


Abstract
In vitro studies were conducted to identify the cytochromes P450 (P450s) involved in the formation of 2- and 3-hydroxycarbamazepine, metabolites that may serve as precursors in the formation of protein-reactive metabolites. Human liver microsomes (HLMs) converted carbamazepine (30–300 μM) to 3-hydroxycarbamazepine at rates >25 times those of 2-hydroxycarbamazepine. Both the 2- and 3-hydroxylation of carbamazepine appeared to conform to monophasic Michaelis-Menten kinetics in HLMs (apparentKm values, ∼1640 and ∼217 μM; apparentVmax values, ∼5.71 and ∼46.9 pmol/mg of protein/min, respectively). Rates of carbamazepine 2- and 3-hydroxylation correlated strongly with CYP2B6 activity (r ≥ 0.757) in a panel of HLMs (n = 8). Carbamazepine 3-hydroxylation also correlated significantly with CYP2C8 activity at a carbamazepine concentration of 30 μM. Formation of 2- and 3-hydroxycarbamazepine did not correlate significantly with any other P450 activities. The chemical inhibitors ketoconazole (CYP3A) and 7-EFC (CYP2B6) inhibited both 2- and 3-hydroxycarbamazepine formation whereas 4-methylpyrazole (CYP2E1) markedly decreased 2-hydroxycarbamazepine formation. Several recombinant P450s catalyzed carbamazepine 2- and 3-hydroxylation, but after adjustment for relative hepatic abundance, CYP3A4 and CYP2B6 appeared to be the major catalysts of carbamazepine 3-hydroxylase activity, and at least five P450s were significant contributors to 2-hydroxycarbamazepine formation; CYP2E1 made the greatest contribution to the Clint of carbamazepine 2-hydroxylation (∼30%), but P450s CYP1A2, 2A6, 2B6, and 3A4 also made significant contributions (∼13–18%). These results suggest that CYP2B6 and CYP3A4 are largely responsible for the formation of 3-hyrdoxycarbamazepine, whereas multiple P450s (CYP1A2, 2A6, 2B6, 2E1, and 3A4) contributed to 2-hydroxycarbamazepine formation.
Carbamazepine is one of the most commonly prescribed drugs for the treatment of epilepsy. Its use, however, has been associated with a variety of idiosyncratic adverse reactions that include cutaneous, hematological, immunological, renal, and hepatic disorders (Shear and Spielberg, 1988; Vittorio and Muglia, 1995). Although the mechanism behind these adverse reactions is unknown, it has been proposed that these adverse reactions may result from the formation of chemically reactive metabolites, the identities of which have not been determined to date. An arene oxide metabolite was originally proposed as the reactive species responsible for the idiosyncratic toxicity of carbamazepine (Spielberg et al., 1981), however, several alternative protein-reactive metabolites have been proposed including 9-acridine carboxaldehyde (Furst et al., 1995), ano-quinone metabolite (Lillibridge et al., 1996), and an iminoquinone metabolite (Ju and Uetrecht, 1999) derived from the carbamazepine metabolite, 2-hydroxyiminostilbene (Fig.1).

Proposed scheme for the formation of reactive metabolites from carbamazepine in humans.
In vitro studies using [14C]carbamazepine have demonstrated that human liver microsomes and cDNA-expressed P450s1 CYP1A1, 1A2, 2C8, and 3A4 are capable of generating protein-reactive carbamazepine metabolites, although the identity of this metabolite(s) remains unknown (Wolkenstein et al., 1998). In vitro bioactivation data with phenytoin, another aromatic anticonvulsant, the use of which is associated with idiosyncratic adverse reactions similar to those observed with carbamazepine, have demonstrated that the formation of a catechol metabolite (3′, 4′-dihydroxyphenytoin) is an essential step leading to the covalent binding of phenytoin to human liver microsomal proteins (Munns et al., 1997). The carbamazepine metabolite corresponding to 3′,4′-dihydroxyphenytoin is 2,3-dihydroxycarbamazepine, which can be derived from either 2-hydroxycarbamazepine or 3-hydroxycarbamazepine (Lertratanangkoon and Horning, 1982). It has been proposed that 2, 3-dihydroxycarbamazepine undergoes nonenzymatic rearrangement to form an o-quinone metabolite that is capable of binding to microsomal proteins. 2-Hydroxycarbamazepine may also be involved in generating an additional protein-reactive species via further metabolism to 2-hydroxyiminostilbene (Lertratanangkoon and Horning, 1982), which can be readily oxidized to a reactive iminoquinone species (Ju and Uetrecht, 1999).
Despite the evident role of cytochrome P450 enzymes in the formation of protein-reactive carbamazepine metabolites, relatively little is known regarding the involvement of P450 enzymes in the metabolism of carbamazepine beyond those responsible for the formation of carbamazepine 10,11 epoxide, the major metabolite formed from carbamazepine (Lertratanangkoon and Horning, 1982; Kerr et al., 1994). One recent in vitro study (Pelkonen et al., 2001) did examine the role of P450 enzymes in the formation of several carbamazepine metabolites. The authors reported that 3-hydroxycarbamazepine was principally formed by CYP2A6, with minor contributions by CYP1A2, CYP2C8, CYP2D6, and CYP3A4. The enzymes responsible for 2-hydroxycarbamazepine formation were not reported in this study. Unfortunately, carbamazepine appears to have been added to incubation mixtures in concentrations of DMSO (0.5–1%) that considerably inhibit the activities of several P450 enzymes (Easterbrook et al., 2000). Because of the role that 2-hydroxycarbamazepine and 3-hydroxycarbamazepine may play as precursors to the formation of protein-reactive metabolites and because previous in vitro studies have not convincingly identified the P450 enzymes involved in the formation of these metabolites, in vitro studies were undertaken to identify the P450 enzymes involved in the 2- and 3-hydroxylation of carbamazepine.
Materials and Methods
Chemicals.
