


Abstract
The goals of the present study were to identify the enzyme responsible for metabolism of itopride hydrochloride (itopride) and to evaluate the likelihood of drug interaction involving itopride. In human liver microsomes, the involvement of flavin-containing monooxygenase in N-oxygenation, the major metabolic pathway of itopride, was indicated by the following results: inhibition by methimazole and thiourea, heat inactivation, and protection against heat inactivation by NADPH. When the effects of ketoconazole on the metabolism of itopride, cisapride, and mosapride citrate (mosapride) were examined using human liver microsomes, ketoconazole strongly inhibited the formation of the primary metabolites of cisapride and mosapride, but not itopride. Other cytochrome P450 (CYP) 3A4 inhibitors, cimetidine, erythromycin, and clarithromycin, also inhibited the metabolism of cisapride and mosapride. In an in vivo study, itopride (30 mg/kg), cisapride (1.5 mg/kg), or mosapride (3 mg/kg) was orally administered to male rats with or without oral pretreatment with ketoconazole (120 mg/kg) twice daily for 2 days. The ketoconazole pretreatment significantly increased the area under the serum concentration curve and the maximum serum concentration of cisapride and mosapride but had no significant effect on the pharmacokinetics of itopride. In addition, itopride did not inhibit five specific CYP-mediated reactions of human liver microsomes. These results suggest that itopride is unlikely to alter the pharmacokinetics of other concomitantly administered drugs.
A novel gastroprokinetic agent, itopride hydrochloride (itopride), stimulates gastrointestinal motor activity through synergistic effects of dopamine D2receptor blockade and acetylcholine esterase inhibition (Iwanaga et al., 1990, 1991, 1993). Itopride undergoes extensive hepatic metabolism in humans. The primary metabolite in humans is the N-oxide, generated by oxidation of the tertiary amine N-dimethyl group (see Fig. 1) (Nakashima et al., 1993). The urinary excretions of itopride and its N-oxide were 3.7% and 75.4%, respectively, in healthy subjects after a single oral administration at a therapeutic dose (Nakashima et al., 1993). However, the enzymes involved in the metabolic pathway have not been fully characterized.

Major metabolic pathways of itopride, cisapride, and mosapride in humans.
Recently, potentially life-threatening ventricular arrhythmias have been associated with concurrent administration of cisapride, a widely used drug in the same therapeutic class as itopride, and imidazole antifungals or macrolide antibiotics (Ahmad and Wolfe, 1995; Wysowski and Bacsanyi, 1996). Cisapride is mainly metabolized to theN-dealkylated form (norcisapride) in humans, as shown in Fig. 1 below (Meuldermans et al., 1988), principally by cytochrome P450 (CYP)1 3A4 (Bedford and Rowbotham, 1996). The imidazole antifungals, such as ketoconazole, and macrolides are potent and relatively specific inhibitors of CYP3A4 activity (Newton et al., 1995). Coadministration of cisapride with such CYP3A4 inhibitors impairs CYP3A4-dependent presystemic extraction, causing greatly elevated plasma concentrations of unchanged cisapride compared with those after administration of cisapride alone (Bedford and Rowbotham, 1996; Haarst et al., 1998). The resultant high levels of cisapride produce adverse effects, such as QT interval prolongation, leading to the risk of ventricular arrhythmias (Bedford and Rowbotham, 1996).
This study, therefore, aimed to identify the enzyme responsible for metabolism of itopride and to investigate the likelihood of drug interaction involving itopride in vitro and in vivo compared with other gastroprokinetic agents, cisapride and mosapride citrate (mosapride, Fig. 1).
Experimental Procedures
Materials.
Itopride, cisapride, mosapride, and clarithromycin were synthesized in our laboratory. These four compounds were identified by1H NMR and IR, and a purity was confirmed by elemental analysis (within ±0.3% of the theoretical value) or thin layer chromatography (>99.5%). Ketoconazole was purchased from Funakoshi (Tokyo, Japan). Erythromycin, cimetidine, SKF-525A, 1-methylxanthine, 7-(β-hydroxypropyl)theophylline, and chlorpropamide were from Sigma Chemical Co. (St. Louis, MO). Methimazole was from Aldrich (Milwaukee, WI). Thiourea was from Kanto Chemical (Tokyo, Japan). n-Octylamine was from Tokyo Chemical Industry (Tokyo, Japan). Theophylline, phenobarbital, and testosterone were from Waco Pure Chemicals (Tokyo, Japan). Tolbutamide, hydroxytolbutamide,S-mephenytoin, 4′-hydroxymephenytoin, bufuralol, hydroxybufuralol, and 6β-hydroxytestosterone were from Daiichi Pure Chemicals (Tokyo, Japan). All other chemicals were of analytical grade. Pooled microsomes from human livers were purchased from Gentest (Woburn, MA) and Human Biologics, Inc. (Phoenix, AZ). Pooled microsomes from rat livers were from Charles River Japan (Tokyo, Japan). Microsomes prepared from human lymphoblastoid cells expressing human CYP1A2, -2A6, -2B6, -2C8, -2C9, -2C19, -2D6, -2E1, or -3A4 and microsomes prepared from baculovirus-infected insect cells expressing human flavin-containing monooxygenase 1 (FMO1), FMO3, or FMO5 were purchased from Gentest.
