


Abstract
The ratio of drug levels in cerebrospinal fluid (CSF) to plasma (CSF/plasma) at equilibrium has been viewed as in vivo free fraction (fp) in plasma [CSF/plasma = fp], if no active transport is involved in brain penetration. We determined the CSF/plasma level following oral administration in rats and in vitro rat plasma protein binding for 20 compounds that were synthesized in our institute and have similar physicochemical properties. However, results indicated that the CSF/plasma was not only poorly correlated with fp but remarkably lower than fp in most of the compounds tested, suggesting that certain transporters such as P-glycoprotein (P-gp) located in blood-brain barrier (BBB) may decrease the unbound drug concentration in the brain. We evaluated P-gp-mediated transport activity of the 20 compounds with P-gp (mdr1a)-transfected LLC-PK1 cells and calculated P-gp efflux index (PEI), indicating the extent of P-gp-mediated transport. A plot of the CSF/plasma versus fp/PEI showed a strong correlation (r = 0.93), and the absolute values were almost identical [CSF/plasma = fp/PEI]. These results suggest that P-gp quantitatively shifts the equilibrium of unbound drugs across the BBB. Although we cannot rule out the possibility that endogenous transporters other than P-gp on BBB and/or blood-CSF barrier may affect CSF levels of compounds, the present study indicated that fp and PEI measurements may be useful in predicting in vivo CSF/plasma fractions for central nervous system-targeting drugs.
Since cerebrospinal fluid (CSF1) is a very low protein fluid, and drug in CSF is considered to be almost unbound, the ratio of drug concentration in CSF to plasma (CSF/plasma) in an equilibrium state has been viewed as in vivo free fraction in plasma (fp) (CSF/plasma = fp), if no active transport is involved in brain penetration (Lin and Lu, 1997).
Consistent with this notion, it has been reported that the in vitro free fraction of phenytoin in serum (0.155) is almost equal to the CSF/serum drug ratio (0.185) (Chou and Levy, 1981), and the in vitro fp of demethylchlorimipramine (0.035) is similar to the CSF/plasma ratio (0.026) (Bertilsson et al., 1979). Furthermore, equilibrium CSF/plasma ratios of eight benzodiazepines are known to be highly correlated with fp (r = 0.93, regression line slope = 0.98) (Arendt et al., 1983). These results suggest that in vitro fp may accurately reflect CSF/plasma. However, CSF/plasma can be lower than fp, if certain active transporters such as P-glycoprotein (P-gp) are involved in brain penetration.
P-gp is an ATP-dependent efflux pump that transports a variety of amphiphilic and hydrophobic drugs and plays a major role in multidrug resistance in cancer cells. P-gp is also expressed in normal tissues, including the apical membranes of intestinal and renal epithelia and the endothelial cells of the blood-brain barrier. The putative function of P-gp in normal tissues is to act as a functional barrier for endogenous and exogenous products that may be toxins or carcinogens. A more interesting function of this protein is the expression in the brain, especially on the endothelium of capillary blood vessels, which makes up so-called “blood-brain barrier” (BBB) that limits the entry of many compounds into the brain.
In this way, P-gp seems to be one of the most important transporters responsible for lower CSF/plasma ratio than expected from fp. In fact, the HIV-1 protease inhibitor, indinavir, which is a well known P-gp substrate (Kim et al., 1998), showed much lower CSF/plasma (Stahle et al., 1997; Letendre et al., 2000; Zhou et al., 2000) than in vitro fp (Vacca et al., 1994). In addition, CSF concentrations of quinidine, a typical P-gp substrate (Kusuhara et al., 1997), in human subjects were lower than unbound levels in serum and plasma (Ochs et al., 1980; Sindrup et al., 1996).
It is considered that there are many cases other than indinavir and quinidine where CSF/plasma is lower than fp, because quite a few drugs are known to be P-gp substrates. In the present study, we hypothesized that the lower CSF/plasma ratio can be quantitatively explained by P-gp-mediated transport. To prove the hypothesis, we chose 20 compounds that were synthesized in our institute and have similar physicochemical properties, and examined their P-gp-mediated transport with P-gp (mdr1a)-transfected LLC-PK1 cells as well as their CSF/plasma ratio and plasma protein binding in rats.
