


Abstract
The pharmacokinetics of a 2′-O-(2-methoxyethyl)-ribose modified phosphorothioate oligonucleotide, ISIS 104838 (human tumor necrosis factor-α antisense), have been characterized in mouse, rat, dog, monkey, and human. Plasma pharmacokinetics after i.v. administration exhibited relatively rapid distribution from plasma to tissues with a distribution half-life estimated from approximately 15 to 45 min in all species. Absorption after s.c. injection was high (80-100%), and absorption after intrajejunal administration in proprietary formulations was as high as 10% bioavailability compared with i.v. administration. Urinary excretion of the parent drug was low, with less than 1% of the administered dose excreted in urine after i.v. infusion in monkeys at clinically relevant doses (≤5 mg/kg). ISIS 104838 is highly bound to plasma proteins, likely preventing renal filtration. However, shortened oligonucleotide metabolites of ISIS 104838 lose their affinity to bind plasma proteins. Thus, excretion of radiolabel (mostly as metabolites) in urine (75%) and feces (5-10%) was nearly complete by 90 days. Elimination of ISIS 104838 from tissue was slow (multiple days) for all species, depending on the tissue or organ. The highest concentrations of ISIS 104838 in tissues were seen in kidney, liver, lymph nodes, bone marrow, and spleen. In general, concentrations of ISIS 104838 were higher in monkey tissues than in rodents at body weight-equivalent doses. Plasma pharmacokinetics scale well across species as a function of body weight alone. This favorable pharmacokinetic profile for ISIS 104838 provides guidance for clinical development and appears to support infrequent and convenient dose administration.
The antisense oligonucleotide chemistry that is most advanced in development today for medical treatment is that of the phosphorothioate oligodeoxynucleotides (PS-ODNs1). PS-ODNs differ from natural DNA in that one of the nonbridging oxygen atoms in the phosphodiester linkage is substituted with sulfur. PS-ODNs are commercially available and easily synthesized, support RNase H activity, exhibit acceptable pharmacokinetics for systemic and local delivery, and have not exhibited major toxicities with use in humans (Glover et al., 1997; Crooke, 1998; Nemunaitis et al., 1999; Stevenson et al., 1999; Cunningham et al., 2000; Yuen et al., 2000).
Significant resources have been applied to identify chemical modifications to further improve upon the properties of PS-ODNs. The primary objectives of this medicinal chemistry effort are to increase potency, decrease toxicity, enhance pharmacokinetics, and decrease cost of treatment.
Modifications in the ribofuranosyl moiety backbone of the oligonucleotide have provided substantial improvements in oligonucleotide drug properties. In particular, 2′-O-(2-methoxyethyl) (2′-MOE) modifications have greatly increased binding affinity to the target mRNA, improved nuclease resistance (Cook, 1998), altered pharmacokinetics (Bennett et al., 2000; Geary et al., 2001a), and shown potentially improved safety profiles (Henry et al., 2001).
It is well known that oligonucleotides that are uniformly modified with 2′-O-modifications do not support an RNase H mechanism of target RNA hydrolysis (Inoue et al., 1987). Since RNase H is an important terminating mechanism for antisense compounds, this has led to the development of a chimeric oligonucleotide strategy. One such chimeric strategy uses 2′-O-(2-methoxyethyl) modifications on the 3′- and 5′-ends, and a “gapped” region remains in the center with a contiguous sequence of 2′-deoxy phosphorothioate nucleotides (Inoue et al., 1987; Lamond and Sproat, 1993; Monia et al., 1993; Yu et al., 1996; Dean and Griffey, 1997). This approach has led to the development of potent, pharmacologically active, specific antisense oligonucleotides, one of which is ISIS 104838.
ISIS 104838 is a 20-base hybrid 2′-O-(2-methoxyethyl)/2′-deoxy phosphorothioate antisense oligonucleotide that represents the first compound of this chemical class to be dosed in humans (Sewell et al., 2002). ISIS 104838 targets the human TNF-α mRNA. Murine-specific homologs of ISIS 104838, targeting murine TNF-α mRNA, have exhibited potent pharmacology in preclinical animal models for inflammatory disease including collagen-induced inflammatory arthritis (internal data, unpublished). The pharmacokinetics of ISIS 104838 have been characterized in mice, rats, dogs, and monkeys during preclinical development. A compilation of the results of these important studies is described in this report, representing the first complete preclinical pharmacokinetic profile for a 2′-O-(2-methoxyethyl) partially modified antisense phosphorothioate oligonucleotide. Furthermore, the ability to predict pharmacokinetics in humans utilizing these data provides a map for the development of future oligonucleotides in this chemical class.
Materials and Methods
Test Compound. ISIS 104838 is a 20-base phosphorothioate oligonucleotide with a total of ten 2′-O-methoxyethyl-modified ribofuranosyl nucleotides, five on each end of the oligonucleotide (Fig. 1). ISIS 104838 targets exon 2 of human mRNA for TNF-α. Full-length purity of the test compound (liquid chromatography/mass spectrometry) was 93.2% with 1.8% of the impurities associated with N-1 (deletion sequence). Dosing was performed based on the quantity of full-length 20-mer oligonucleotide.

Sequence of ISIS 104838 fully thioated 20-mer.
Bold and underlined nucleotides are 2′-O-(2-methoxyethyl)-modified. Center gap, deoxynucleotide (DNA). Stable tritium (3H) label incorporated on 5′ carbon of a 2′-O-(2-methoxyethyl)-modified thymidine residue in ISIS 104838 oligonucleotide.
Radiolabeled ISIS 104838 was mixed together with “cold” ISIS 104838 for radiolabel disposition and mass balance studies in rat. The radiolabel was tritium incorporated in the nonexchangeable 5-carbon position of the ribose sugar of one of the 2′-O-methoxy-modified thymidine nucleotides in the 5′ portion of the oligonucleotide (Fig. 1). The radiolabel purity of [3H]ISIS 104838 was >99% by HPLC. The specific activity of [3H]ISIS 104838 ranged from 2 to 7 μCi/mg.
Animals. Male and female Hsd ICR(CD-1) mice (Harlan, Indianapolis, IN), male Sprague-Dawley rats [Crl:CD(SD)IGS BR; Charles River Laboratories, Inc., Wilmington, MA), male and female beagle dogs (Charles River Laboratories), male and female cynomolgus monkeys (Macaca fascicularis; Sierra Biomedical Animal Colony, Sparks, NV), and male rhesus monkeys (Macaca mulatta; Sierra Biomedical Animal Colony) received either intravenous infusion, subcutaneous injection, or intrajejunal administration of ISIS 104838 at doses ranging from 1 to 50 mg/kg. All studies were conducted utilizing protocols and methods approved by the Institutional Animal Care and Use Committee and carried out in accordance with the Guide for the Care and Use of Laboratory Animals adopted and promulgated by the U.S. National Institutes of Health.
