of R(+)- and S(−)-gallopamil in the incubation mixture was performed by an HPLC-fluorescence detection method. The HPLC system consisted of a model LC-6A pump (Shimadzu, Kyoto, Japan), model SIL-6A autosampler (Shimadzu), model CTO-6A column oven (Shimadzu), model RF-535 fluorescence detector (Shimadzu), model C-R3A integrator (Shimadzu), and J’sphere ODS-L80 column (150 × 2.0 mm internal diameter, YMC Co., Ltd., Kyoto, Japan). The mobile phase consisted of 0.12% perchloric acid and acetonitrile (68:32 v/v) delivered at 0.3 ml/min. Column temperature was maintained at 40°C. The excitation and emission wavelengths of the fluorescence detector were set at 235 and 315 nm, respectively. Analytes were quantified by comparison with the standard curve generated from peak area ratios of gallopamil against the internal standard. Intra- and interassay coefficients of variation were <5%.
Intrinsic Clearance of R(+)- andS(−)-Gallopamil.
The intrinsic clearances (CLint) of gallopamil enantiomers in nine different human liver microsomes were estimated from the volume of medium (V) and the half-life of substrate disappearance (T1/2) with the following equation: CLint = V × 0.693/T1/2 (Chenery et al., 1987). The half-life of R(+)- and S(−)-gallopamil in the incubation medium was calculated by the regression analysis of semilogarithmic plots.
Correlation Study.
Correlations between the metabolic activities of substrates toward the respective CYP isoforms and those of gallopamil enantiomers and between the metabolic activities of R(+)- andS(−)-gallopamil were studied for nine different human liver microsomes. Phenacetin O-deethylation (CYP1A2) (Tassaneeyakul et al., 1993), coumarin 7-hydroxylation (CYP2A6) (Yamano et al., 1990), diclofenac 4′-hydroxylation (CYP2C9) (Leemann et al., 1992), S-mephenytoin 4′-hydroxylation (CYP2C19) (Wrighton et al., 1993), desipramine 2-hydroxylation (CYP2D6) (Birgersson et al., 1986), chlorzoxazone 6-hydroxylation (CYP2E1) (Peter et al., 1990), and testosterone 6β-hydroxylation (CYP3A4) (Waxman et al., 1988) were used for assessing the respective CYP-catalytic probe activities. Metabolites of the above in vitro probe substrates were determined with the respective HPLC assay methods, as reported elsewhere (Leemann et al., 1992; Chiba et al., 1993b; Tassaneeyakul et al., 1993; Yoshimoto et al., 1995; Zhao et al., 1996). Correlation coefficients (r) were determined by the least-squares linear regression analysis (REG procedure, SAS Institute, Inc., Cary, NC).
Inhibition Study.
The effects of CYP3A4-selective inhibitors on the metabolism of gallopamil enantiomers were investigated with a microsomal preparation obtained from human liver sample HL-38. Ketoconazole and TAO were used as CYP3A4-selective inhibitors (Newton et al., 1995). Ketoconazole (1 μM) was coincubated with each gallopamil enantiomer (0.5 μM) under the incubation conditions described above. TAO (1 μM) was preincubated with microsomes and the NADPH-generating system for 15 min, before addition of each enantiomer of gallopamil (0.5 μM) to initiate the reaction. The effects of inhibitors on the metabolic clearance of gallopamil enantiomers were compared with the control values and expressed as percentages of the respective control values.
Immunoinhibition Study.
The inhibitory antibody to human CYP3A4/5 used in this study was a polyclonal antibody raised in rabbits against rat CYP3A1 with ∼95% inhibition of the testosterone 6β-hydroxylation catalyzed by the rat and human CYP3A subfamily, but this antibody does not inhibit the activities of CYP1A2, CYP2A6, CYP2B6, CYP2C8/9, CYP2D6, and CYP2E1 (Amersham).
The immunoinhibition of metabolic clearance for gallopamil enantiomers was examined by preincubating human liver microsomal samples (HL-38, 0.1 mg/ml) with various concentrations of anti-CYP3A4/5 antibodies (0–5 mg IgG/mg microsomal protein) in 0.1 mM potassium phosphate buffer (pH 7.4) for 30 min on ice. Each enantiomer of gallopamil (0.5 μM) and other components of the incubation medium were added, and the reaction was assessed as described above.