Experimental Animals.
Male Sprague-Dawley rats at 6 weeks old were obtained from Charles River Japan and acclimatized for at least 7 days in a controlled animal area maintained at 20–26°C and 30% to 70% relative humidity, with 12-h light/dark cycles. They were all deprived of food from approximately 24 h before drug administration to 4 h after administration.
Analytical Procedures for Itopride N-Oxygenation.
A typical incubation mixture consisted of 100 mM phosphate buffer (pH 7.4), 0.05 mM EDTA, an NADPH-generating system (0.8 mM NADP+, 6 mM MgCl2, 8 mM glucose 6-phosphate, and 1 U/ml glucose-6-phosphate dehydrogenase) and human liver microsomes (0.5 mg protein/ml) in a final volume of 0.2 ml. The incubation mixture was preincubated in the presence of the NADPH-generating system at 37°C for 3 min, and the reaction was initiated by the addition of itopride. After incubation for 30 min, ice-cold CH3CN (0.2 ml) was added to terminate the reaction. The mixture was centrifuged at 10,000 rpm for 10 min to precipitate the protein. An aliquot of the supernatant (20 μl) was subjected to HPLC with a pump (LC-10AD; Shimadzu, Kyoto, Japan), a WISP 717 autosampler (Waters, Milford, MA), a TSKgel ODS-80Tm column (5 μm, 150 × 4.6 mm i.d., Tosoh, Tokyo, Japan), and an 821-FP fluorescence detector (Jasco, Tokyo, Japan) set at an excitation wavelength of 308 nm and an emission wavelength of 344 nm. The mobile phase, consisting of 50 mM phosphate buffer (pH 2.5):CH3CN (80:20, v/v), was delivered at a flow rate of 1 ml/min. The peak areas were obtained from a Shimadzu C-R4AX integrator. The retention time for itoprideN-oxide was 9.6 min.
Analytical Procedures for N-Dealkylation of Cisapride and Mosapride.
The primary metabolites of cisapride (N-dealkylcisapride) and mosapride (N-dealkylmosapride) were determined by an HPLC method. A typical incubation mixture was the same as the measurement of itopride N-oxygenase activity, whereas the incubation mixture was preincubated in the presence of substrate at 37°C for 3 min and the reaction was initiated by the addition of the NADPH-generating system. After incubation for 30 min, ice-cold CH3CN or CH3OH (0.2 ml) was added to terminate the reaction. The mixture was centrifuged at 10,000 rpm for 10 min. An aliquot of the supernatant (20 μl) was subjected to HPLC. The HPLC system for cisapride was the same as the measurement of itopride N-oxygenase activity except for the use of an excitation wavelength of 295 nm and an emission wavelength of 350 nm. The mobile phase, consisting of 20 mM phosphate buffer (pH 7.0):CH3CN (60:40, v/v), was delivered at a flow rate of 1 ml/min. The HPLC system for mosapride was composed of a Jasco BIP-I pump, a Waters WISP 710 autosampler, a TSKgel ODS-80Tm column (5 μm, 150 × 4.6 mm i.d., Tosoh), and a Jasco UVIDEC 100-V UV spectrometer set at a wavelength of 307 nm. The mobile phase, consisting of 50 mM phosphate buffer (pH 2.5):CH3OH (60:40, v/v), was delivered at a flow rate of 1 ml/min. The peak areas were obtained from a Shimadzu C-R4AX integrator. The retention times for N-dealkylated cisapride and mosapride were 4.3 and 5.3 min, respectively.
Studies on Inhibition of Itopride, Cisapride, and Mosapride Metabolism.