Mouse. Male and female CD-1 mice were administered intravenous bolus doses of 0, 1, 5, 20, or 50 mg/kg every other day for four doses (loading regimen), followed by every fourth day dosing for the duration of a 13-week toxicology study. Drug concentrations were measured in plasma for animals receiving the 5 and 20 mg/kg doses. Blood samples were collected for ISIS 104838 quantitation in plasma by cardiac puncture at sacrifice in tubes containing EDTA at 2, 10, 30, and 60 min, and 2, 3, and 24 h after the first dose and last dose of the 13-week dosing period (three male mice per time point).
The half-life of ISIS 104838 in kidney, liver, and spleen tissues was calculated by measuring drug concentration in tissues collected at 24 h and 7, 14, 28, and 90 days after a single i.v. dose of 5 or 20 mg/kg. Concentration of drug was also measured in liver, kidney, spleen, lymph nodes, brain, heart, lung, intestine, skeletal muscle, skin, ovaries, testes, and thymus collected, at sacrifice approximately 24 h after the last dose (day 92) and after a 12-week recovery period for all dose groups (1-50 mg/kg). Urine collection over a 24-h period immediately after the first and last dose of administration was done at all dose levels.
Rat. Single-dose i.v. injections of 5 and 20 mg/kg [3H]ISIS 104838 were administered to male Sprague-Dawley rats. The dose volume was 1 ml/kg, and the total amount of radioactivity administered was 24 μCi/kg. Before dosing, each animal selected for blood collection had a jugular cannula implanted. After a single i.v. dose of 5 mg/kg, whole blood was collected in tubes containing EDTA at 2, 10, 30, and 60 min, and 2, 3, 4, 8, 12, 24, and 48 h.
Mass balance recovery of radiolabel in urine, feces, tissue, carcass, and cage wash was assessed at 1, 7, 28, 56, and 90 days after single-dose administration of 5 mg/kg. Half-lives of total radiolabel were calculated in multiple tissues at both doses of 5 mg/kg and 20 mg/kg over the 90 days following radiolabeled ISIS 104838 administration.
Excretion was assessed over collection periods of 0 to 6 h and 6 to 24 h for both dose groups. Additional urine and feces excretion data were compiled over 90 days following the single 5 mg/kg injection for mass balance assessment.
Dog. Male beagle dogs were administered bolus intravenous doses of ISIS 104838 at 0.5 and 2 mg/kg. Blood was collected for quantitation of ISIS 104838 in plasma by peripheral venipuncture into tubes containing EDTA at 2, 5, 10, 20, 30, 45, 60, and 90 min, and at 2, 2.5, 3, and 4 h after administration.
Monkey. Single- and multiple-dose pharmacokinetic studies were conducted in male and female cynomolgus monkeys. Dose levels for ISIS 104838 included 0.125, 0.5, 1, and 2 mg/kg administered as a bolus i.v. injection; 1, 3, and 10 mg/kg administered via 60-min i.v. infusion; and 20 mg/kg administered by s.c. injection. Doses were administered either as a single injection or every other day for four doses (1 week), followed by dosing once every fourth day for the remainder of a 13-week dosing period. On days 3 (second dose) and 91 (last dose), blood was collected for quantitation of ISIS 104838 in plasma by peripheral venipuncture into EDTA-containing Vacutainers just before dosing and at 60, 75, and 90 min, and 2, 3, 4, 6, and 24 h after i.v. infusion, or at 5, 20, 30, 40, 60, and 90 min and 2, 3, 4, 6, and 24 h after s.c. injection. Urine was collected for a 0- to 24-h period after treatment on days 1, 7, 35, and 87. After the final dose of ISIS 104838 in the 10 mg/kg dose group, additional plasma samples were taken 15, 20, 27, 48, and 91 days after the last dose to assess the terminal elimination half-life in plasma using a sensitive hybridization ELISA method.
ISIS 104838 concentrations were measured in tissues collected at sacrifice (liver, kidney cortex, kidney medulla, spleen, lymph nodes, bone marrow, colon, jejunum, heart, lung, ovaries, testes, pancreas, skeletal muscle, brain, and skin) approximately 24 h after the final dose at all doses. Tissues were also collected at several time points after a single dose and at the end of the loading regimen to allow an estimate of tissue elimination half-life and accumulation ratios in tissues. For this group, two monkeys per time point (one male and one female) were sacrificed 24 h after a single dose, and 24 h and 7, 14, 28, and 54 days after the fourth loading dose. Urinary excretion was monitored for 24 h after single and multiple dose administration (0- to 24-h interval).
In a separate study, male rhesus monkeys received both i.v. and intrajejunal (i.j.) doses of ISIS 104838 to explore the potential for oral absorption of ISIS 104838. Several proprietary formulations containing varying compositions of medium-chain fatty acids were introduced by bolus injection into the jejunum via a surgically implanted subcutaneous ported catheter. The i.j. dose was 10 mg/kg ISIS 104838. The control intravenous dose of ISIS 104838 was administered in sterile saline by bolus injection at a dose of 1 mg/kg.
Human. Phase I clinical studies have been conducted in normal male volunteers. The i.v. and s.c. studies have been published previously (Sewell et al., 2002). Selected pharmacokinetic data from the Phase I studies have been compared with preclinical data and findings summarized.
Analytical Methods.Capillary gel electrophoresis (CGE). Validated CGE methods were used to measure unlabeled drug concentrations in mouse, dog, and monkey plasma, as well as mouse and monkey urine and tissues. The methods for ISIS 104838 are modifications of previously published methods (Leeds et al., 1996; Geary et al., 1999). An internal standard (ISIS 13866, a 27-mer 2′-O-methoxyethyl-modified phosphorothioate oligonucleotide) was added prior to extraction.
Briefly, plasma and urine extraction involved a two-step solid phase extraction in series (strong anion exchange followed by reverse-phase C18) before electrokinetic injection. Tissues were minced, weighed, homogenized, and extracted using a phenol/chloroform liquid-liquid extraction method. This was followed by solid phase extraction of the supernatant on a phenyl-bonded column before CGE electrokinetic injection.