Carbamazepine, carbamazepine 10,11-epoxide, α-naphthoflavone, coumarin, ketoconazole, 4-methylpyrazole, omeprazole, quinidine, sulfaphenazole, glucose-6-phospate, glucose-6-phosphate dehydrogenase, NADP, and EDTA were purchased from Sigma-Aldrich (St. Louis, MO). 7-EFC was purchased from Molecular Probes (Eugene, OR). All other reagents were of analytical grade. 3-Hydroxycarbamazepine and 10,11-dihydro-10,11-trans-dihydroxycarbamazepine were kindly provided by S. Nelson (University of Washington, Seattle, WA) and R. Heckendorn (Ciba Geigy, Toms River, NJ), respectively.
Synthesis of 2-Hydroxycarbamazepine.
2-Hydroxycarbamazepine (6) was synthesized using a modification of the procedure described by Chang (1983), according to the scheme depicted in Fig. 2.Dibenzo[b,f]azepin-2-one(2); Fremy's salt (5 g) and disodium hydrogen phosphate (3.6 g) were dissolved in 190 ml of deionized water. Iminostilbene (1) (1.1 g; 5.31 mmol) was separately dissolved in acetone. The aqueous solution was added slowly to the acetone solution in portions slowly with constant stirring over a period of 25 min. The solution was then filtered and kept in the refrigerator overnight (2–8°C). Acetone was removed from the solution by evaporation (≤20°C). The resulting reddish brown aqueous solution was extracted with ethyl acetate (50-ml portions) until no red color was seen. Ethyl acetate was removed from the combined extracts by evaporation. The resulting residue was purified by flash column chromatography using a gradient solvent system (100% hexane → 60% hexane and 40% ethyl acetate). The eluate containing the desired ketone (2) was evaporated to dryness to give 253 mg (23% yield) of dibenzo[b,f]azepin-2-one as a deep reddish solid. m.p. = 141°C; 1H NMR (Chloroform-d): δ 6.57 (s, 1H), 6.74–6.79 (d,1H, J = 12.5 Hz), 6.93–6.99 (t, 2H), 7.49–7.63 (m, 4H), 7.92–7.95 (d,1H,J = 7.5); 13C NMR (chloroform-d): δ 127.41, 130.98, 132.03, 132.50, 132.84, 134.55, 136.29, 137.53, 139.92, 144.67, 186.69(C = O); MS: 208 [M + H]+, 230 [M + Na]+, 415 [2M + H]+.2-(tert-Butyl-dimethyl-silanyloxy)-5H-dibenzo[b,f]azepin-2-one(4); 2 (360 mg; 1.74 mmol) was dissolved in methylene chloride (100 ml) and shaken in a separating flask with saturated aqueous sodium dithionite solution until the deep reddish color changed to golden yellow. The methylene chloride layer was collected through a funnel containing anhydrous sodium sulfate and concentrated (at a temperature less than 20°C with minimal exposure to light and air). The concentrated solution was purged with nitrogen.tert-Butyl dimethyl silyl chloride (4 g; 8 mmol), imidazole (0.4 g; 2 mmol), and triethylamine (3 ml) were added to the solution and stirred overnight. The solution was evaporated to dryness under reduced pressure and extracted with hexanes. The hexanes were then evaporated to yield (4). The yield was approximately 100% (584 mg); 1H NMR (Chloroform-d): δ 0.19 (s, 6H), 0.98 (s, 9H), 4.7 (s, 1H, NH), 6.79–6.89 (m, 4H), 7.30–7.44 (m, 5H).2-(tert-Butyl-dimethyl-silanyloxy)-5H-dibenzo[b,f]azepine-5-carboxylic acid 2,4-dimethoxy-benzylamide (5). Compound4 (660 mg; 1.59 mmol) was added to a solution of diimadazolecarbonyl (320 mg), dissolved in methylene chloride (10 ml), and stirred for 20 min at room temperature. Dimethoxybenzylamine (0.4 ml) was added to the reaction, and the solution was stirred overnight. The resulting solution was analyzed by thin layer chromatography using hexane/ethyl acetate (1:1 v/v) and theRf was determined to be 0.5. Additional methylene chloride was added to the solution, which was then washed with a solution of saturated ammonium chloride (2 × 50 ml) and saturated brine (2 × 50 ml). The solvent was removed, and the residue was isolated by flash column chromatography using a gradient solvent system (100% hexane → 60% hexane and 40% ethyl acetate). The eluate that contained 5 was evaporated to dryness under reduced pressure to give 445 mg (54% yield); 1H NMR (Chloroform-d): δ 0.24 (s, 6H), 1.02 (s, 9H), 3.60 (s, 3H, OMe), 3.75(s, 3H, OMe), 4.28–4.30 (d, 2H, J = 5Hz), 6.40 (s, 2H), 6.75–6.92 (m, 2H), 7.11–7.42 (m, 8H).6; a solution containing compound 5 (445 mg; 0.86 mmol) and trifluoroacetic acid (5 ml) was stirred for 24 h. The reaction was stopped and the trifluoroacetic acid was removed by evaporation under reduced pressure. The resulting residue was purified by flash column chromatography using 100% ethyl acetate and evaporated to dryness to give 180 mg (83% yield) of 6 as a white solid; m.p. = 230–234°C; 1H NMR (DMSO-d6): δ 5.51(s, Br, 2H, CONH2), 6.78–7.38 (m, 9H), 9.64 (s 1H, ArOH);13C NMR (DMSO-d6): δ 114.4, 116.4, 126.69, 128.24, 128.37, 128.90, 129.80, 130.14, 132.19, 134.61, 135.67, 140.98, 156.10 (C-OH), 156.69 (C = O); MS: 253 [M + H]+, 275 [M + Na]+, 505 [2M + H]+, 527 [2M + Na]+.

Scheme for the synthesis of 2-hydroxycarbamazepine.
TBSCl, tert-butyl dimethyl silyl chloride; CDI,1,1′-carbonyldiimidazole; TFA, trifluoroacetic acid.
Analysis of Synthesized Chemicals.