Contributions of CYP3A4 to Overall Metabolic Clearance ofR(+)- and S(−)-Gallopamil in Human Liver Microsomes.
The percent contribution of CYP3A4 to the total clearance ofR(+)- and S(−)-gallopamil in human liver microsomes was estimated with data for recombinant CYP3A4 and relative activity factor (RAF) proposed by Crespi (1995). Because the overall metabolic clearance of gallopamil in human liver microsomes (CLHLM) is the sum of the metabolic clearances mediated by multiple enzymes contributing to the microsomal metabolism of gallopamil enantiomers, the contribution of CYP3A4 to CLHLM can be estimated as follows:
Data Analysis.
Results are expressed as means ± S.D. throughout the text. Correlations between the metabolite formation rates of the respective CYP isoform-specific substrates and the CLintvalues for the disappearance of gallopamil enantiomers were examined by the least-squares linear regression analysis.Km and Vmaxvalues were estimated by the nonlinear least-squares regression analysis. The mean kinetic values were compared by Student’st test for unpaired data (SAS system for Windows, v.6.10).p values <.05 were considered statistically significant.
Results
Intrinsic Clearance of R(+)- andS(−)-Gallopamil.
The individual and mean (±S.D.) CLint values determined for the disappearance of R(+)- andS(−)-gallopamil at 0.5 μM in nine different human liver microsomes are given in Table 1. Although there was ∼5- to 7-fold interindividual variability, the CLint was greater for R(+)-gallopamil than for S(−)-gallopamil in all the human liver microsomes, and the respective mean values of CLint were 0.320 ± 0.165 ml/min/mg protein for R(+)- and 0.205 ± 0.107 ml/min/mg protein for S(−)-gallopamil (S/R ratio = 0.64 ± 0.12).
Intrinsic clearance for R (+)- and S (−)-gallopamil in nine different human liver microsomes
Correlation Study.
The CLint values of gallopamil enantiomers correlated well with each other (r = 0.938,p < .001) in nine different human liver microsomes, suggesting the same CYP isoform(s) are involved in the metabolism of both enantiomers. In addition, the CLint values of gallopamil enantiomers exhibited significant correlations (r = 0.977, p < .001 forR(+)- and r = 0.900, p < .001 for S(−)-gallopamil) with the testosterone 6β-hydroxylation activity (Fig. 2). The CLint of R(+)-gallopamil also exhibited a significant correlation with the S-mephenytoin 4′-hydroxylation activity (r = 0.769,p < .05). However, there were no significant correlations between the CLint of gallopamil enantiomers and any other metabolic activities of probe substrates used in the present study (Table 2).

Relationship between the intrinsic clearances of gallopamil enantiomers and testosterone 6β-hydroxylase activity in nine different human liver microsomes.
Correlations between the intrinsic clearance of gallopamil enantiomers and metabolic activities (Vmax/Km) of seven selective substrates of distinct human CYP isoforms
Inhibition Study.
The effects of ketoconazole (1 μM) and TAO (1 μM) on the CLint of gallopamil enantiomers at 0.5 μM in microsomes obtained from human liver sample HL-38 are shown in Fig.3. These two widely used selective inhibitors of CYP3A4 strongly reduced the CLintof gallopamil enantiomers. The reductions of CLint for R(+)- andS(−)-gallopamil by ketoconazole were 98 and 91%, and those by TAO were 79 and 76%, respectively.

Effects of CYP3A4 inhibitors on the intrinsic clearances of gallopamil enantiomers in human liver microsomes (HL-38).
Immunoinhibition Study.
Figure 4 shows the reduction of CLint of gallopamil enantiomers by anti-CYP3A4/5 polyclonal antibody. The addition of antibody to the incubation medium reduced the CLint of R(+)- andS(−)-gallopamil in a concentration-dependent manner, and at a concentration of 5 mg IgG/mg microsomal protein, the reductions were >99% for R(+)- and >91% for S(−)-gallopamil.