Materials and Methods
Chemicals. Vincristine was purchased from Sigma-Aldrich (St. Louis, MO). Compounds A to T were synthesized at Banyu Tsukuba Research Institute. (We cannot show the chemical names of compounds A to T in this article due to the restriction of the disclosure of their chemical structures.) All other reagents were of analytical grade.
Cell Lines and Cell Cultures. Human MDR1 transfectants L-MDR1, mouse mdr1a transfectants L-mdr1a, and their parental cell line LLC-PK1 porcine kidney epithelial cells were kindly provided by Dr. Alfred H. Schinkel (The Netherlands Cancer Institute, Amsterdam, The Netherlands) and used under license agreement. Cells were cultured in Medium 199 supplemented with 1 mM l-glutamine, penicillin (50 units/ml), streptomycin (50 μg/ml), and 10% (v/v) fetal calf serum. For L-MDR1 and L-mdr1a, cells were maintained in the continuous presence of vincristine (640 nM). Confluent monolayers were subcultured every 3 to 4 days by treatment with 0.25% trypsin/1 mM EDTA in Ca2+- and Mg2+-free Hanks' balanced salt solution. All cultures were incubated at 37°C in a humidified atmosphere of 5% CO2/95% air (Schinkel et al., 1995; Yamazaki et al., 2001).
Transport Studies. Transepithelial transport study was carried out as described previously (Kim et al., 1998; Yamazaki et al., 2001), with minor modifications. L-MDR1, L-mdr1a, and LLC-PK1 cells were plated at a density of 4 × 105 cells/12-mm well on porous (3.0-μm) polycarbonate membrane filters (Transwell; Costar, Cambridge, MA). Cells were supplemented with fresh media every 2 days and used in the transport studies on the fifth to sixth day after plating. Transepithelial resistance was measured in each well using a Millicell ohmmeter (model ERS; Millipore Corporation, Bedford, MA); wells registering a resistance of 300 Ω or greater were used in the transport experiments. About 1 to 2 h before the start of the transport experiments, the medium in each compartment was replaced with serum-free Hanks' balanced salt solution with 10 mM Hepes (pH 7.4). The transport experiment was then initiated (t = 0) by replacing the medium in each compartment with 700 μl of transport medium with and without each test compound (1 μM). After 3 h, 100-μl aliquots were taken from the opposite compartment into a 96-well plate. An equivalent volume of ethanol containing the internal standard was added to each well and mixed well. Samples were stored at -20°C until use.
Plasma Protein Binding. Binding of compounds to rat plasma was determined by an ultrafiltration method (n = 3). The compounds were added to plasma to yield a final concentration of 1 μM. After incubation of plasma samples for 15 min, 0.4 ml of plasma was immediately transferred to a Centri-free tube (Millipore Corporation). The tube was then centrifuged at 1,500g for 10 min. The unbound fraction of the compound was estimated directly from the ratio of drug concentration in the ultrafiltrate to the total drug concentration in the original plasma samples before centrifugation.
Animal Study. Each compound at 10 mg/kg was given by gavage as a suspension in 0.5% methylcellulose (5.0 ml/kg) to fed male Sprague-Dawley rats (n = 3). The rats were anesthetized by intraperitoneal injection of pentobarbital (50 mg/kg), and blood samples were obtained from the abdominal aorta at 2 h postdose. Plasma was prepared by immediate centrifugation at 2,000g for 15 min at 4°C. CSF was obtained by cisternal puncture of the atlanto-occipital membrane with a 30-gauge needle that had a 40-cm length of polyethylene tubing attached to a syringe. From 50 μl to 100 μl of CSF was then collected through the tubing into a micro test tube. Collection was terminated as soon as blood appeared in the tubing and the blood-tainted portion of CSF was prevented from entering the collection. Immediately after CSF collection, the brain was removed. Plasma, brain, and CSF were stored at -80°C until analysis. All animal care and treatment procedures were approved by the Banyu Tsukuba Research Institutional Animal Care and Use Committee prior to initiating these studies.
Sample Preparation. Plasma and CSF samples were extracted with 3 volumes of ethanol containing an internal standard and then centrifuged at 10,000g for 10 min to obtain supernatant. Brain samples were homogenized with equivalent or 4 volumes of water. An aliquot of homogenates was deproteinized with 3 volumes of ethanol and then centrifuged at 10,000g for 10 min to obtain supernatant.