A Beckman P/ACE model 5010 or MDQ capillary electrophoresis instrument (Beckman Coulter, Fullerton, CA) was used for gel-filled capillary electrophoretic analysis. Oligonucleotide peaks were detected by UV absorbance at 260 nm. For plasma, urine, and tissues, the limits of quantitation (LOQs) were 0.154 μg/ml, 0.039 μg/ml, and 0.39 μg/g, respectively.
LC/radiometric and HPLC-ES/MS metabolite identification. Selected plasma, urine, and tissue samples from mouse, rat, and monkey were analyzed for metabolite identification purposes by HPLC-ES/MS. Separation was accomplished using a Hewlett-Packard 1100 HPLC system consisting of a quaternary pump, a variable-wavelength UV detector, a column oven, and an autosampler. Typically, 100 μl of each sample was injected directly on a Jupiter C18 column (250 mm × 2.0 mm; 5-μm particles; Phenomenex, Torrance, CA). The column was maintained at 50°C and flow rate on the column was 0.25 ml/min. The column was equilibrated with 10% acetonitrile in 10 mM tributylammonium acetate, pH 7.0. A gradient from 10 to 55% acetonitrile over 60 min was used to separately elute ISIS 104838 and shortened oligonucleotide metabolites. Absorbance was measured at 260 nm. Under these condition, oligonucleotides differing by a single nucleotide could be resolved. This resolution was resolved using synthetic standards.
Fractions were collected from these separations on HPLC for subsequent liquid scintillation counting. Identification of putative metabolites was confirmed using synthetic metabolite standards and HPLC-ES/MS identification. Mass measurements were made on-line using an LCQ mass spectrometer. During the first 35 min of each analysis, the mass spectrometer was set to single ion monitoring (629.9, the -1 charge state observed for a methoxyethyl T monomer and two noncovalent acetate groups). From 35 to 80 min, the mass spectrometer was set to scan a m/z window of 500 to 2000. Mass spectra were obtained using a spray voltage of 3.4 kV, a sheath gas flow of 100 (arbitrary units), an auxiliary gas flow rate of 60 (arbitrary units), a capillary temperature of 220°C, a capillary voltage of -25 V, and a tube lens offset of 40 V. Masses were identified using the Qual browser of Xcalibur software v. 1.0 SR1 (Thermoquest, San Jose, CA). Potential metabolite peaks were identified either from the total ion current trace or from the UV trace. Each peak was manually averaged for its m/z value, and the results were compared with a table containing the calculated m/z values of expected metabolites.
Radiometric analysis (LSC). Radioactivity in all rat samples was quantitated by liquid scintillation counting using a Beckman 6001C Counter (Beckman Coulter). Urine and plasma were combined with Ultima Gold LSC cocktail (PerkinElmer Life Sciences, Boston, MA). For solubilized tissue samples, Hionic Fluor LSC cocktail was used (PerkinElmer Life Sciences). Oxygen combustion of feces and bone was carried out using a biological oxidizer (PerkinElmer Life Sciences). Ultima Gold cocktail was used to trap the 3H2O generated from each sample.
The scintillation counter was operated in the background subtract mode and samples were counted for a minimum of 2 min. Quench curve correction was used to convert cpm to dpm. The calibration of the scintillation counter was verified daily.
Hybridization ELISA. Selected monkey and human plasma samples were also analyzed using a quantitative, sensitive hybridization ELISA method, which is a variation on the method reported by Yu et al. (2002). Briefly, the assay involves binding of the analyte antisense drug to the sense DNA probe and subsequent detection of the double-strand hybridized drug using an antibody that recognizes a 3′-linked digoxigenin. Selection to the double-strand hybrid is accomplished by elimination of single-strand (nonhybridized) probe using S1 nuclease digestion. The assay was validated for precision, accuracy, selectivity, sensitivity, and stability of ISIS 104838 quantitation before analysis of monkey samples. Assay studies conducted with synthesized putate shortened oligonucleotide metabolite standards showed no measurable cross-reactivity confirming the assay's specificity for the parent oligonucleotide. The LOQ was determined to be 0.78 ng/ml, approximately 2 orders of magnitude more sensitive than the CGE method.
Protein Binding Assay. An ultrafiltration method (T. Watanabe, R. Geary, and A. Levin, manuscript in preparation) was used to assess whole plasma and individual protein binding characteristics of ISIS 104838. Briefly, Millipore Corporation (Bedford, MA) Ultrafree-MC filters (mol. wt. cutoff of 30,000) were used. ISIS 104838 was labeled with 32P before addition of the oligonucleotide to plasma (Maniatis et al., 1982). Whole plasma binding of four species (mouse, rat, monkey, and human) was evaluated over the plasma concentration range of 0.7 to 770 μg/ml. The protein binding of isolated protein solutions of human serum albumin (39.6 g/l; mol. wt. 69,000 g/mol), α2-macroglobulin (1.64 g/l; mol. wt. 750,000 g/mol), and α1 acid glycoprotein (0.7 g/l; mol. wt. 72,000 g/mol) were evaluated at physiological concentrations. The protein binding effects of four ISIS 104838 metabolites (n-9 to n-12) were evaluated in human plasma. The base length study was conducted over the concentration range of 0.1 to 50 μM ISIS 104838 (equivalent to approximately 0.8 -390 μg/ml ISIS 104838) or its metabolites. Due to differences in the molecular weight of the ISIS 104838 metabolites, the plasma concentrations were prepared as μmol-Eq concentrations. Three to five replicates were used to calculate average radiolabel counts and standard deviations.
Pharmacokinetic Analysis. Both compartmental and noncompartmental analysis methods were used for pharmacokinetic characterization of the plasma concentration data (WinNonlin 3.1; Pharsight, Mountainview, CA). Areas under the plasma concentration curve (AUCs) were estimated using the linear trapezoidal rule extrapolated to infinity by adding Clast/β to the AUC0-tlast, where Clast is the plasma concentration at the last time point measured and β is the slope of the apparent terminal disposition phase. Plasma clearance (CL) was calculated by dividing the dose of ISIS 104838 by AUC. The area under the first moment curve (AUMC; integral of C · t versus t) was used to calculate the apparent volume of distribution as follows: Vss = CL · (AUMC/AUC).
First-order elimination rates for ISIS 104838 in tissue were calculated using noncompartmental nonlinear regression of the decay curves for the respective tissues. A minimum of four time points and a regression coefficient >0.75 were required to accept the estimate. Half-life was calculated by dividing 0.693 by the first-order elimination rate.