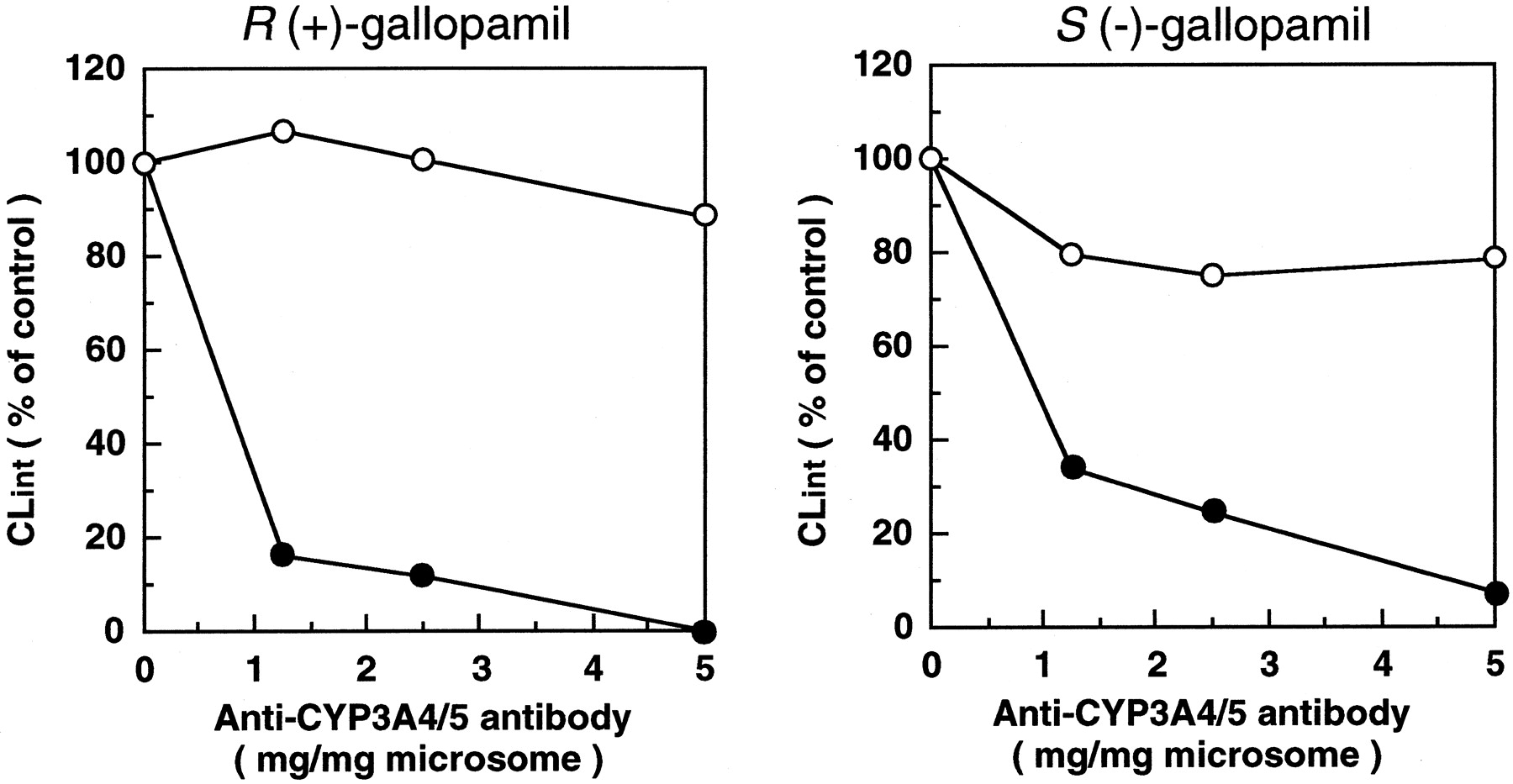
Effects of anti-CYP3A4/5 (●) and preimmune rabbit IgG (○) on the intrinsic clearances of gallopamil enantiomers in human liver microsomes (HL-38).
Recombinant CYP Isoform Study.