Quantification. Quantification was achieved by liquid chromatography/tandem mass spectrometry with a PerkinElmerSciex (Boston, MA) API 300/365/3000 triple quadrupole mass spectrometer and a Waters 2690/2790 HPLC (Waters, Milford, MA) equipped with an Inertsil ODS-3 column (2.1 × 150 mm, 5 μm; GL Sciences Inc., Tokyo, Japan). Isocratic elution of the mobile phase was carried out with 50 to 80% acetonitrile containing 10 mM ammonium acetate, at a flow rate of 0.2 ml/min. TurboIonSpray was used as an ion source. The turbo probe was set at 425°C, with the nebulizing gas pressure and turbo gas flow set at 50 p.s.i. and 7.0 l/min, respectively. Other mass spectrometric conditions such as orifice potential were optimized for each compound. Detection of all compounds was carried out by multiple reaction monitoring, whereby the first quadrupole (Q1) transmitted the [M + H]+ ions of the compound. After collision-induced dissociation of the [M + H]+ ion in the second quadrupole (Q2), an appropriate product ion was selected in the third quadrupole (Q3).
Results and Discussion
We chose 20 compounds for this study. The calculated properties of the compounds are shown in Table 1. Molecular weight ranges from 350 to 450 and calculated logP ranges from 0.9 to 3.9, which indicates that the compounds tested are relatively lipophilic and have suitable molecular weight for good membrane permeability. We determined CSF and plasma levels at 2 h after oral administration in rats and calculated CSF/plasma concentration ratio for the 20 compounds (Table 2). Rat plasma protein binding was also evaluated with an ultrafiltration method to obtain in vitro fp (Table 2).
Calculated properties of compounds tested in the present study
In vivo and in vitro results for compounds tested in the present study