Ketoconazole, cimetidine, erythromycin, clarithromycin, SKF-525A, methimazole, thiourea, and n-octylamine were added to incubation mixtures at 10 μM, 100 μM, 1 mM, or 3 mM. A typical incubation mixture contained 100 mM phosphate buffer (pH 7.4), 0.05 mM EDTA, the NADPH-generating system, and liver microsomes from humans or rats (0.5 mg of protein/ml) in a final volume of 0.2 ml. The reaction mixtures containing erythromycin or clarithromycin were preincubated in the presence of the NADPH-generating system at 37°C for 15 min, and the reaction was initiated by addition of the substrate. Ketoconazole and cimetidine were dissolved in CH3OH. Erythromycin and clarithromycin were dissolved in dimethyl sulfoxide. SKF-525A, methimazole, and thiourea were dissolved in CH3OH:water (10:90, v/v). n-Octylamine was dissolved in CH3OH:1 M HCl:water (10:10:80, v/v/v). The final concentration of dimethyl sulfoxide or CH3OH was adjusted to 1% (v/v).
In Vivo Pharmacokinetics Studies.
Rats were divided into two groups (four animals per group). In the ketoconazole-pretreated group, rats were administered ketoconazole orally at a dose of 120 mg/kg twice before drug administration with a 12-h dosing interval. In the control group, rats received the vehicle (0.05 M HCl) in the same procedure. Each group of rats was given a single dose of itopride (30 mg/kg), cisapride (1.5 mg/kg), or mosapride (3 mg/kg) orally 1 h before the final administration of ketoconazole. Blood samples (about 0.4 ml) were collected from the tail vein at desired time points after drug administration. The blood samples were coagulated and centrifuged at 3000 rpm for 10 min to obtain serum. The serum samples were stored at −20°C until analysis. Serum itopride concentrations were determined by an HPLC method using a column switching technique (Takahara et al., 1992). Serum was directly subjected to HPLC (a Waters 600 pump and a Shimadzu LC-10AD pump, a Waters WISP 717 plus autosampler, and Waters six-port automated switching valves). A Nucleosilc CN column (25–40 μm, 10 × 4.0 mm i.d., Macherey-Nagel, Duren, Germany) and a TSKgel ODS-80Tm(5 μm, 150 × 4.6 mm i.d., Tosoh) were used for the preseparation and the analytical separation, respectively. A Shimadzu RF-10Axl fluorescence detector was set at an excitation wavelength of 308 nm and an emission wavelength of 344 nm. Two mobile phases, 0.1 M phosphate buffer (pH 7.0) for the preseparation and 0.05 M phosphate buffer (pH 5.5):CH3CN (80:20, v/v) for the analytical separation, were delivered at a flow rate of 1 ml/min. The peak areas were obtained from a Shimadzu C-R7A plus integrator. The retention time for itopride was 12.4 min. Serum concentrations of cisapride or mosapride were determined by liquid chromatography-tandem mass spectrometry (LC/MS/MS). For the quantification of cisapride, mosapride was used as an internal standard (I.S.) and, conversely, cisapride was used as an I.S. for the quantification of mosapride. A serum sample (0.1 ml) was alkalinized with 0.07 ml of 1 M NaOH, and then 0.1 ml of the I.S. solution (mosapride, 100 ng/ml; cisapride, 50 ng/ml) and 2.5 ml of CH3Cl:2-propanol (90:10, v/v) were added. The mixture was shaken for 5 min then centrifuged at 3000 rpm for 5 min at room temperature, and the organic layer (2 ml) was taken and evaporated to dryness under vacuum. The residue was dissolved in 0.2 ml of mobile phase and a 20-μl (cisapride) or 50-μl (mosapride) aliquot of the solution was subjected to LC/MS/MS. The chromatographic separation was achieved with a Waters 2690 separation module HPLC system and a TSKgel ODS-80Tm column (5 μm, 50 × 4.6 mm i.d., Tosoh) using a mobile phase of 10 mM ammonium acetate:CH3CN (40:60, v/v) at a flow rate of 1 ml/min. The column eluate was split, and approximately 0.2 ml/min was introduced into the electrospray ionization source. The mass spectrometer was a Quattro II (Micromass, Manchester, UK) equipped with an electrospray ionization interface. Multiple positive ion monitoring was employed for the quantification of cisapride at m/z 466 > 184 and at m/z 422 > 198 for the I.S., and for the quantification of mosapride at m/z422 > 198 and at m/z 466 > 184 for the I.S. The retention times for cisapride and mosapride were 1.9 and 1.8 min, respectively.
Studies on Inhibition of CYP-Mediated Reactions.