Flash chromatography was performed with Grade 3 × 0–799 silica gel (particle size 32–36; Selecto Scientific Inc., Suwanee, GA). Thin-layer chromatography was performed on flexible plates for thin layer chromatography (AL SIL G/UV; Whatman International Ltd., Maidstone, Kent, UK). Solvents were removed by a BüchiR-114 rotovaporator (Brinkman Instruments, Inc., Westbury, NY). Melting points were determined on a MELT-TEMP II apparatus (Laboratory Services, Inc., Holliston, MA) and are reported as uncorrected values. 1H and13C NMR spectra were recorded on an AC250 Bruker Instrument (FT-NMR; IBM Instruments Co., Danbury, CT) at 250 MHz and at 62 MHz, respectively. Electrospray ionization mass spectra were obtained on a Finnigan (Bremen, Germany) TSQ-700 triple quadrupole mass spectrometer equipped with an electrospray source. The spray voltage was maintained at 4.5 kilovolts, and the capillary temperature was set at 200°C. Dry nitrogen was used as the sheath gas and auxiliary gas for the analyses. Aliquots (0.2 μl) of analytes dissolved in methanol were introduced into the electrospray source via a mobile phase consisting of methanol and 0.1% (v/v) acetic acid. Full scan spectra (from m/z 100 to 600) were generated by collision-induced dissociation. Mass spectra were acquired at 3 s per scan, and 3 to 4 scans were averaged.
Human Liver Microsomes and cDNA-Expressed Enzymes.
Microsomes prepared from eight different human livers and from baculovirus-infected insect cells (SUPERSOMES) expressing human P450 enzymes (CYP1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 3A4, 3A5, and 3A7) or control vector were purchased from BD Gentest Corp. (Woburn, MA). All recombinant enzymes were co-expressed with human NADPH-cytochrome P450 reductase; some enzymes (CYP2B6, CYP2C19, CYP2E1, CYP3A4, and CYP3A7) were also coexpressed with human cytochrome b5 . The manufacturer provided protein concentrations, P450 contents, and P450 enzyme activities. Vials of microsomes were stored at −70°C until use. Microsomes were rapidly thawed in room temperature water and placed on ice prior to use.
Incubation of Carbamazepine with Human Liver Microsomes.
In vitro enzyme assays were performed in 96-well microtiter plates at 37 ± 0.1°C in a Thermo Forma (Marietta, OH) Benchtop Orbital Shaker incubator. Standard incubation mixtures (100-μl final volume) contained human liver microsomes (25 μg of microsomal protein), potassium phosphate buffer (50 mM, pH 7.4), MgCl2(3 mM), EDTA (1 mM), and carbamazepine (10–2000 μM) dissolved in methanol (≤1% v/v final concentration) at the final concentrations listed. After a 3-min preincubation at 37 ± 1°C, reactions were initiated by the addition of an NADPH-generating system, consisting of NADP (1 mM), glucose 6-phosphate (1 U/ml), glucose-6-phosphate dehydrogenase (5 mM), and terminated after 30 min by the addition of 100 μl of ice-cold methanol. Protein was precipitated by centrifugation at 10,000 rpm for 10 min. An aliquot (50–75 μl) of the supernatant was analyzed by HPLC/MS via direct injection. Under these conditions, the rates of carbamazepine 2- and 3-hydroxylation were proportional to incubation time and protein concentration, and metabolism of the parent compound did not exceed 20%. Unless otherwise noted, experiments were performed with two replicate samples per condition in triplicate (n = 6 determinations).
Correlation Experiments.
Rates of 2- and 3-hydroxycarbazepine formation were determined in microsomes prepared from eight different human livers at concentrations of carbamazepine (30, 100, and 300 μM) that spanned the average plasma Cmax (∼60 μM). The rates of carbamazepine 2- and 3-hydroxylation were then compared with the following P450 enzyme activities provided by BD Gentest: phenacetinO-deethylation (CYP1A2), coumarin 7-hydroxylation (CYP2A6),S-mephenytoin N-demethylation (CYP2B6), paclitaxel 6α-hydroxylation (CYP2C8), diclofenac 4′-hydroxylation (CYP2C9), S-mephenytoin 4′-hydroxylation (CYP2C19), bufurolol 1′-hydroxylation (CYP2D6), chlorzoxazone 6-hydroxylation (CYP2E1), testosterone 6β-hydroxylation (CYP3A4/5), and lauric acid 12-hydroxylation (CYP4A9/11).
Chemical-Inhibition Experiments.
Formation of 2- and 3-hydroxylated metabolites from carbamazepine (30 and 300 μM) by human liver microsomes was evaluated in the presence or absence (i.e., control) of known P450 isoform-selective inhibitors. The following inhibitors were examined at the indicated concentrations: α-naphthoflavone (CYP1A2, 1 μM), coumarin (CYP2A6, 10 μM), orphenadrine (CYP2B6, 100 μM), 7-EFC (CYP2B6, 10 μM), sulfaphenazole (CYP2C9, 10 μM), omeprazole (CYP2C19, 10 μM), quinidine (CYP2D6, 1 μM), 4-methylpyrazole (CYP2E1, 1 μM), and ketoconazole (CYP3A4/5, 1 μM). Inhibitors were dissolved in methanol and diluted in the incubation mixtures to a final solvent concentration of 1% (v/v). Control incubations contained an equal volume of methanol.
Experiments with cDNA-Expressed Human P450 Enzymes.
Incubations of carbamazepine with microsomes containing cDNA-expressed P450 enzymes were performed as described for liver microsomes except that the amount of enzyme used was 5 pmol/incubation, and incubations were terminated after 60 min (30 min for CYP3A4). Rates of carbamazepine 2- and 3-hydroxylation by recombinant P450 enzymes are reported as background (control) corrected rates.
HPLC/MS Analysis.