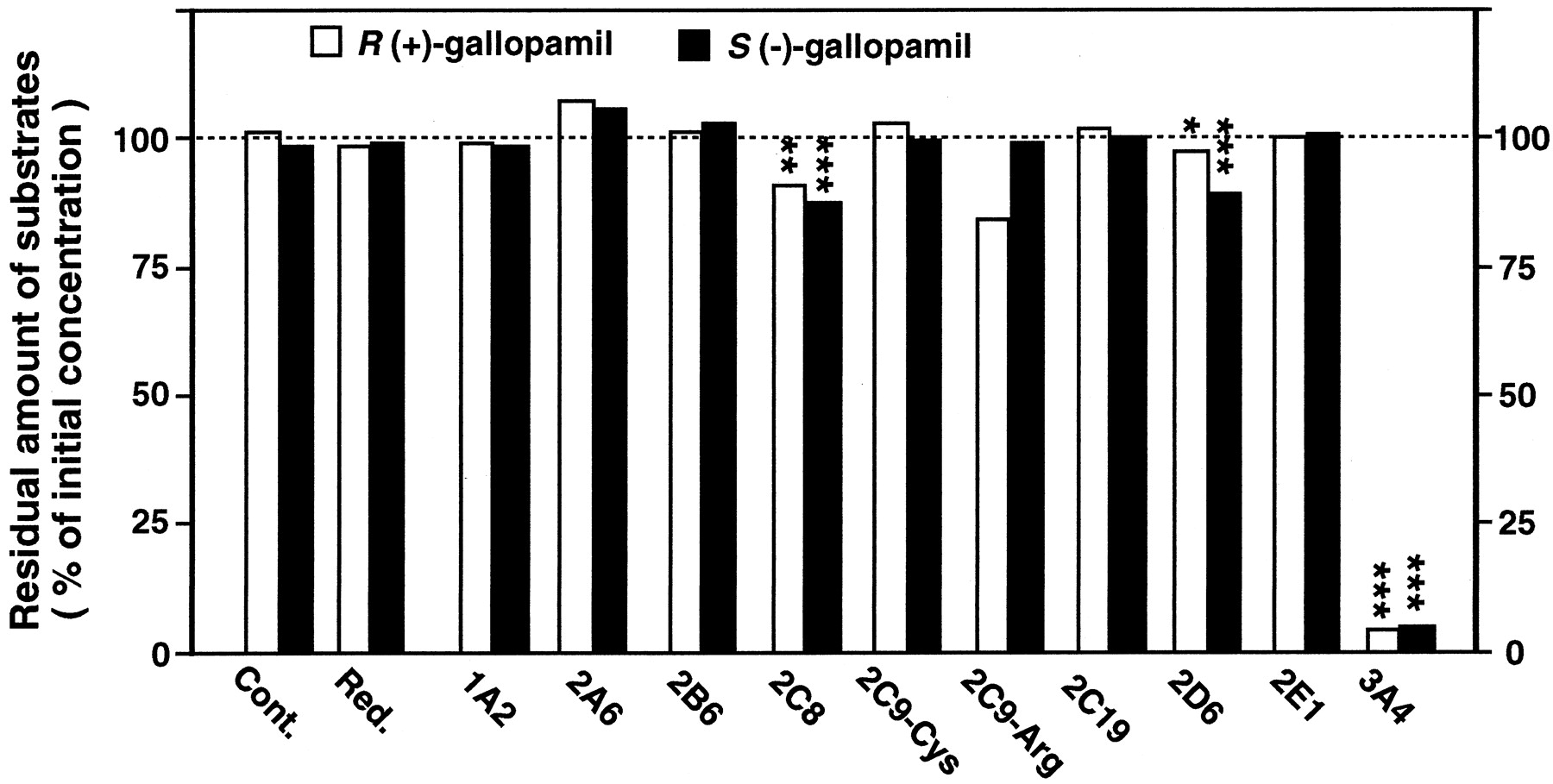
CYP isoforms expressed in human B lymphoblastoid cell lines were studied for their abilities to metabolize gallopamil enantiomers (Fig.5). CYP3A4 revealed the highest metabolic activity for the disappearance of both enantiomers, whereas CYP2C8 and CYP2D6 also exhibited a significant metabolic activity. However, the other CYP isoforms exhibited no significant metabolic activity.

Metabolic activities of recombinant human CYP isoforms expressed in human B lymphoblastoid cells for gallopamil enantiomers.
Data are expressed as the percentages of substrate concentration remaining in the incubation mixture at 120 min compared with the initial value. Each column represents the mean values obtained from triplicate experiments. *p < .05, **p < .01, and ***p < .001.
Contributions of CYP3A4 to Overall Metabolic Clearance ofR(+)- and S(−)-Gallopamil in Human Liver Microsomes.