The effects of itopride on theophylline 1-demethylation, tolbutamide hydroxylation, S-mephenytoin 4′-hydroxylation, bufuralol 1′-hydroxylation, and testosterone 6β-hydroxylation were studied using human liver microsomes. Incubations and subsequent HPLC analysis were conducted as described (Kronbach et al., 1987; Reinerink et al., 1991; Sarkar et al., 1992; Chiba et al., 1993; Sharer et al., 1995) with some modifications. The substrate concentrations used for each assay were 2 mM theophylline, 100 μM tolbutamide, 40 μMS-mephenytoin, 20 μM bufuralol, or 100 μM testosterone. Itopride was added to incubation mixtures at a concentration of 1, 10, or 100 μM. The incubation mixtures containing 100 mM phosphate buffer (pH 7.4), 0.05 mM EDTA, the NADPH-generating system, and human liver microsomes (0.5 or 1 mg protein/ml) in a final volume of 0.25 ml were preincubated at 37°C for 5 min, and the reactions were initiated by addition of the NADPH-generating system. Incubations were performed at 37°C for 20 to 30 min, and the reactions were terminated by adding 0.4 ml of 2% (w/v) ZnSO4 for theophylline 1-demethylation, 0.2 ml of CH3Cl for tolbutamide hydroxylation, 0.1 ml of CH3CN forS-mephenytoin 4′-hydroxylation, and 0.025 ml of 60% HClO4 for bufuralol 1′-hydroxylation. Testosterone 6β-hydroxylation was terminated by cooling with ice. Samples for evaluation of S-mephenytoin and bufuralol metabolism were centrifuged at 10,000 rpm for 10 min. An aliquot of the supernatant (10 μl) was subjected to HPLC. Samples for evaluation of theophylline metabolism were saturated with 500 mg of (NH4)2SO4, and 25 μl of 5 μg/ml 7-(β-hydroxypropyl)theophylline was added as an I.S. The mixture was extracted with 5 ml of CH2Cl2:2-propanol (80:20, v/v). The organic layer was evaporated to dryness under vacuum. Extraction was repeated with another 5 ml of CH2Cl2:2-propanol. The organic layer combined with the residue was evaporated to dryness. The residue was dissolved in 0.2 ml of the mobile phase, and a 50-μl aliquot of the solution was subjected to HPLC. Samples for evaluation of tolbutamide metabolism were spiked with 10 μl of 10 μg/ml chlorpropamide as an I.S. After the addition of 20 μl of 35% H3PO4, the mixture was extracted with 1 ml of CH3Cl. The organic layer (1 ml) was taken and evaporated to dryness under vacuum. The residue was dissolved in 0.1 ml of the mobile phase, and a 50-μl aliquot of the solution was subjected to HPLC. Samples for evaluation of testosterone metabolism were extracted with 6 ml of CH2Cl2. The organic layer (5 ml) was taken and evaporated to dryness under vacuum. The residue was dissolved in 0.2 ml of CH3OH:water (1:1, v/v), and a 10-μl aliquot of the solution was subjected to HPLC. The HPLC system was composed of a Shimadzu LC-10AD or LC-9A pump, a Waters WISP 717 plus autosampler, a TSKgel ODS-80Tmcolumn (5 μm, 150 × 4.6 mm i.d., Tosoh), a Shimadzu SPD-10A or Jasco UV-970 UV spectrometer, and a Jasco 821-FP fluorescence detector. The peak areas were obtained from a Shimadzu C-R7A plus or C-R7Ae plus integrator. The mobile phase for theophylline was 10 mM acetate buffer (pH 4.5):CH3CN at a flow rate of 1 ml/min. Isocratic elution with 2% CH3CN:98% acetate buffer was run for 17 min, followed by linear gradients from 2 to 5% CH3CN in 12 min and 5 to 9% CH3CN in 18 min. Subsequently, isocratic elutions of 9% CH3CN for 8 min and 40% CH3CN for 5 min were run. UV detection was performed at 290 nm. The product of tolbutamide methylhydroxylation was eluted isocratically with 50 mM phosphate buffer (pH 7.0):CH3CN (85:15, v/v) at a flow rate of 1 ml/min. UV absorption was monitored at 230 nm. The mobile phase for evaluation of S-mephenytoin 4′-hydroxylation was 50 mM phosphate buffer (pH 4.0):CH3CN (74:26, v/v) at a flow rate of 0.8 ml/min under isocratic conditions, and UV detection was performed at 204 nm. Bufuralol 1′-hydroxylation was analyzed using 20 mM NaClO4 buffer (pH 2.5):CH3CN (65:35, v/v) at a flow rate of 1 ml/min. Spectrofluorometric detection was performed at an excitation wavelength of 252 nm and an emission wavelength of 302 nm. For evaluation of testosterone 6β-hydroxylation, gradient elution was performed with mixture A (acetic acid solution, pH 4.5:CH3OH; 70:30, v/v) and mixture B (CH3OH:CH3CN; 90:10, v/v) as the mobile phases at a flow rate of 0.8 ml/min. Isocratic elution with 11% B was run for 10 min, followed by a linear gradient from 11% B to 40% B in 35 min and isocratic elution with 40% B for 5 min. UV detection was performed at 240 nm. The column temperature was 50°C.