Carbamazepine and its metabolites were resolved by reverse-phase HPLC with a Hewlett Packard HP1100 HPLC system equipped with a HP1100 de-gasser, binary pump, auto-sampler, column heater, diode array detector, and mass spectral detector (Hewlett Packard Instruments, Santa Clara, CA). Separation of carbamazepine and its metabolites was achieved on a Phenomenex (Torrance, CA) Luna C8(2) column (4.6 mm × 15 cm, 3-μm particle size) connected in series with a Hewlett Packard Zorbax XDBC8 column (4.6 mm × 7.5 cm, 3.5-μm particle size). The analytical columns were preceded by a Phenomenex C8 guard column (4 × 3 mm i.d., 5-μm particle size). The mobile phase was a 53:47 mixture of methanol/water containing 0.1% acetic acid and was delivered at a constant flow of 0.5 ml/min. The column temperature was maintained at 40°C. Under these conditions, carbamazepine 10,11-dihydrodiol, 2-hydroxycarbamazepine, carbamazepine 10,11-epoxide, 3-hydroxycarbamazepine, and carbamazepine eluted at ∼9.5, 10.4, 10.9, 12.8, and 19.6 min, respectively. The column effluent was monitored by UV detection (290 nm; for verification of metabolite identity) and by atmospheric pressure chemical ionization detection with a mass spectrometer operating in a selective positive ion-monitoring mode. Ion detection was optimized for 2-OH-carbamazepine detection. The drying gas temperature and flow were maintained at 300°C and 3.5 l/min, respectively, and the nebulizer pressure was set at 20 psig. The vaporizer temperature was maintained at 350°C. The capillary voltage was set at 4 kV, and the corona current was set at 4 μΑ. Under these conditions, 2-and 3-hydroxycarbamazepine yielded [MH]+ ions at m/z 253, whereas carbamazepine 10,11-epoxide (which elutes close to 2-hydroxycarbamazepine) yielded small amounts of an [MH]+ ion at m/z 253 (∼20%) and significant amounts of an [MH-HNCO]+ ion atm/z 208 (∼80%). Data were collected and integrated with Hewlett Packard Chemstation V A.0401 software. Carbamazepine and its metabolites were quantified by comparison of their peak areas (determined by mass spectral analysis) with those of analytical standards. The lower limit of quantification for the assay was 187.5 fmol for 2-and 3-hydroxycarbamazepine. The analytical method was linear over a standard concentration range of 5 nM to 10 μM (r2 > 0.99).
Data Analysis.
Data from kinetic studies using human liver microsomes were analyzed by nonlinear regression (GraFit 5; Erithacus Software Ltd., Surrey, UK) to estimate apparent Km andVmax values. After initial kinetic estimates were obtained, the data were also analyzed by linear transformation (Eadie-Hofstee plots) to confirm enzyme models. Kinetic parameters for the formation of 2- and 3-hydroxycarbamazepine by cDNA-expressed human P450 enzymes were estimated from the best-fit line(s) using least-squares linear regression analysis of Lineweaver-Burk plots. Regression coefficients (r2) between the rates of carbamazepine 2- and 3-hydroxylation and the activities of cytochrome P450 enzymes were also determined using least-squares regression analysis. Significance was determined by Pearson's regression analysis from two-tailed t tables.
Results
Metabolism of Carbamazepine by Human Liver Microsomes.
A representative chromatogram from an incubation of carbamazepine with NADPH-fortified human liver microsomes is shown in Fig.3. As expected, the predominant metabolite formed by human liver microsomes was identified as carbamazepine 10,11-epoxide. Human liver microsomes converted carbamazepine to at least five other minor metabolites, two of which were identified as 2- and 3-hydroxycarbamazepine (based on retention times, absorbance properties at 290 nm and positive ion mass of authentic standards). 3-Hydroxycarbamazepine was formed by human liver microsomes at each of the carbamazepine concentrations examined in this study and was at least 25 times more prevalent than was 2-hydroxycarbamazepine. In contrast, 2-hydroxycarbamazepine typically was not detected in incubations conducted at substrate concentrations below 100 μM. In the absence of NADPH or human liver microsomes, no carbamazepine metabolites were detected under these experimental conditions.

HPLC/MS chromatogram obtained by APCI of [MH]+ ions (m/z 253) of carbamazepine and metabolites formed by human liver microsomes.
1, 2-hydroxycarbamazepine (10.4 min); 2, carbamazepine 10,11-epoxide (10.9 min); 3, 3-hydroxycarbamazepine (12.8 min). APCI, atmospheric pressure chemical ionization.
The formation of 3-hydroxycarbamazepine by human liver microsomes conformed to monophasic Michaelis-Menten kinetics, and a representative plot is presented in Fig. 4. Eadie-Hofstee plots (inset, Fig. 4) of the rates of carbamazepine 3-hydroxylation appeared linear over the concentration range examined (10–2000 μM), which suggests that this reaction is catalyzed predominantly by a single P450 enzyme. In general, insufficient data were generated to permit reliable kinetic analyses of 2-hydroxycarbamazepine formation by human liver microsomes. Unfortunately, concentrations of carbamazepine >2000 μM could not be achieved due to the solubility characteristics of carbamazepine in the incubation mixtures. However, in a single experiment, a sufficient number of data points were obtained with two preparations of human liver microsomes (HLM 56 and 112) to examine the kinetics of 2-hydroxycarbamazepine formation, and a representative plot is shown in Fig. 4. Eadie-Hofstee plots of these data had a pattern consistent with monophasic Michaelis-Menten kinetics. The apparent kinetic parameters derived from microsomal preparations of four individual livers are shown in Table 1.

Rates of formation of 2- and 3-hydroxycarbamazepine by human liver microsomes (HLM 056).
Insets, Eadie-Hofstee plots.
Apparent kinetic parameters for the formation of 2-hydroxycarbamazepine and 3-hydroxycarbamazepine in human liver microsomes
Correlation Experiments.
Human liver microsomes prepared from eight donors were examined for their ability to catalyze the 2- and 3-hydroxylation of carbamazepine at three substrate concentrations (30, 100, and 300 μM). All eight microsomal preparations converted carbamazepine to 3-hydroxycarbamazepine. The rate of carbamazepine 3-hydroxylation varied <5-fold among the microsomal samples [range (rates in pmol/mg of protein/min ± S.D.), at respective substrate concentration: 3.05 ± 0.93 to 14.8 ± 2.6, 30 μM; 8.26 ± 1.35 to 31.4 ± 2.9, 100 μM; and 15.7 ± 1.0 to 41.5 ± 4.1, 300 μM]. The sample-to-sample variation in the rates of 3-hydroxycarbamazepine formation correlated significantly with CYP2B6 activity (r ≥ 0.756, P < 0.05) and with CYP2C8 activity at a substrate concentration of 30 μM (r = 0.751, P < 0.05) (Table2). 3-Hydroxcarbamazepine formation was not significantly correlated with any other cytochrome P450 activities. With the exception of a significant correlation between CYP1A2 and CYP4A activities (r = 0.826) in the panel of human liver microsomes, none of the activities selective for other P450 isoforms were significantly correlated with one another.