Results indicated that the CSF/plasma was not only poorly correlated with fp but remarkably lower than fp in most of the compounds tested (Fig. 1), suggesting that certain transporters such as P-gp located in BBB may decrease the unbound drug concentration in the brain extracellular cerebral fluid, which is in equilibrium with CSF.
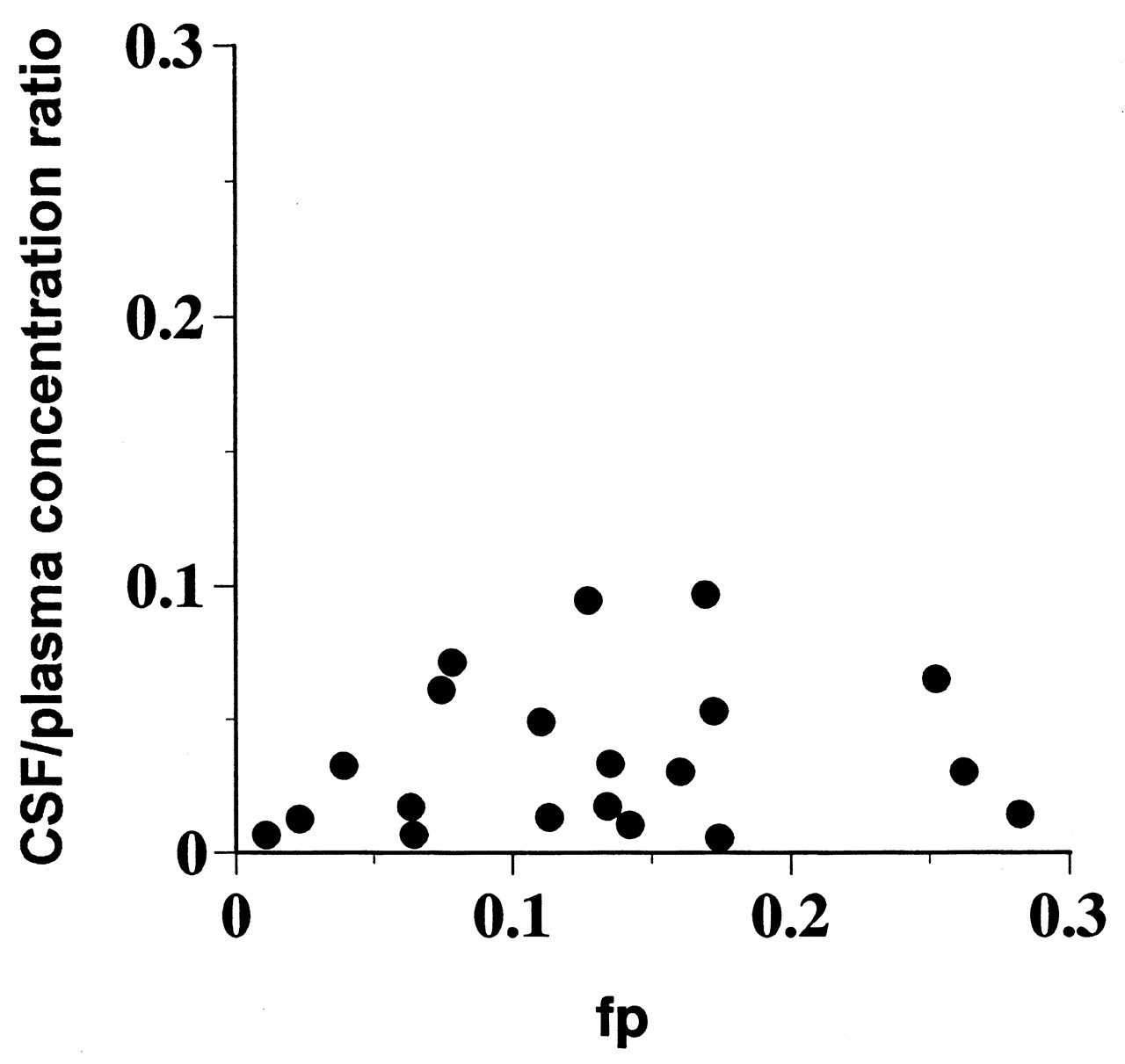
A plot of in vivo CSF/plasma concentration ratio versus in vitro fp.
We examined the extent of P-gp-mediated transport by using the polarized pig kidney epithelial cell line LLC-PK1 and L-mdr1a, a subclone stably transfected with mouse P-gp (mdr1a) cDNA (Table 2). For most of the compounds, the basolateral-to-apical (B-to-A) flux in L-mdr1a exceeded that in the opposite direction, whereas no B-to-A-directed transport was observed in the parental LLC-PK1 cells. From this experiment, P-gp efflux index (PEI) was calculated as B-to-A flux/A-to-B flux. PEI is the relative rate of polarized transport of each compound; therefore, the value might represent an equilibrium constant across P-gp-expressing cell monolayers at equilibrium.
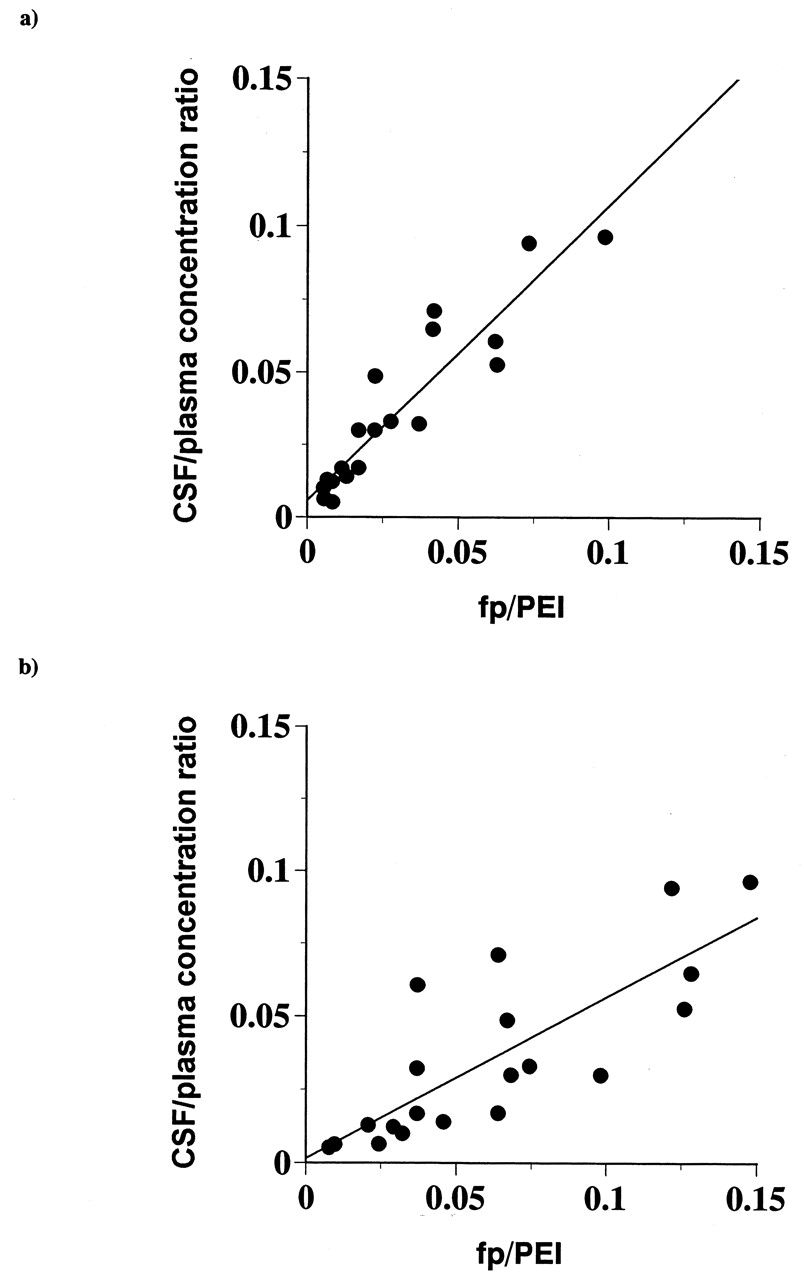
A plot of the CSF/plasma versus fp divided by PEI in L-mdr1a showed an excellent correlation (r = 0.93), and the absolute values were almost identical (regression line slope = 1.0) (Fig. 2a). These results demonstrate that P-gp quantitatively shifts the equilibrium of unbound drugs across the BBB. In contrast, the division by PEI in L-MDR1, a subclone stably transfected with human P-gp (MDR1) cDNA, did not lead to a good correlation (r = 0.80), and the slope was 0.55 (Fig. 2b), which implies that the species difference in P-gp susceptibility between humans and rats might be greater than that between mice and rats, at least in the compounds used in this study.

A plot of in vivo CSF/plasma concentration ratio versus in vitro fp/PEI.