Data Analysis.
The pharmacokinetics parameters were calculated based on the time course data of serum concentrations, as follows. Maximum concentration in the serum after drug administration (Cmax) and the time to reachCmax (Tmax) were obtained from the data. The elimination rate constant (Kel) was estimated by least-squares regression of the terminal log-linear portion of the serum concentration-time profile. Terminal half-life (t1/2) was estimated as the ratio of the natural logarithm of 2 divided by Kel. The area under the serum concentration-time curve up to the terminal measurement time “t ” (AUC0–t) was calculated by a trapezoidal method. The AUC0–∞ value was calculated as the sum of AUC0–t and the area fromt to infinite time (AUCinf). The AUCinf value was calculated by extrapolation using Kel and the serum concentration at the final data point.
Statistical Method.
The statistical comparisons between pharmacokinetic parameters with and without ketoconazole administration were made using Dunnett's test. The criterion of statistical significance was P < .05.
Results
Itopride N-Oxygenation by Human Liver Microsomes.
Eadie-Hofstee plots for the itopride N-oxygenation indicated that this reaction exhibited monophasic enzyme kinetics.Km and Vmaxvalues for the itopride N-oxygenation were 67 μM and 2.24 nmol/min/mg of protein, respectively. To identify the enzyme responsible for the itopride N-oxygenation, we examined the effects of inhibitors and an enhancer of FMO and CYP. FMO in liver microsomes is known to be inhibited by methimazole (Tugnait et al., 1997) and thiourea (Cashman et al., 1993a), and its activity is not affected by SKF-525A, a known inhibitor of CYP-dependent monooxygenase (Ziegler and Mitchell, 1972). As shown in Fig.2, the itopride N-oxygenation by human liver microsomes was inhibited by methimazole and thiourea but was not affected by SKF-525A. n-Octylamine, which is an enhancer of FMO activity in mouse liver (Prough and Ziegler, 1977) but has little effect or is inhibitory in certain other species (Ziegler, 1988), did not affect the itopride N-oxygenation. FMO in human liver microsomes is known to be unstable to heat, and this is probably one of the reasons why it is difficult to purify FMO from liver microsomes. Kitchell et al. (1978) reported that this enzyme could be stabilized by NADPH. Thus, we examined the effect of NADPH on FMO in human liver microsomes using itopride as a substrate. In the absence of NADPH, the N-oxygenase activity was rapidly lost within 5 min at 45°C, whereas it was stable for 10 min when NADPH was present (Fig. 3). These results suggest that the N-oxygenation of itopride is catalyzed by FMO, not by CYP.

Effects of inhibitors of FMO or CYP on itopride N-oxygenation by human liver microsomes.
Each column represents the mean from duplicate incubation. Itopride was added to the incubation mixture at 10 μM. *Standard incubation mixture consisted of 100 mM phosphate buffer (pH 7.4), 0.05 mM EDTA, the NADPH-generating system, and 0.1 mg of microsomal protein as described under Experimental Procedures.

Effects of heat treatment on itopride N-oxygenation by human liver microsomes.
Each data point represents the mean from duplicate incubation. The human liver microsomes were treated at 45°C in the absence or presence of the NADPH-generating system for the indicated periods. Itopride was added to the incubation mixture at 10 μM.
Itopride N-Oxygenation by Microsomes Expressing a Human CYP or FMO.
To clarify further the possible involvement of FMO in itoprideN-oxygenation, the activities of theN-oxygenation by microsomes from genetically engineered cells expressing human CYP or FMO were investigated (Fig.4). FMO1 and FMO3 exhibitedN-oxygenase activity (3.64 and 4.10 nmol/min/mg of protein, respectively). FMO5 also exhibited N-oxygenase activity, but the activity was low (0.01 nmol/min/mg of protein). However, no activity was detectable with CYP1A2, -2A6, -2B6, -2C8, -2C9, -2C19, -2D6, -2E1, or -3A4.

Itopride N-oxygenation by microsomes expressing a human CYP or FMO.