Plasma bioavailability (F) following subcutaneous and intrajejunal administration in monkey was calculated by the ratio of the dose-normalized plasma AUCs. There were no gender differences noted for pharmacokinetics of ISIS 104838 in monkeys using two-way analysis of variance (SAS Version 8.0; SAS Institute, Cary, NC) and thus, male and female data were pooled for all analyses.
Cross-species regression utilizing allometry was performed by linear regression of clearance (CL) and apparent volume of distribution (Vss) versus body weight (W). The equation used to relate the pharmacokinetic parameters (Y) to body weight (W) was as follows: Y = aWb.
Thus, a log-log regression of a pharmacokinetic parameter, Y (i.e., clearance or volume of distribution), versus body weight (W) will yield a y-intercept value of a and a slope of b. The sign and magnitude of the exponent (b) indicate how the physiological variable is changing as a function of body weight (W) (Boxenbaum, 1982). Clearance and volume of distribution estimates were adjusted by mean body weight for mouse and rat studies and by individual animal body weight for monkeys, dogs, and humans.
Finally, a liver tissue model was developed for monkey liver simulation of ISIS 104838 concentrations as a function of time and multiple dosing. The model was constructed using liver tissue half-life determined in monkey (13.3 days) following a week of load dosing (every other day dosing of 3 mg/kg for one week, total of 4 doses). The model that provided reasonable prediction of both loading and multiple dose accumulation (3 months of dosing) was a simple first order linear model with first order input from blood (WinNonlin 3.1, Pharsight Corporation, Mountainview, CA).
Results
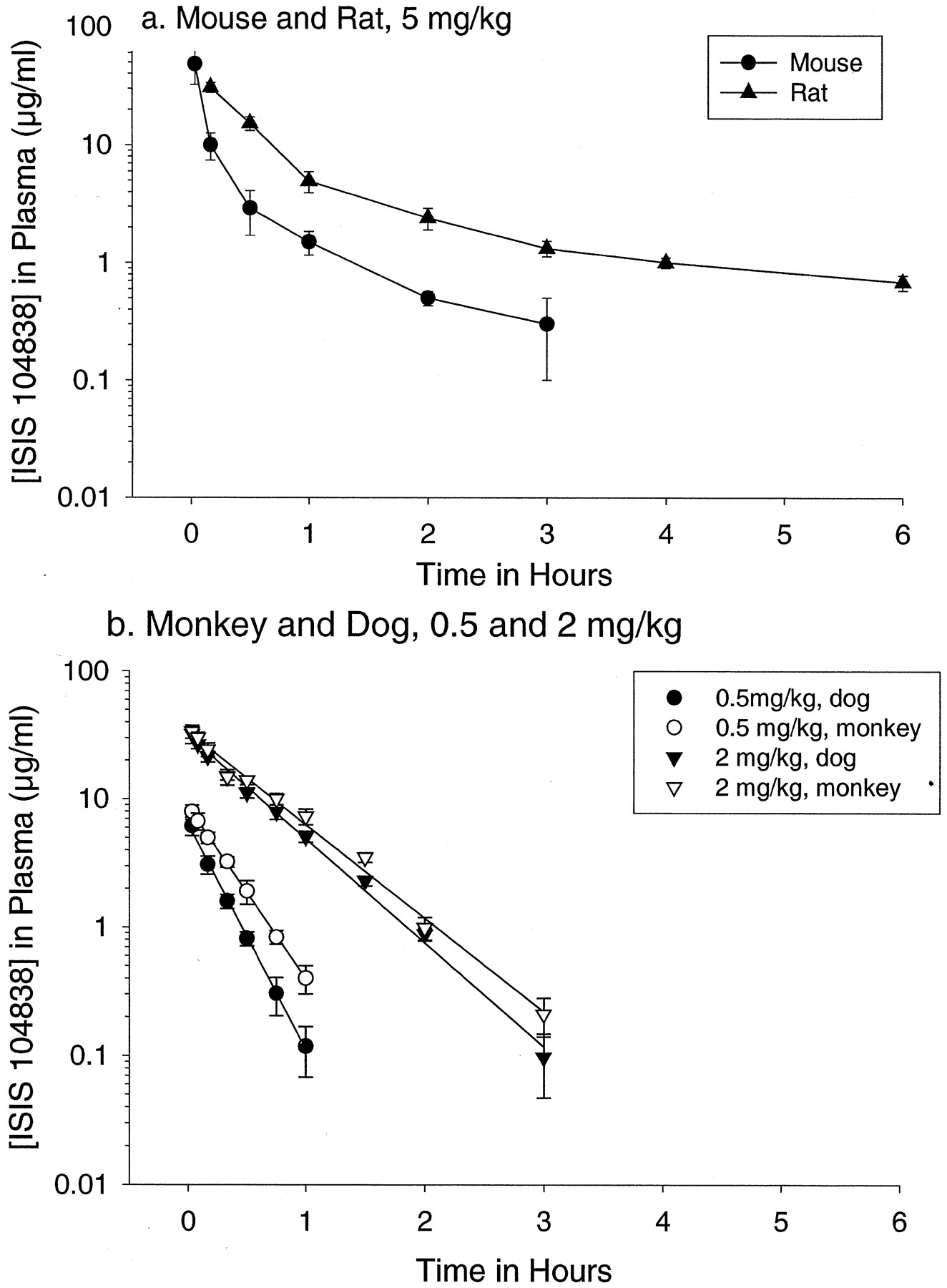
Plasma Pharmacokinetics. The plasma concentration-time distribution profile decreased rapidly in a polyphasic manner for mouse and rat after i.v. bolus administration, but concentrations of ISIS 104838 declined in a monoexponential manner in dog and monkey (Fig. 2; Table 1). The initial distribution half-life after i.v. bolus administration was relatively fast, generally less than 30 min. Distribution rates increased with increasing dose in dog and monkey, but not in mouse (not evaluated in rat). That is, higher doses resulted in somewhat slower distribution rates suggesting a saturable component to the distribution process in dog and monkey (Fig. 2b; Table 2). Interestingly, the apparent saturable component was observed at relatively low doses (≤2 mg/kg) in monkey and dog. At higher doses, ≥3 mg/kg in monkey (higher doses were not tested in dog), the distribution and clearance kinetics appeared to be similar (Table 2). Consistent with relatively rapid distribution from plasma, there was no evidence of measurable accumulation in plasma following repeat dose administration on an every-other-day or every fourth day schedule (data not shown).

Pharmacokinetic profiles of ISIS 104838 in plasma after i.v. bolus injection in mouse, rat, dog, and monkey.