The mean CLrec-CYP3A4 values for R(+)- and S(−)-gallopamil were 0.107 and 0.049 ml/min/mg microsomal protein, respectively. TheVmax/Km values of testosterone 6β-hydroxylation ranged from 0.030 to 0.139 ml/min/mg microsomal protein in nine different human liver microsomes. TheVmax/Km value of testosterone 6β-hydroxylation in the recombinant CYP3A4 was 0.031 ml/min/mg microsomal protein. Thus, the RAFCYP3A4values ranged from 0.97 to 4.48 (Table3). Based on these values and CLHLM obtained from nine different human liver microsomes, the contributions of CYP3A4 to the CLHLM of gallopamil enantiomers were estimated (Table 3). The mean ± S.D. contributions of CYP3A4 were 92 ± 18 and 68 ± 19% for R(+)- andS(−)-gallopamil, respectively.
Contribution of CYP3A4 to the overall metabolic clearance of R (+)- and S (−)-gallopamil estimated by RAF in human liver microsomes
Discussion
Several lines of evidence obtained from the present study suggest that CYP3A4 is the principal isoform involved in the metabolic clearance of R(+)-and S(−)-gallopamil. This contention is based on the following observations: 1) the metabolic clearances of gallopamil enantiomers in nine different human liver microsomes were highly correlated with the testosterone 6β-hydroxylation activity, known as a marker for CYP3A4 (Fig. 2); 2) ketoconazole and TAO, known as selective inhibitors of CYP3A4, strongly inhibited the metabolic clearance of gallopamil enantiomers (Fig. 3); 3) the metabolic clearance of gallopamil enantiomers was almost completely inhibited by the addition of anti-CYP3A4/5 antibodies (Fig.4); and 4) recombinant human CYP3A4 exhibited the most dominant activity in the metabolism of gallopamil enantiomers (Fig. 5).
Although recombinant CYP2C19 was not capable of metabolizing gallopamil enantiomers (Fig. 5), there was a significant correlation between the metabolic clearance of R(+)-gallopamil and 4′-hydroxylation activity of S-mephenytoin, a specific substrate of CYP2C19 (Table 2). This may be derived from the fact that there was a significant correlation between CYP2C19 and CYP3A4 activities (r = 0.732, p < .05, data not shown) in the panel of liver microsomes. Therefore, CYP2C19 does not appear to be involved in the clearance of gallopamil. Similarly, CYP2D6 appears to play minor role in the clearance of gallopamil enantiomers, although recombinant CYP2D6 and CYP2C8 were capable of metabolizing gallopamil enantiomers in addition to CYP3A4 (Fig. 5). This is because there was no significant correlation between the CLint of gallopamil enantiomers and 2-hydroxylation activity of desipramine, a marker of CYP2D6 (Table 2). In addition, CYP2D6 in the recombinant system used in the present study is overexpressed and its content is >10-fold that present in average human liver microsomal preparations (data provided by Gentest).
However, Busse et al. (1995) reported that verapamil, a more widely used gallopamil analog that differs from gallopamil by only one aromatic methoxy group at position 5 in the chemical structure, is biotransformed to O-demethylated metabolites by more than one CYP2C isoform. Similarly, we detected O-demethylated metabolites of gallopamil in the incubation mixture in the presence of recombinant CYP2C8 (A.S., I.I., F.T., M.A., K.F., M.T., T.I. and K.C., unpublished data). However, because O-demethylation is a relatively minor pathway in gallopamil metabolism (Mutlib and Nelson, 1990a,b) and expression of CYP2C8 protein appears to be low in human liver microsomes (Imaoka et al., 1996), the involvement of CYP2C8 in the overall metabolism of gallopamil would be of a minor importance, if any, in human liver microsomes. Collectively, the findings of the present study indicate that CYP3A4 is the predominant, if not sole, CYP isoform involved in the overall metabolic clearance of bothR(+)- and S(−)-gallopamil in human liver microsomes.
In this study, we identified the human CYP isoform(s) responsible for the metabolism of gallopamil enantiomers based on the disappearance rate of parent drug from an incubation mixture. This approach appears to have some advantages over the traditional one for the following reasons: 1) the metabolic profile of a drug is not completely elucidated and all the metabolites formed from the parent drug are sometimes not available in an early stage of new drug development; 2) it is difficult to measure the metabolite production rate when the primary metabolite is unstable or sequentially metabolized to a secondary metabolite; and 3) determining the metabolite production rate is troublesome when the metabolic pathway is complex and yields a large number of metabolites. However, it should be noted that the traditional approach is essential when active or toxic metabolite is formed from parent compound.