Each column represents the mean from duplicate incubation. Itopride was added to the incubation mixture at 10 μM. ND, not detectable. The detection limit for the formation of the N-oxide was 0.007 nmol/min/mg of protein.
Effects of CYP3A4 Inhibitors on Metabolism of Itopride, Cisapride, and Mosapride.
To predict drug-drug interaction of gastroprokinetic agents, the effects of CYP3A4 inhibitors on itopride N-oxygenation, cisapride N-dealkylation, and mosaprideN-dealkylation were examined, using liver microsomes of humans and rats (Fig. 5). Ketoconazole, a potent inhibitor of CYP3A4, inhibited the N-dealkylation of cisapride and mosapride to the extent of 100% and 90%, respectively. Furthermore, cimetidine, erythromycin, and clarithromycin, all CYP3A4 inhibitors, also inhibited the N-dealkylation of cisapride and mosapride. However, these inhibitors showed little or no inhibitory effect on itopride N-oxygenation.

Effects of inhibitors of CYP3A4 on itopride N-oxygenation, cisapride N-dealkylation, and mosapride N-dealkylation by liver microsomes from humans (A) and rats (B).
Each column represents the mean from duplicate incubation. Standard incubation mixture consisted of 100 mM phosphate buffer (pH 7.4), 0.05 mM EDTA, the NADPH-generating system, and 0.1 mg of microsomal protein as described under Experimental Procedures. ▪, itopride (10 μM); ▨, cisapride (10 μM); ■, mosapride (10 μM); ND, not detectable.
Ketoconazole and cimetidine effectively inhibited theN-dealkylation of cisapride and mosapride in rat liver microsomes, as observed in human microsomes, exhibiting inhibition ratio values ranging from 74 to 100%. However, erythromycin and clarithromycin had little effect.
Next, a pharmacokinetic study was conducted in rats to evaluate the inhibitory effect of a CYP3A4 inhibitor on the in vivo metabolism of itopride, cisapride, and mosapride. We used ketoconazole, because it strongly inhibited the metabolism of cisapride and mosapride in liver microsomes of both species.
Effect of Ketoconazole on Pharmacokinetics of Itopride, Cisapride, and Mosapride in Rats.
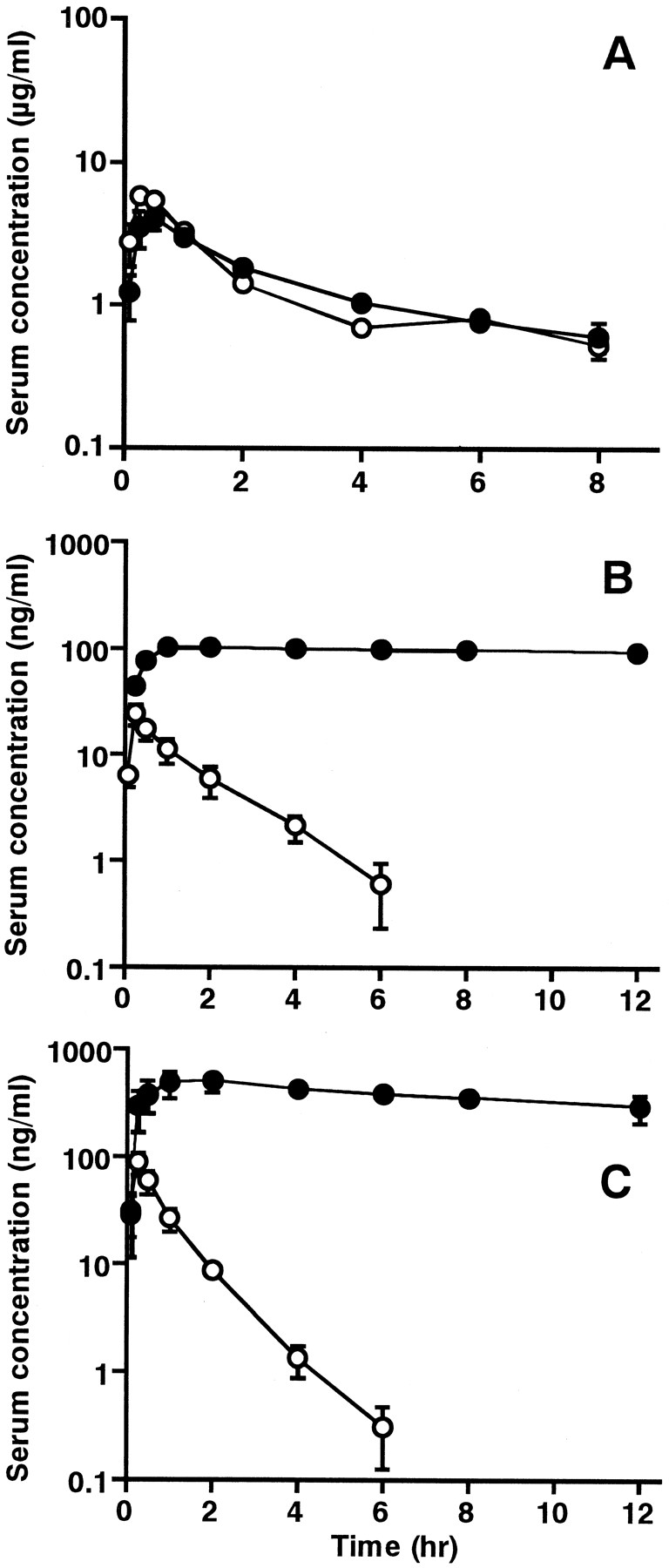
The recommended therapeutic doses for itopride, cisapride, mosapride, and ketoconazole are 50, 2.5, 5, and 200 mg, respectively. In the present study, we administered these drugs at a 30-fold higher than therapeutic dose, because of the low serum concentrations obtained after dosing of cisapride and mosapride to rats. The serum concentration versus time curves of itopride (30 mg/kg), cisapride (1.5 mg/kg), and mosapride (3 mg/kg) after oral administration with or without ketoconazole (120 mg/kg) are shown in Fig.6. Pharmacokinetic parameters of itopride, cisapride, and mosapride are listed in Table1. The serum concentrations of cisapride and mosapride were greatly increased by coadministration of ketoconazole. The Cmax and AUC of cisapride were 6 and 38 times greater, respectively, than those of the drug given alone. The Cmax and AUC of mosapride were 6 and 58 times greater, respectively, than those of the drug given alone. On the other hand, the serum concentrations of itopride after coadministration with ketoconazole were comparable with those of the drug given alone.

Serum concentrations of itopride (A), cisapride (B), and mosapride (C) after an oral administration to rats with (●) or without (○) ketoconazole.
Rats were administered a single oral dose of itopride (30 mg/kg), cisapride (1.5 mg/kg), or mosapride (3 mg/kg) concomitantly with, or without, ketoconazole (120 mg/kg) as described underExperimental Procedures. Each point represents the mean ± S.E. of four rats.
Pharmacokinetic parameters for itopride, cisapride, and mosapride after an oral administration to rats with or without ketoconazole
Effect of Itopride on CYP-Mediated Reactions of Human Liver Microsomes.
To assess the inhibitory effect of itopride on CYP-mediated metabolism, theophylline 1-demethylation, tolbutamide hydroxylation,S-mephenytoin 4′-hydroxylation, bufuralol 1′-hydroxylation, and testosterone 6β-hydroxylation were assayed in human liver microsomes as indicators of the activities of CYP1A2, -2C8/9, -2C19, -2D6, and -3A4, respectively. Even at the highest concentration examined (100 μM), itopride failed to inhibit these reactions.
Discussion
Our results support a predominant role of FMO in the formation of itopride N-oxide in human liver microsomes. The involvement of both CYP- and FMO-dependent N-oxygenation is frequently observed in the metabolism of the tertiary amines (Cashman et al., 1993a; Blake et al., 1995). Therefore, the contributions of the CYP and FMO families to itopride N-oxygenation in human liver microsomes were investigated using metabolic inhibitors and heat-treatment techniques. Itopride N-oxide formation was inhibited in the presence of methimazole and thiourea, alternative substrate-competitive inhibitors of FMO (Fig. 2). In addition, the decrease in itopride N-oxide formation to 0% of the control caused by treatment at 45°C for 5 min suggests a heat-mediated inactivation of FMO that was delayed when NADPH was included (Fig. 3).