Each symbol is the average of three to six animals. Error bars are standard deviation of the mean. ISIS 104838 concentrations were measured using capillary gel electrophoresis with UV detection at 260 nm (LLOQ = 120 ng/ml).
Plasma pharmacokinetic parameter estimates for ISIS 104838 after i.v. bolus administration and compared across species
Standard error of the estimate values are shown in parentheses.
Plasma pharmacokinetic parameter estimates for ISIS 104838 after i.v. administration in monkey and compared across dose
Standard deviation of individual estimates (n = 3-6) are represented in parentheses.
Use of an ultrasensitive hybridization ELISA provided characterization of a slow terminal elimination phase in monkey and human (Fig. 3). The ISIS 104838 concentration decline in plasma slowed dramatically after nearly complete distribution, reflected by a 3- to 4-log decline of ISIS 104838 concentration in plasma over a period of approximately 24 h. The apparent terminal elimination rate in monkey plasma, with a half-life of approximately 27 days, was consistent with the slow elimination of ISIS 104838 from monkey tissues and, thus, appears to reflect an equilibrium with oligonucleotide in tissue.

Polyphasic pharmacokinetic profiles of ISIS 104838 in plasma after 1-h i.v. infusion in monkey and human.
ISIS 104838 concentrations in plasma were measured using a hybridization ELISA method (LLOQ = 0.7 ng/ml).
Allometric comparison of clearance and volume of distribution estimated at doses ranging from 2 to 5 mg/kg across all species shows a linear (slope of 0.98-1.01) relationship based on body weight alone across species from rat to human (Fig. 4). Mouse appears to be the single “outlier,” with 5- to 10-fold more rapid weight-normalized plasma clearance.

Allometric relationship of plasma clearance (CL) and volume of distribution at steady state (Vss).
Each point represents the average of three to six measurements. Doses ranged from 2 to 5 mg/kg.
Excretion. The fraction of ISIS 104838 excreted intact was low in all species (Table 3). The highest rate of excretion occurred shortly after dosing (0-24 h), with the greatest percentage of dose excreted after bolus injection seen in rodents (as high as 17% of the administered 50 mg/kg dose in mouse). However, after 1-h infusion in monkeys, less ISIS 104838 and fewer related oligonucleotide metabolites were excreted in urine. In monkey, less than 5% of the administered dose was excreted in urine as the parent drug (20-mer oligonucleotide, ISIS 104838) and less than 10% of the total dose was recovered as parent and oligonucleotide metabolites (total oligonucleotide recovered in urine). The fraction of the dose excreted in monkey urine was somewhat lower after s.c. injection at 20 mg/kg when compared with i.v. infusion at 10 mg/kg (4.5% after s.c. versus 7.7% after i.v.). This phenomenon is likely due to lower plasma concentrations after s.c. injection and, subsequently, less free drug available for filtration.
Fraction of ISIS 104838 dose excreted in urine as ISIS 104838 (parent drug) and its metabolites over the period of 0 to 24 h after intravenous and subcutaneous injection compared across species
Whereas excretion of intact oligonucleotide was low, urinary excretion ultimately accounted for approximately 75% of the total radiolabel dose by 90 days after a single dose of 5 mg/kg ISIS 104838 in the rat. Excreted radiolabel was predominantly associated with shorter oligonucleotide metabolites, presumably a product of nuclease digestion in tissues. The remaining radiolabel remained associated with multiple tissues and organs. A small amount of radiolabel was also recovered in feces (<10% of administered dose over 90 days), bringing the total excretion to approximately 80 to 85% after a single dose (Fig. 5).

Mass balance excretion of radiolabel residue associated with [3H]ISIS 104838 over 90 days after a single 5 mg/kg i.v. bolus injection in rats.
Tissue Distribution and Pharmacokinetics. The highest concentrations of ISIS 104838 were measured in kidney, liver, lymph nodes, and spleen (Fig. 6), independent of the species studied. Very low levels were observed in skeletal muscle. Little to no evidence of ISIS 104838 was seen in brain. Consistent with more rapid clearance and larger apparent volume of distribution in mice, the ISIS 104838 concentrations in mouse tissues were generally much lower than those measured in other species. For example, in the monkey liver, concentration of ISIS 104838 was 352 ± 114 μg/g 24 h after 13 weeks of repeated 3 mg/kg dosing (every fourth day), whereas mouse liver concentrations were approximately 8-fold lower (45 ± 14 μg/g), 24 h after an identical dosing regimen of 5 mg/kg ISIS 104838.

Comparison of ISIS 104838 concentrations in tissues taken 24 h after the last dose administered over 3 months to CD-1 mice and cynomolgus monkeys.
Each bar represents the average of three to six measurements (CGE-UV). Error bars are standard deviation of the mean.
Compared with plasma disposition pharmacokinetics, the elimination from tissue was much slower (days versus minutes) in all species (Table 4). The most rapid clearance from tissue was observed in CD-1 mice (3- to 7-day half-lives). However, in rat, the radiolabel was cleared more slowly (18- to 30-day half-lives dependent on organ or tissue). Likewise, monkey exhibited very slow (5- to 29-day half-lives) tissue elimination of parent drug, ISIS 104838. The half-life observed in tissue appears to be reflected in the long plasma terminal half-life.
ISIS 104838 half-lives (in days) in tissues across species
Prolonged tissue residence resulted in accumulation of oligonucleotide in tissues upon repeated dose administration that was consistent with the elimination kinetics (Fig. 7a). Pharmacokinetic models developed for predicting the concentration of ISIS 104838 in the liver over the 13-week repeat dosing period provided reasonable prediction of ISIS 104838 concentrations compared with observed concentrations (Fig. 7b).

Pharmacokinetic simulation of ISIS 104838 concentrations in cynomolgus monkey liver during 1 week of i.v. dosing at 3 mg/kg administered every other day (a), and 3 months of i.v. dosing at 1 and 3 mg/kg with doses administered every other day for the first week and every fourth day thereafter (b).
Simulations are compared with measured concentrations measured at 24 h after first, fourth, and last dose at 3 mg/kg and 24 h after the last 1 mg/kg dose.