When multiple isoforms of CYP are involved in the metabolism of a drug, it is desirable to determine the percentage of contribution of each isoform to the overall metabolism of the drug. In a previous study,Kobayashi et al. (1997) estimated the percentage of contribution of CYP2C19 and CYP3A4 to the overall activities of citalopramN-demethylation in human liver microsomes by applying the RAF concept proposed by Crespi (1995). With the same approach, we assessed the contribution of CYP3A4 to the overall metabolic clearance of R(+)- and S(−)-gallopamil in human liver microsomes. The mean ± S.D. values of the contribution of CYP3A4 to the metabolic clearances of R(+)- andS(−)-gallopamil were 92 ± 18 and 68 ± 19%, respectively (Table 3). These values are almost the same as the rates of inhibition by CYP3A inhibitors or anti-CYP3A4/5 antibody for the metabolic clearance of R(+)- and S(−)-gallopamil in human liver microsomes, and appear to be consistent with the finding that the rates of inhibition by these inhibitors or antibodies are greater for R(+)- than for S(−)-gallopamil in human liver microsomes (Figs. 2 and 3). These findings may suggest that RAF is a useful tool for estimating the contribution of human CYP isoform(s) to the total metabolic clearance of a drug in human liver microsomes. However, further studies are needed to confirm this contention because gallopamil is a relatively straightfoward example, and it may be necessary to validate the applicability of this approach for a drug with more complex metabolism (i.e., a drug metabolized via multiple pathways with similar contributions of different isoforms of CYP).
In Table 3, one liver sample (HL-35) showed an estimated contribution of CYP3A4 >100% for the metabolic clearance ofR(+)-gallopamil. The reason for this phenomenon is unclear; however, a possible explanation is that microsomal enzyme(s) other than CYP3A4 (i.e., CYP3A5) may contribute to testosterone 6β-hydroxylation or gallopamil metabolism in human liver microsomes and activity of such enzyme(s) in HL-35 may be different from those of other liver microsomal samples.
The identification of enzymes involved in the metabolism of gallopamil may permit a prediction of potential interaction of gallopamil with other drugs metabolized by CYP3A4. This is because CYP3A4 metabolizes a broad spectrum of drugs, including calcium channel antagonists, such as nifedipine (Guengerich et al., 1986), diltiazem (Pichard et al., 1990b), and verapamil (Kroemer et al., 1993), and midazolam (Kronbach et al., 1989), cyclosporin (Kronbach et al., 1988), tamoxifen (Jacolot et al., 1991), terfenadine (Yun at al., 1993), and lidocaine (Bargetzi et al., 1989). Many of these drugs competitively inhibit the activity of CYP3A4 (Pichard et al., 1990a). At least on a theoretical basis, oral clearance of gallopamil may be decreased in individuals taking drugs that inhibit the activity of or that are metabolized by CYP3A4 and may be increased in individuals taking drugs that induce CYP3A4 (e.g., rifampicin, phenobarbital, phenytoin, carbamazepine) (Combalbert et al., 1989; Brockmöller and Roots, 1994).
In conclusion, the present study with human liver microsomes and recombinant human CYP isoforms indicates that CYP3A4 is the major isoform involved in the overall metabolic clearance of gallopamil enantiomers. We wish to assert that an approach based on the disappearance rate of parent drug from an incubation mixture is applicable for determining the major isoform(s) of CYP involved in the metabolism of a drug in human liver microsomes. Lastly, RAF, as proposed by Crespi (1995), appears to be a useful tool for estimating the contribution of each CYP isoform to the overall metabolic clearance of a drug when multiple isoforms of CYP are involved in the metabolism of that drug.
Footnotes
-
Send reprint requests to: Kan Chiba, Ph.D., Laboratory of Biochemical Pharmacology and Toxicology, Faculty of Pharmaceutical Sciences, Chiba University, 1–33 Yayoi-cho, Inage-ku, Chiba-shi, Chiba 263-8522, Japan. E-mail: kchiba{at}p.chiba-u.ac.jp
- Abbreviations used are::
- CYP
- cytochrome P-450
- TAO
- troleandomycin
- CLint
- intrinsic clearance
- RAF
- relative activity factor
- Received April 29, 1999.
- Accepted July 23, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}