Regression analysis (r2) of the relationship between the rates of carbamazepine conversion to 2-and 3-hydroxycarbamazepine with the sample-to-sample variation in cytochrome P450 activity in human liver microsomes
At substrate concentrations ≤100 μM, microsomes from four of the eight donors failed to convert carbamazepine to 2-hydroxycarbamazepine in measurable quantities. Seven of the eight microsomal preparations, however, did catalyze the formation of 2-hydroxycarbamazepine at a concentration of carbamazepine equal to 300 μM, hence correlation analyses were performed only with rates obtained at this concentration of substrate. The rate of 2-hydroxycarbamazepine formation varied 5.1-fold (0.309 ± 0.073 to 1.58 ± 0.04 pmol/mg of protein/min) among the microsomal samples and was significantly correlated with CYP2B6 activity (r = 0.862,P < 0.025) but not with any other cytochrome P450 activities.
Chemical Inhibition of Carbamazepine 2- and 3-Hydroxylation.
The effects of various P450 inhibitors on the conversion of carbamazepine (30 and 300 μM) to 2- and 3-hydroxycarbamazepine are illustrated in Fig. 5. Because correlation studies implicated CYP2B6 in the conversion of carbamazepine to both 2- and 3-hydroxycarbamazepine, chemical inhibitors were incubated with human liver microsomes from two donors, one with high CYP2B6 activity (H042, which also possessed high CYP3A4/5 activity) and one with low CYP2B6 activity (H056, which also had low CYP3A4/5 activity). In microsomes with high CYP2B6/3A activity, coumarin (selective for CYP2A6), 7-EFC (a CYP2B6 inhibitor) and ketoconazole (selective for CYP3A enzymes) markedly inhibited the conversion of 30 μM carbamazepine to 3-hydroxycarbamazepine (35–40%), whereas in microsomes with low CYP2B6/3A activity, only coumarin inhibited (57%) 3-hydroxycarbazepine formation substantially. At a substrate concentration of 300 μM, ketoconazole inhibited carbamazepine 3-hydroxylation (30–40%) in both microsomal preparations, and coumarin markedly inhibited 3-hydroxycarbamazepine formation (46%) in microsomes with low CYP2B6/3A activity. Moderate (20–25%) inhibition of 3-hydroxycarbamazepine formation was observed with 7-EFC and 4-methylpyrazole (selective for CYP2E1) in both microsomal preparations, and with coumarin in microsomes with low CYP2B6/3A activity. Interestingly, omeprazole appeared to cause a slight to moderate increase in carbamazepine 3-hydroxylation (∼30–60% and ∼10–30% increase in 3-hydroxylase activity at substrate concentrations of 30 and 300 μM, respectively). The other inhibitors examined had little or no effect (<20%) on the rate of 3-hydroxycarbamazepine formation.

Effects of various P450 isoform-selective inhibitors on the formation of 2- and 3-hydroxycarbamazepine by human liver microsomes.
Human liver microsomes were incubated with carbamazepine (30 or 300 μM) in the presence or absence of various chemicals, as described under Materials and Methods. Final inhibitor concentrations are indicated in the brackets. Each bar represents the mean ± S.D. of six determinations. The uninhibited rates of 2-hydroxycarbamazepine (2OHCBZ; [S] = 300 μM) and 3-hydroxycarbamazepine (3OHCBZ; [S] = 30 or 300 μM) formation were 1.58 ± 0.04, 14.8 ± 2.6, and 41.5 ± 4.1 pmol/mg of protein/min, respectively, in human liver microsomes with high CYP2B6 and CYP3A activity (H042) and 0.81 ± 0.05, 7.73 ± 1.04, and 21.4 ± 4.9 pmol/mg of protein/min, respectively, in human liver microsomes with low CYP2B6 and CYP3A activity (H056). [S] represents the concentration of carbamazepine present in incubation mixtures. ANF, α-naphthoflavone.
To determine the effects of the chemical inhibitors on the formation of 2-hydroxycarbamazepine, only the results obtained at a concentration of 300 μM carbamazepine were evaluated, due to the low apparent rate of carbamazepine 2-hydroxylation at the lower substrate concentration. Under these experimental conditions, the 2-hydroxylation of carbamazepine was extensively inhibited by 4-methylpyrazole (∼66%) and markedly inhibited by 7-EFC (∼47%) in human liver microsomes with either high or low CYP2B6 and CYP3A4/5 activity. Ketoconazole markedly inhibited 2-hydroxycarbamazepine formation (46%) in microsomes with high CYP2B6/3A activity but only moderately inhibited carbamazepine 2-hydroxylation (27%) in microsomes with low CYP2B6/3A activity. α-Naphthoflavone, an inhibitor of CYP1A2, had no apparent effect on carbamazepine 2-hydroxylation. The other chemicals examined caused little or no inhibition of carbamazepine conversion to 2-hydroxycarbamazepine in microsomes with high CYP2B6/3A activity but caused weak to moderate inhibition (20–33%) in microsomes with low CYP2B6/3A activity.
Carbamazepine 2- and 3-Hydroxylation by cDNA-Expressed Human P450 Enzymes.