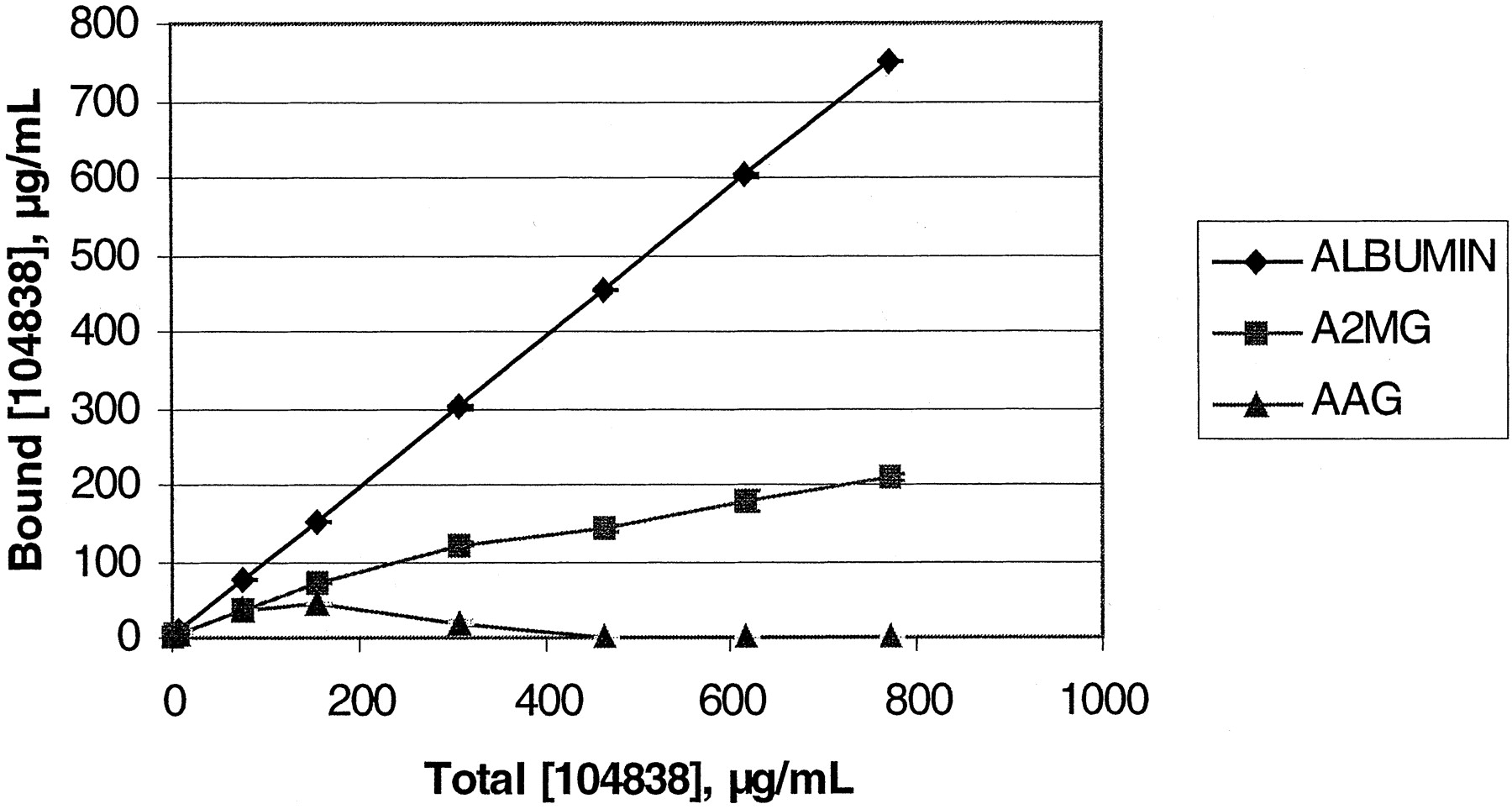
Protein Binding. The percentage of ISIS 104838 bound to plasma proteins was >97% across the concentration range tested (7-77 μg/ml) in all species except mouse (>90%). The highest capacity binder was albumin (Fig. 8). ISIS 104838 was also shown to bind to human α2-macroglobulin and human α1 acid glycoprotein, although to a lesser extent (lower capacity). Shorter synthesized oligonucleotide metabolite standards were less plasma protein bound in whole human plasma (Table 5).

Concentration of [32P]ISIS 104838 bound to albumin, α2-macroglobulin, and α1acid glycoprotein as a function of total concentration added (capacity of binding) to the individual protein solutions.
Each point represents the mean of three to five measurements.
Effect of length of oligonucleotide (ISIS 104838 and metabolites) on protein binding in whole human plasma
Metabolism. No measurable oligonucleotide metabolites were observed in plasma at any time point utilizing CGE methods. A more sensitive LC/radiometric method allowed for identification of shorter oligonucleotide metabolites observed at later time points in plasma, but in low abundance compared with parent ISIS 104838 (20-mer).
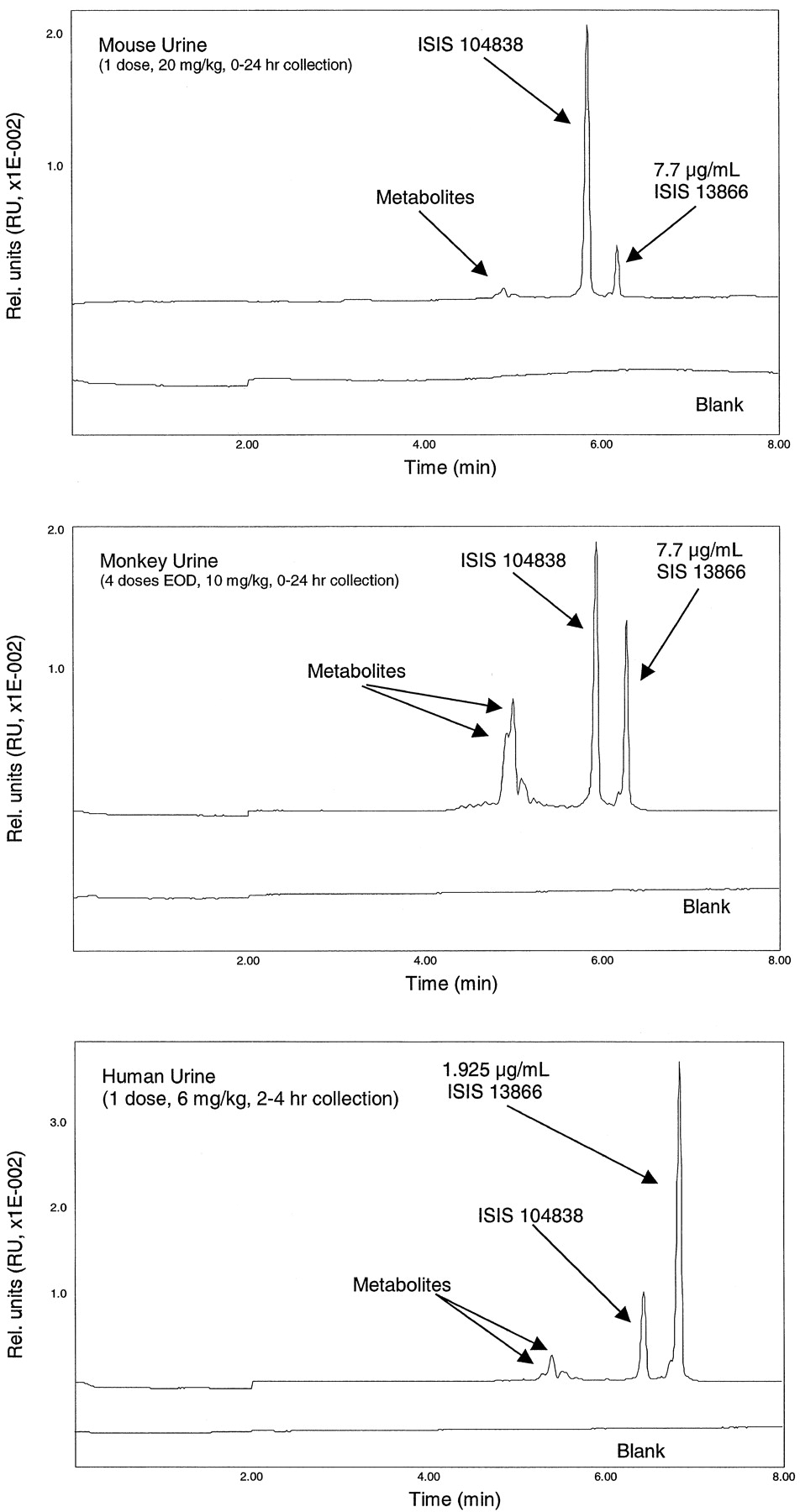
In contrast, ISIS 104838 metabolites were detected in urine by 6 h after injection. These metabolites coeluted on CGE with synthesized shortened oligonucleotides ranging from 8 to 12 nucleotides in length in all species studied (Fig. 9). In addition, the more sensitive HPLCES/MS analysis confirmed the identity of these shorter oligonucleotides in urine (Table 6). In tissues analyzed by both LC/radiometric methods and HPLC-ES/MS, the same metabolites were detected, suggesting that these metabolites are generated in tissues and subsequently excreted in urine.

Representative capillary gel electrophoresis of urine samples collected from mouse, monkey, and human following i.v. administration.
Metabolites coeluted with short oligonucleotides ranging from 8 to 12 nucleotides in length. ISIS 13866 is an internal standard 27-mer oligonucleotide.
Tabular summary of LC-ES/MS results for monkey urine (0-24 h after a single dose of 10 mg/kg)
Alternate Routes of Administration.Subcutaneous injection. A 20 mg/kg subcutaneous injection resulted in essentially complete (100%) bioavailability as measured by plasma and tissue concentrations of ISIS 104838 in monkeys (Table 7). After a s.c. injection of 20 mg/kg in monkeys, Cmax was approximately 2-fold lower than that seen after a 1-h infusion of 10 mg/kg. Tmax (the time Cmax was observed) was approximately 2 to 3 h. The apparent distribution half-life is substantially longer after s.c. injection when compared with i.v. infusion. Taken together, these data suggest complete, but relatively slow absorption from the subcutaneous injection site.
Subcutaneous administration of ISIS 104838 to monkeys resulted in essentially complete bioavailability measured by both single-dose plasma AUC and tissue concentrations of ISIS 104838 measured 24 h after the last dose administered over a 13-week period
Data are presented as mean ± standard deviation (n = 6).