Mammalian FMO comprises a family of xenobiotic-metabolizing enzymes found in many tissues and catalyzes the NADPH-dependent oxygenation of nucleophilic nitrogen, sulfur, phosphorus, and selenium in a structurally diverse range of compounds (Ziegler, 1993). Recently, cDNAs encoding FMOs of pig, mouse, rat, rabbit, guinea pig, and human have been isolated and sequenced. Based on the similarity of the amino acid sequences, FMOs were classified into five gene subfamilies: FMO1 through FMO5 (Lawton et al., 1994). In human liver, FMO1, FMO3, and FMO5 seem to be important isoforms for metabolism of xenobiotics (Cashman, 1995). To investigate the contribution of the FMO isoforms to itopride metabolism, the itopride N-oxygenation capacity of microsomes of cells expressing human CYP or FMO was determined with 10 μM itopride as the substrate. Itopride N-oxygenase activity was detectable only in cells expressing FMOs (Fig. 4). Generally, both FMO and CYP are implicated in the oxygenation of nitrogen- and sulfur-containing compound. Various drugs, including cimetidine (Cashman et al., 1993b; Overby et al., 1997), ranitidine (Overby et al., 1997), imipramine (Lemoine et al., 1990), tamoxifen (Mani et al., 1993), chlorpromazine (Cashman et al., 1993a), (S)-nicotine (Berkman et al., 1995), clozapine (Tugnait et al., 1997), and zotepine (Shiraga et al., 1999) are reported to be substrates for FMO, although the precise contribution of FMO to their metabolic clearance is unknown in most cases. It is of interest to note that itopride N-oxygenation was mediated only by FMO, not by CYP. The involvement of both FMO1 and FMO3 in the itoprideN-oxygenation indicates that the FMO isoforms have overlapping substrate specificity.
We investigated the effect of the CYP3A4 inhibitor ketoconazole on the pharmacokinetics of cisapride, mosapride, and itopride in rats. In the in vivo study, pretreatment with ketoconazole resulted in significantly increased Cmax and AUC of cisapride and mosapride. No statistically significant effect on the pharmacokinetics of itopride was observed (Fig. 6, Table 1). We have also shown that ketoconazole inhibits the N-dealkylation of cisapride and mosapride, but not the N-oxygenation of itopride at a concentration of 10 μM in liver microsomes from rats and humans (Fig. 5). The in vitro data suggest that the increased serum levels of cisapride and mosapride observed in rats after pretreatment with ketoconazole may be attributed to inhibition of CYP3A4-mediated metabolism by ketoconazole. It is well known that oral CYP3A4 inhibitor dosing significantly inhibits the metabolism of cisapride, resulting in markedly elevated plasma concentration of cisapride in patients (Bedford and Rowbotham, 1996; Haarst et al., 1998). This drug-drug interaction may produce adverse effects, such as QT interval prolongation, leading to the risk of ventricular arrhythmias (Bedford and Rowbotham, 1996). Recently, Katoh et al. (1999) reported that theCmax of mosapride increased 1.6-fold andt1/2 was prolonged from 1.6 to 2.4 h by the concomitant use of erythromycin in humans. In the present study, all CYP3A4 inhibitors that we tested strongly inhibited the metabolism of mosapride by human liver microsomes, suggesting that, not only erythromycin, but also other CYP3A4 inhibitors cause accumulation of unmetabolized mosapride in humans.
According to Overby et al. (1997), cimetidine S-oxidase activity was associated with FMO3 in human liver. In the present study, itopride N-oxygenation was not affected by cimetidine even at 3 mM concentration (Fig. 5), which is about 800-fold higher than the steady-state plasma concentration of cimetidine in patients (Somogyi and Gugler, 1983). Furthermore, imipramine, considered to be a selective substrate of FMO1 (Lemoine et al., 1990), did not inhibit itopride N-oxygenation at much higher concentration than the therapeutic level (data not shown). These results suggest that clinically significant drug interaction between itopride and other FMO substrates will not occur.
In conclusion, we demonstrated the involvement of FMOs in the formation of itopride N-oxide in human liver. Ketoconazole, cimetidine, erythromycin, and clarithromycin did not inhibit itoprideN-oxygenation in human liver microsomes. Furthermore, pretreatment with ketoconazole did not alter the metabolism of itopride in rats, whereas it caused accumulation of unmetabolized cisapride and mosapride. In addition, itopride showed no inhibitory effect on five specific CYP-mediated reactions in human liver microsomes. We conclude that itopride is unlikely to cause clinically significant pharmacokinetic drug interactions.
Footnotes
-
Send reprint requests to: Taisei Mushiroda, Research & Development Headquarters, Hokuriku Seiyaku Co., Katsuyama, Fukui, 911-8555, Japan. E-mail: taisei.mushiroda{at}hokuriku-seiyaku.co.jp
- Abbreviations used are::
- CYP
- cytochrome P450
- FMO
- flavin-containing monooxygenase
- LC/MS/MS
- liquid chromatography-tandem mass spectrometry
- SKF-525A
- proadifen hydrochloride
- I.S.
- internal standard
- Received March 8, 2000.
- Accepted July 17, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}