An initial screen with microsomes containing 1 of 14 cDNA-expressed human P450 enzymes or a vector control was conducted at two carbamazepine concentrations (30 and 300 μM) to evaluate the ability of these enzymes to catalyze the 2- and 3-hydroxylation of carbamazepine. As demonstrated in Fig. 6, all of the recombinant P450 enzymes were capable of converting carbamazepine to 2-hydroxycarbamazepine and 3-hydroxycarbamazepine at the highest substrate concentration examined. However, at a substrate concentration of 30 μM, P450s CYP1B1, 2A6, 3A4, and 3A5 failed to produce detectable quantities of 2-hydroxycarbamazepine and CYP3A5 formed no detectable quantities of 3-hydroxycarbamazepine. The highest rates of carbamazepine 3-hydroxylation were catalyzed by CYP2B6, followed by CYP3A4, CYP1A1 and CYP1A2, whereas the highest rates of carbamazepine 2-hydroxylation were catalyzed by CYP2E1, followed by CYP2B6, CYP1A1, and CYP1A2. Based on the results obtained with the screen, kinetic studies were subsequently performed with those enzymes catalyzing the 2- or 3-hydroxylation of carbamazepine at rates greater than 2 or 20 pmol/nmol P450/min, respectively.
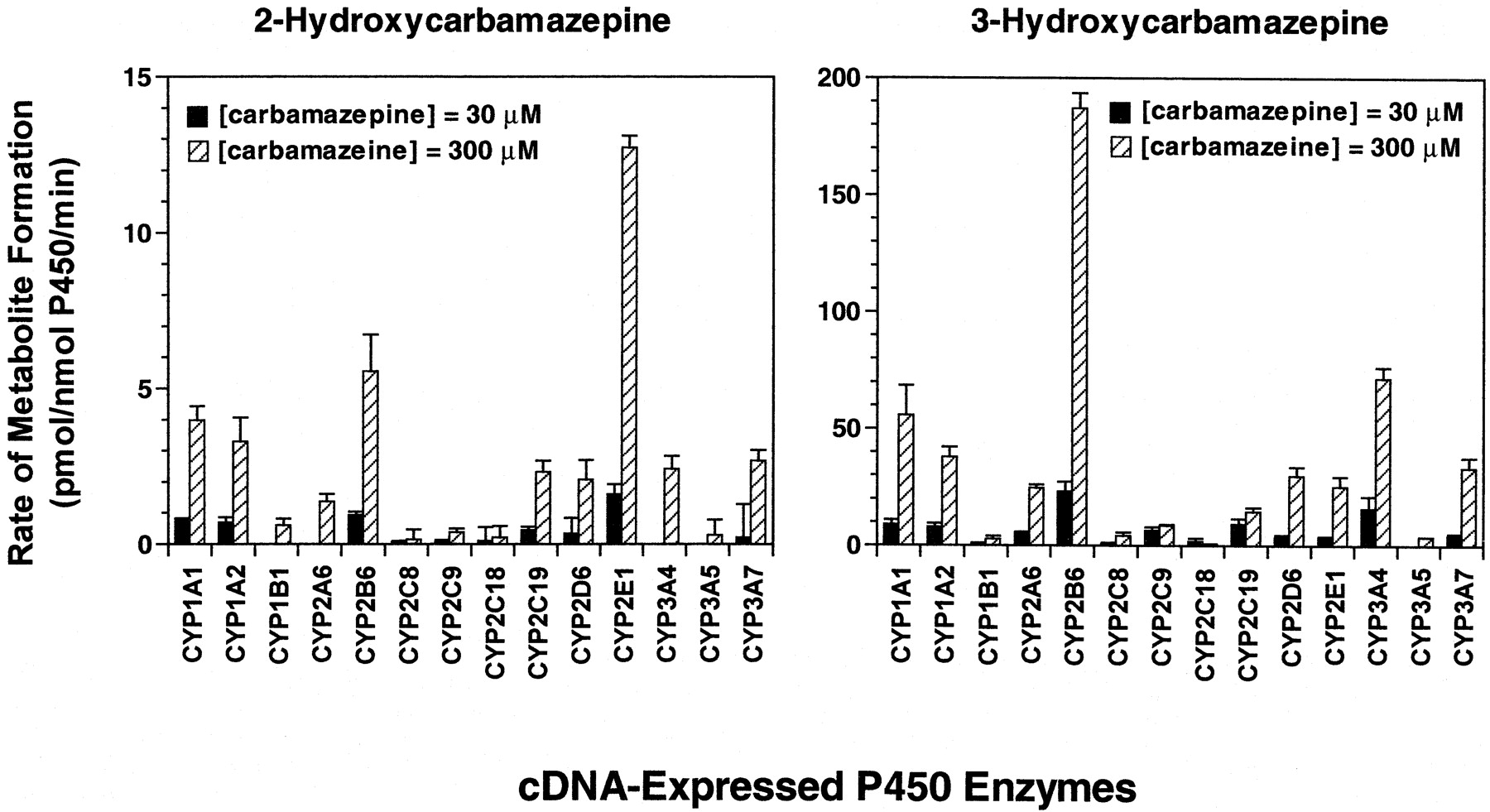
Carbamazepine 2- and 3-hydroxylation by human recombinant cytochrome P450 enzymes.
Carbamazepine (30 and 300 μM) was incubated with heterologously expressed human P450 enzymes as described under Materials and Methods. Each bar represents the mean ± S.D. of six determinations.
Formation of 2- and 3-hydroxycarbamazepine by the recombinant human P450 enzymes conformed to typical Michaelis-Menten kinetics, based on Lineweaver-Burk plots (plots not shown). Apparent kinetic parameters determined from the plots are listed in Table3. ApparentKm values for the formation of 3-hydroxycarbamazepine by recombinant CYP2C19 and CYP3A4 (∼239 and 283 μM, respectively) were similar to the mean apparentKm value obtained for human liver microsomes (Table 1). Three other P450 enzymes (CYP1A2, CYP2A6, and CYP2B6) had apparent Km values for the formation of 3-hydroxycarbamazepine that were within ∼2-fold those of CYP2C19 and CYP3A4. When calculated on the basis of picomoles of P450, the in vitro intrinsic clearance (Clint; defined asVmax/Km) of carbamazepine by 3-hydroxylation was dominated by CYP2B6 and CYP3A4. On normalization of the data with respect to the abundance of each enzyme present in human liver microsomes (Rodrigues, 1999), it was found that ∼47% and ∼29% of carbamazepine 3-hydroxylase activity was attributable to CYP3A4 and CYP2B6, respectively. CYP1A2, CYP2A6, and CYP2E1 were responsible for most of the remaining carbamazepine 3-hydroxylase activity.