Intrajejunal (i.j.) administration. Administration of proprietary formulations of ISIS 104838 containing medium-chain fatty acids into the jejunal section of the small intestine of rhesus monkeys has resulted in average absolute plasma bioavailability (compared with i.v. injection) of ISIS 104838 as high as 10 to 12% (Table 8). Absorption of ISIS 104838 after 10 mg/kg doses was rapid, with Cmax concentrations being in the range of 5 to 10 μg/ml. Tmax was achieved in 15 to 30 min, consistent with the transient and reversible enhancement associated with fatty acid solutions.
Systemic (plasma) bioavailability of ISIS 104838 (10 mg/kg) after intrajejunal administration to monkeys in medium-chain fatty acid aqueous solutions (50 mg/kg)
Data are presented as mean ± standard deviation (n = 6).
Discussion
Plasma pharmacokinetics of the partially modified ISIS 104838 oligonucleotide were similar to those observed for unmodified phosphorothioate oligodeoxynucleotides with the marked exception of very little evidence of measurable metabolites in plasma. Like unmodified predecessors, ISIS 104838 is rapidly distributed to tissues, and the clearance of the oligonucleotide from plasma is largely dominated by the rate of this distribution process (Geary et al., 1997a, 2001b; Phillips et al., 1997). In addition, the distribution to tissues appears to have a saturable component as previously reported for unmodified phosphorothioate oligodeoxynucleotides (Rifai et al., 1996; Phillips et al., 1997). Interestingly, the saturable distribution kinetics for ISIS 104838 were observed at the lower doses and appear to be quite linear at doses ≥2 to 3 mg/kg in animals and ≥0.5 mg/kg in humans (Sewell et al., 2002). Thus, the nonlinearity in distribution kinetics appears to be an effect of a relatively low-capacity distribution site/tissue which, once saturated, no longer impacts the overall distribution kinetics. The observed nonlinear component observed in distribution of oligonucleotide to tissue is similar in some regards to product inhibition nonlinear kinetics. However, the nonlinearity observed due to saturation of distribution differs from product inhibition nonlinearity in that the effect on kinetics continues to be manifested in the latter case, even at higher doses, whereas the saturable distribution case resumes linearity once the organ of distribution has reached its maximal saturation. Saturable uptake in organs of high affinity to the oligonucleotide results in the oligonucleotide being redistributed to other organs (Phillips et al., 1997) that are able to clear the drug from plasma. Alternatively, in the case of product inhibition, there are generally no alternative clearing mechanisms to compensate for the inhibition.
Allometric comparison across species for phosphorothioate oligodeoxynucleotides has been successful in accurately predicting human pharmacokinetics for first-generation compounds (Geary et al., 1997b, 2001b). These pharmacokinetic analyses for ISIS 104838 suggest that the same paradigm is true for the 2′-methoxyethyl-modified, second-generation oligonucleotides. Thus, plasma and, by inference, tissue pharmacokinetics measured in preclinical animal models can be scaled according to body weight to allow prediction of ISIS 104838 pharmacokinetics in humans.