PEI (P-gp efflux index) was calculated as the ratio of basolateral-to-apical permeability versus apical-to-basolateral permeability in the monolayer efflux assay. a, PEI in L-mdr1a was used; b, PEI in L-MDR1 was used.
Thus, the greater 1:1 correlation between CSF/plasma and fp/PEI clearly demonstrates that it is desirable to consider PEI as well as fp to more precisely predict CSF/plasma.
In conclusion, although we cannot rule out the possibility that endogenous transporters other than P-gp on BBB and/or blood-CSF barrier may affect CSF levels of compounds, the present study demonstrated that in vitro measurements of both fp and PEI may be necessary for predicting in vivo CSF/plasma fractions in an equilibrium state. As described above, the compounds used in this study have similar lipophilicity and molecular weight. In fact, most of the compounds originate from a lead compound. Therefore, it cannot be denied that our finding may be true only in a special case. However, in some cases, our theory should be useful for explaining CSF levels of various drugs which are known to be related to their pharmacological (side) effects on the central nervous system. Therefore, the findings in the present study are of great importance for the discovery/development of central nervous system-targeting drugs.
Acknowledgments
We gratefully thank Dr. Alfred H. Schinkel from The Netherlands Cancer Institute for permission to use MDR1/mdr1a-transfected LLC-PK1 cell lines. We are grateful to Tomoko Iguchi, Kazuo Marutsuka, and Yoshio Sawazaki in our institute for their technical support.
Footnotes
-
↵1 Abbreviations used are: CSF, cerebrospinal fluid; fp, free fraction in plasma; P-gp, P-glycoprotein; BBB, blood-brain barrier; mdr1a, multidrug resistance 1a P-gp; B, basolateral; A, apical; PEI, P-gp efflux index.
- Received March 18, 2003.
- Accepted July 7, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}