Estimated kinetic parameters for the formation of 2-Hydroxycarbamazepine and 3- Hydroxycarbamazepine by cDNA-expressed human P450 enzymes
Among the cDNA-expressed isoforms examined, CYP2B6 showed the highest affinity (Km = ∼700 μM) for catalyzing the formation of 2-hydroxycarbamazepine (Table 3). The other recombinant enzymes examined formed 2-hydroxycarbamazepine with lower affinity (apparent Km range: 933-3160 μM) than CYP2B6. Overall, the apparentKm values for the formation of 2-hydroxycarbamazepine by the recombinant enzymes were similar to the mean apparent Km value (i.e., within 2-fold) obtained for human liver microsomes (Table 1). CYP2E1 had the highest Clint for carbamazepine by 2-hydroxylation but was closely followed by that of CYP2B6 (0.041 and 0.032 μl/nmol P450, respectively) when these values were calculated on the basis of picomoles of P450. When the data were normalized with respect to the abundance of each isoform, CYP2E1 still made the greatest contribution to the Clint for carbamazepine 2-hydroxylation (∼30%), but P450s CYP1A2, 2A6, 2B6, and 3A4 also made significant contributions to the intrinsic clearance of 2-hydroxycarbamazepine.
The extent to which any of the P450 enzymes catalyzing carbamazepine 2-hydroxylation contribute to the overall formation of this metabolite depends on the level of expression of the various isoforms in a given individual. When rates of 2-hydroxycarbamazepine formation were calculated for the recombinant enzymes (with the Michaelis-Menten equation using the derived apparent Kmand Vmax values) and subsequently adjusted for the content of individual P450 isoforms present in the human liver microsomes used in this study, it was apparent that the enzyme dominating the formation of 2-hydroxycarbamazepine was highly variable among individuals and dependent on the P450 expression profile of each individual. As demonstrated in Table4, CYP1A2 is the dominant enzyme catalyzing carbamazepine 2-hydroxylation in human liver microsomes from donor H030, whereas the principal catalysts of this reaction are CYP3A4 and CYP2B6 in sample H042 and CYP2E1 in sample H093. None of these isoforms, however, appears to contribute more than 50% to the total production of 2-hydroxycarbazepine.
Abundance normalized rates of carbamazepine (30 and 300 μM) conversion to 2-hydroxycarbamazepine by cDNA-expressed human P450 enzymes
Discussion
Hypersensitivity reactions associated with therapeutic drug use have been proposed to result from the oxidative metabolism of the drug to reactive, potentially cytotoxic intermediates capable of binding covalently with cellular proteins (Park et al., 1987). These drug-modified proteins (or haptens) generally are believed to be capable of functioning as antigens with the potential to initiate immune responses against drug-modified or native proteins. However, the processes by which drug-modified macromolecules are processed and presented to the immune system have not been identified to date.
Some important new clues may be derived from the results of studies investigating the consequences of P450 inactivation by mechanism-based enzyme inhibitors. For example, a series of studies have demonstrated that the nifedipine analog, 3,5-dicarbethoxy-2,6-dimethyl-4-ethyl-1,4-dihydropyridine, inactivates CYP3A1 through heme alkylation of the apoprotein. A major consequence of this process is structural damage that exposes targetable lysine residues for ubiquitin conjugation followed by rapid degradation mediated by hepatic ubiquitin—dependent proteasomal proteases (Correia et al., 1987, 1992a,b)—the hallmark of a major histocompatibility complex class I antigen processing and presentation pathway (Maffei et al., 1997). Additional studies have revealed that inactivation of CYP3A4 by cumene hydroperoxide is accompanied by heme fragmentation and modification of amino acid residues localized to the K-helix and the proximal l-helix/conserved cysteine domain (He et al., 1998). Interestingly, we have reported that antibodies in the sera of patients experiencing a hypersensitivity reaction to phenytoin or carbamazepine recognize a linear amino acid sequence localized to the CYP3A K-helix (Leeder et al., 1996). The central nature of the K-helix has been confirmed by Beaune's group in Paris who have mapped conformational epitopes on CYP2C9 (tienilic acid hepatitis) and CYP1A2 (dihydralazine hepatitis) to regions containing the K-helix as well as parts of the J- and l-helices (Bourdi et al., 1995; Lecoeur et al., 1996). Thus, the key to linking drug bioactivation and the subsequent immune response requires identification of the cellular targets of reactive drug metabolites and a better understanding of the cellular fate of proteins damaged in this manner.
Although the antigen to which the immune response is directed has not been identified, bioactivation of both phenytoin and carbamazepine has been demonstrated, either by the generation of cytotoxic metabolites (Riley et al., 1988; Pirmohamed et al., 1992b) or the formation of covalent adducts with microsomal proteins (Pirmohamed et al., 1992a;Lillibridge et al., 1996; Munns et al., 1997; Cuttle et al., 2000). Munns et al. (1997) demonstrated that phenytoin can be converted to a protein-reactive species via a two-step process. In the first step, phenytoin is hydroxylated by P450 enzymes to its primary, phenol metabolite, 5-(p-hydroxyphenyl-),5-phenylhydantoin. 5-(p-hydroxyphenyl-),5-phenylhydantoin can in turn be converted to a catechol product, 3′,4′-dihydroxyphenytoin, that spontaneously oxidizes to semiquinone and quinone species capable of forming covalent adducts with human liver microsomal proteins. Furthermore, the P450 enzymes responsible for catalyzing the formation of the catechol metabolite, namely CYP2C19, CYP2C9, CYP3A4, CYP3A5, and CYP3A7, have also been shown to be the major targets of covalent adduct formation (Cuttle et al., 2000).