Plasma distribution kinetics of ISIS 104838 are rapid and, as such, are unlikely to influence dosing frequency strategies. Rather, frequency of dosing should be established based on the slow elimination from tissue with the goal to minimize accumulation and to ensure continued exposure for efficacy. The ability to characterize a terminal plasma elimination phase in plasma using a sensitive hybridization/immunobinding assay has important implications for development of this compound in the clinic, where tissue sampling will likely be prohibitive. Nevertheless, care must be taken in the interpretation of the terminal elimination rate since it is, by definition, a composite of multiple tissues with the slowest eliminating tissue ultimately dominating the composite plasma elimination rate. The observed organ to organ differences in ISIS 104838 clearance, together with previously described differences in suborgan cellular elimination rates (Yu et al., 2001), indicates that it is at least possible that the elimination rate described by the terminal plasma phase will overestimate the half-life at the target tissue and/or cell. Ultimately, a combination of clinical testing of multiple dosing frequencies and careful pharmacokinetic analysis should be used to fully characterize the optimal dosing frequency required to ensure continued pharmacological activity at the target. Nevertheless, these data, taken together with recently published pharmacodynamic modeling for this chemical class of antisense oligonucleotide, suggest that infrequent dosing strategies will be successful in providing sustained target suppression (Yu et al., 2001).
Tissue distribution and tissue half-life data for ISIS 104838, taken together with the excretion and metabolism data seen for this oligonucleotide, suggest that 2′-MOE-modified oligonucleotides are slowly cleared from tissue predominantly by slow nuclease metabolism and efflux into plasma, followed by urinary excretion of the shortened metabolite products. Protein binding capacity in plasma is remarkably high for parent drug, similar to that observed for unmodified oligonucleotides (T. Watanabe, R. Geary, and A. Levin, manuscript in preparation). The reduction in protein binding observed for shorter oligonucleotide metabolites of ISIS 104838 suggests a mechanism for preferential excretion of the shorter metabolites compared with parent 20-mer. Thus, as the drug is metabolized in tissue, the shortened metabolic products bind with less avidity and are eliminated from tissue and, ultimately, the body by urinary excretion.
The metabolites observed in tissue and urine are evidence that the earliest cleavage occurs in the 10-base 2′-deoxy gap, effectively splitting the oligomer in half. These data implicate endonuclease digestion as the initial cleavage event. These data are consistent with the nuclease resistance of 2′-MOE-modified nucleotides placed in the 3′- and 5′-terminal portions of ISIS 104838 (Zhang et al., 1995). There was a notable absence of metabolites ranging from 16 to 19 nucleotides in length, suggesting little to no exonuclease products, which are often seen with unmodified phosphorothioate oligodeoxynucleotides (Temsamani et al., 1994; Gaus et al., 1997; Griffey et al., 1997).
Alternative routes of administration appear promising with nearly complete absorption of ISIS 104838 after s.c. injection in monkeys. Follow-up studies in humans confirm excellent bioavailability after s.c. injection of ISIS 104838 in normal volunteers (Sewell et al., 2002). Since absorption is relatively slow after s.c. injection, peak plasma concentrations are lower when compared with equivalent i.v. doses. Thus, s.c. injection may represent a safe alternative for administration of relatively high doses of ISIS 104838, minimizing the high plasma concentrations generated by both i.v. injection and infusion and, at the same time, reducing loss by excretion. The marked reduction in excretion is likely due to the overall lower ISIS 104838 plasma concentrations generated after s.c. injection, with a higher fraction bound to plasma proteins that may exclude ISIS 104838 from renal filtration.
Early work with 2′-methoxy-modified oligonucleotides suggested that second-generation oligonucleotides may have improved permeability across the gastrointestinal tract in rodent models (Agrawal et al., 1995). Although these early studies suffered from the lack of drug-specific assay methods and represented an overestimate of the oral bioavailability, the improved gastrointestinal stability of the 2′-MOE-modified oligonucleotides (Agrawal et al., 1995; Geary et al., 2001a) provides the foundation for the possibility for oral administration of ISIS 104838. Indeed, with the aid of appropriate oral formulations, the initial promising results achieved in monkeys further confirm the potential. Whereas 10% bioavailability represents a significant reduction in efficiency of delivery compared with s.c. injection, the convenience of the oral route may play a significant role in the development of ISIS 104838.
Thus, the pharmacokinetics of ISIS 104838 were characterized across species and have been shown to predict pharmacokinetics in humans. The remarkable similarity of the pharmacokinetics across species is attributed to common distribution pathways for clearance from plasma. Thus, taken together with the similarity in organ distribution across species, the pharmacokinetic profile in humans infers organ distribution similar to that in the preclinical animal models. Ultimately, ISIS 104838 is slowly cleared from multiple organs and tissues by endonucleolytic cleavage and excretion of shorter oligonucleotide metabolites in urine. Tissue residence times are long, with half-lives generally greater than 1 week. This pharmacokinetic profile supports relatively infrequent dosing regimens in the clinic and provides a basis for studying pharmacodynamic effects of antisense therapy in human disease. The promise of more convenient dosing routes such as s.c. injection and oral administration is exciting and warrants additional research.
Antisense oligonucleotides with the same configuration and chemistry as ISIS 104838, but different sequence, are expected to have similar pharmacokinetic properties. Given this expectation, it is not difficult to envision the use of modified 2′-MOE gap-mer oligonucleotides for a wide range of disease indications including more chronic disorders such as diabetes, hyperlipidemia, and cardiovascular indications.
Acknowledgments
We thank Drs. Lea Sewell, Andrew Dorr, and Stanley Crooke for scientific discussion and critical review of the manuscript. Finally, this article would not be possible without the administrative support provided by Paradorn Thiel, for which we are grateful.
Footnotes
-
↵1 Abbreviations used are: PS-ODN, phosphorothioate oligodeoxynucleotide; HPLC, high-performance liquid chromatography; ELISA, enzyme-linked immunosorbent assay; i.j., intrajejunal; CGE, capillary gel electrophoresis; LOQ, limit of quantitation; LLOQ, lower LOQ; ES/MS, electrospray ionization-mass spectrometry; LSC, liquid scintillation counting; AUC, area under the plasma concentration curve; CL, clearance; AUMC, area under the first moment curve; 2′-MOE, 2′-O-(2-methoxyethyl).
- Received May 12, 2003.
- Accepted August 19, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}