The carbamazepine metabolite corresponding to 3′,4′-dihydroxyphenytoin is 2,3-dihydroxycarbamazepine, which is present in human urine and can be derived from either 2-hydroxycarbamazepine or 3-hydroxycarbamazepine (Lertratanangkoon and Horning, 1982). It is possible that 2,3-dihydroxycarbamazepine may be oxidized further to ano-quinone metabolite, just as the catechol product formed from phenytoin is oxidized further to semiquinone and quinone species. It is important to note that previous in vitro studies (Pirmohamed et al., 1992b; Lillibridge et al., 1996) have implicated a quinone metabolite as a major reactive metabolite of carbamazepine. Protein reactive quinone species may also be generated via a pathway involving conversion of 2-hydroxycarbamazepine to 2-hydroxyiminostilbene (Lertratanangkoon and Horning, 1982), which in turn can be readily oxidized to a reactive iminoquinone species (Ju and Uetrecht, 1999). Ju and Uetrecht further demonstrated that this iminoquinone species was capable of reacting with sulfhydryl-containing nucleophiles, such as glutathione and N-acetyl cysteine, and that a metabolite with a mass and fragmentation pattern consistent with that of the glucuronidated conjugate of 4-methylthio-2-hydroxyiminostilbene (which the authors propose is a further metabolite of an iminoquinone-glutathione conjugate) was present in human urine, thus providing additional support for the relevance of this pathway in the formation of reactive intermediates in vivo.
The production of protein-reactive metabolites by human liver microsomes appears to be quite low when carbamazepine is the substrate (Pirmohamed et al., 1992b) suggesting that studies conducted with a more proximal substrate may facilitate identification of the relevant protein targets of carbamazepine-derived reactive metabolites. Results of the studies conducted with phenytoin metabolites discussed earlier provide a compelling argument to pursue similar studies with carbamazepine. Therefore, the present study was undertaken to identify the P450 enzymes involved in the formation of 2- and 3-hydroxycarbamazepine, potentially the first step in the formation of protein-reactive carbamazepine metabolites. Human liver microsomes catalyzed the formation of both 2- and 3-hydroxycarbamazepine, albeit at relatively slow rates of formation. 3-Hydroxycarbamazepine was formed at rates ∼25 times greater than those of 2-hydroxycarbamazepine. 2-Hydroxycabamazepine formation also was generally not detectable at low substrate concentrations [i.e., those bracketing the mean plasma Cmax (60 μM; Anttila et al., 1979)]. Results from correlation analyses, inhibition experiments, and studies with recombinant cDNA-expressed human P450 enzymes suggest that CYP2B6 plays a major role in the in vitro formation of 3-hydroxycarbamazepine. Although correlation experiments did not support a role for CYP3A4, recombinant CYP3A4 catalyzed carbamazepine 3-hydroxylation at rates second only to those of CYP2B6 (∼40%) and the CYP3A inhibitor, ketoconazole, inhibited 3-hydroxycarbamazepine formation up to ∼40% in human liver microsomes. These results suggest that CYP3A4 makes at least a minor contribution to the formation of 3-hydroxycarbamazepine in vitro. The cumulative results also suggest that CYP1A2 and CYP2A6 may also play a minor role in the formation of this metabolite. In contrast, several P450 enzymes (CYP1A2, CYP2A6, CYP2B6, CYP2E1, and CYP3A4) appeared to contribute to the intrinsic clearance of carbamazepine via 2-hydroxycarbamazepine formation, the relative importance of each enzyme depending upon its level of expression in individual livers. Based on kinetic parameters derived from cDNA-expressed P450 enzymes, the contribution of any individual isoform does not appear to exceed 50% of the total formation of 2-hydroxycarbamazepine.
In general, the results of this study are consistent with those reported from previous in vitro studies (Kerr et al., 1994; Pelkonen et al., 2001). The major discrepancy between the present study and previous in vitro studies involves the role of CYP3A4 in carbamazepine 2- and 3-hydroxylation. In contrast to the results presented here, Kerr et al. (1994) found no evidence for CYP3A4-mediated 2- or 3-hydroxylation of carbamazepine in correlation experiments with a larger panel of human liver microsomes than that used in this study, in antibody and chemical inhibition experiments using liver microsomes with good CYP3A4/5 activity or in experiments with recombinant CYP3A4 (although it should be noted that this was an older, less active system lacking b5 supplementation). AlthoughPelkonen et al. (2001) did not find that CYP3A4 was a major catalyst of 3-hydroxycarbamazepine, they reported that CYP3A4 did play a minor role in the formation of this metabolite. It is possible that CYP3A4 activity was compromised in these experiments, however, by the concentration of DMSO included in the incubation mixtures (0.5 to 1.0%). Addition of 1% DMSO to incubations containing human liver microsomes has been shown to inhibit CYP3A4 activity up to 50% (Easterbrook et al., 2000).
Clearly, additional studies will be required to determine whether protein-reactive metabolites may be derived from 2-hydroxycarbamazepine and/or 3-hydroxycarbamazepine. Future studies in this laboratory will examine the further metabolism of these compounds, in an attempt to determine the identity and route of formation of the protein-reactive metabolite(s).
Acknowledgments
The contributions made by Dr. Thomas C. Boge, formerly of the University of Missouri at Kansas City, in the synthesis of 2-hydroxycarbamazepine are gratefully acknowledged. Sincere thanks are also extended to Paul Brown of Quintiles for performing the mass spectrometric analysis of dibenzo[b,f]azepin-2-one and 2-hydroxycarbamazepine.
Footnotes
-
Supported in part by Grant R01GM58883–02 (J.S.L.), National Institute of General Medical Sciences.
- Abbreviations used are::
- P450
- cytochrome P450
- DMSO
- dimethyl sulfoxide
- HPLC
- high-performance liquid chromatography
- MS
- mass spectometry
- 1
- iminostilbene
- 2
- dibenzo[b,f]azepin-2-one
- 3
- 5H-dibenzo[b,f]azepin-2-ol
- 4
- 2-(tert-butyl-dimethyl-silanyloxy)-5H-dibenzo[b,f]azepin-2-one
- 5
- 2-(tert-butyl-dimethyl-silanyloxy)-5H-dibenzo[b,f]azepine-5-carboxylic acid 2,4-dimethoxy-benzylamide
- 6
- 2-hydroxycarbamazepine
- 7-EFC
- 7-ethoxy-4-trifluorocoumarin
- Received April 29, 2002.
- Accepted July 21, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}