


Abstract
Conversion of the carbamazepine metabolite, 2-hydroxycarbamazepine, to the potentially reactive species, carbamazepine iminoquinone (CBZ-IQ), has been proposed as a possible bioactivation pathway in the pathogenesis of carbamazepine-induced hypersensitivity. Generation of CBZ-IQ has been proposed to proceed through the intermediate, 2-hydroxyiminostilbene (2-OHIS); however, data suggested that 2-hydroxycarbamazepine is oxidized by cytochromes P450 (P450s) directly to CBZ-IQ, followed by NADPH-mediated reduction to 2-OHIS. In vitro studies were conducted to identify the P450s responsible for converting 2-hydroxycarbamazepine to 2-OHIS and to determine functional consequences of this bioactivation pathway. Formation of 2-OHIS in human liver microsomes (HLMs) was consistent with monophasic, Michaelis-Menten kinetics. The sample-to-sample variation in the rate of 2-OHIS formation correlated significantly (r2 ≥ 0.706) with CYP3A4/5 and CYP2B6 activities in a panel of HLMs (n = 10). Studies with a panel of cDNA-expressed enzymes revealed that CYP3A4 preferentially catalyzed 2-OHIS formation; CYP3A4 formed 2-OHIS at a rate >10 times that of other enzymes capable of forming 2-OHIS (CYP1A1, CYP2C19, and CYP3A7). Inhibitors of CYP3A enzymes markedly impaired 2-OHIS formation in HLMs, whereas inhibitors of other P450s resulted in ≤20% inhibition. Although CYP3A4 was primarily responsible for converting 2-hydroxycarbamazepine to 2-OHIS, neither 2-hydroxycarbamazepine, 2-OHIS, nor CBZ-IQ caused time-dependent inactivation of CYP3A activity. No thiol adducts were formed directly from 2-hydroxycarbamazepine. However, glutathione- and N-acetylcysteine-conjugates were formed with 2-OHIS or CBZ-IQ as substrates. Thus, CYP3A4-dependent secondary oxidation of 2-hydroxycarbamazepine represents a potential carbamazepine bioactivation pathway leading to the formation of thiol-reactive metabolites, intermediates that may play a role in the etiology of idiosyncratic toxicity attributed to carbamazepine.
Carbamazepine (CBZ) and other aromatic anticonvulsants, such as phenytoin and phenobarbital, have been associated with a number of toxic effects, including a drug-induced hypersensitivity syndrome that manifests with a triad of fever, rash, and lymphadenopathy (Shear and Spielberg, 1988; Vittorio and Muglia, 1995). In a proportion of affected individuals, these symptoms may also be accompanied by eosinophilia, atypical lymphocytosis, or evidence of secondary organ involvement including hepatic, renal, and more severe cutaneous, hematological, and immunological disorders. Although anticonvulsant-induced hypersensitivity reactions are relatively rare events, occurring with an incidence of between 1:1000 and 1:10,000 exposures (Vittorio and Muglia, 1995), these idiosyncratic reactions can be potentially life-threatening.
The clinical manifestation of “anticonvulsant hypersensitivity syndrome” is consistent with an immune etiology; symptoms typically appear 2 to 8 weeks after the initiation of therapy, generally abate upon discontinuation of the offending compound(s), and reoccur much sooner following rechallenge than with the primary exposure (Shear and Spielberg, 1988). In addition, antibodies that recognize a linear amino acid sequence localized to the CYP3A K-helix have been observed in the sera of patients experiencing a hypersensitivity reaction to phenytoin or carbamazepine (Leeder et al., 1996). Unfortunately, the mechanism by which anticonvulsants induce hypersensitivity reactions has not been well characterized. Although one recent report has suggested that the immune response elicited by CBZ may (at least in part) be caused by CBZ directly (Naisbitt et al., 2003), biotransformation of aromatic anticonvulsants into chemically reactive metabolites is widely believed to be the critical step in the initiation of downstream pathogenic events such as formation of protein adducts and subsequent immune responses (Park et al., 1987).
Bioactivation of CBZ has been demonstrated by both the generation of cytotoxic metabolites (Riley et al., 1988; Pirmohamed et al., 1993) and the formation of covalent adducts with microsomal proteins (Pirmohamed et al., 1992; Lillibridge et al., 1996). In vitro studies using [14C]CBZ and cDNA-expressed human P450 enzymes have demonstrated that 65% and 31% of covalent adduct formation can be attributed to CYP3A4 and CYP1A2, respectively, after adjustment for relative hepatic abundance. Although the identity of the actual protein-reactive metabolite(s) remains unknown, the mass of peak radioactivity (∼50 kDa) is consistent with that of a P450 (Wolkenstein et al., 1998). Identification of the reactive metabolite(s) has been difficult because the rate at which human liver microsomes convert carbamazepine to metabolites capable of binding to proteins is quite low (Pirmohamed et al., 1993).

Proposed bioactivation scheme for the bioactivation pathway of carbamazepine to the reactive metabolite, CBZ-IQ. The original, two-step bioactivation described by Ju and Uetrecht (1999) is indicated by the solid arrows. An alternative pathway leading directly from 2-OHCBZ to CBZ-IQ is indicated by the open arrows.
Although an arene oxide was originally proposed as the reactive species, carbonium ion, o-quinone metabolites, and an iminoquinone metabolite derived from the CBZ metabolite, 2-hydroxyiminostilbene (2-OHIS), have also been proposed (Spielberg et al., 1981; Furst et al., 1995; Lillibridge et al., 1996; Ju and Uetrecht, 1999). The latter two species are particularly attractive candidates for the reactive metabolite, because quinone- and iminoquinone-type metabolites are considered to be the active intermediates in drug-induced hepatotoxicity caused by phenytoin, troglitazone, acetaminophen, and diclofenac (Dahlin et al., 1984; Shen et al., 1992; Munns et al., 1997; Yamazaki et al., 1999). Previous in vitro studies have also implicated quinone-type metabolites in the production of CBZ-derived protein adducts (Pirmohamed et al., 1992; Lillibridge et al., 1996). Furthermore, Ju and Uetrecht (1999) have identified a metabolite present in patient urine with a mass and fragmentation pattern consistent with that of the glucuronidated conjugate of 4-methylthio-2-hydroxyiminostilbene, which the authors propose is a further metabolite of glutathione-conjugated carbamazepine iminoquinone (CBZ-IQ) (Ju and Uetrecht, 1999). CBZ-IQ readily forms glutathione (GSH) and N-acetylcysteine (NAC) conjugates in vitro (Ju and Uetrecht, 1999) and is derived from 2-hydroxycarbamazepine (2-OHCBZ), a primary oxidative metabolite of CBZ whose formation is catalyzed by several P450 enzymes, including CYP1A2, 2A6, 2B6, 2E1, and 3A4 (Pearce et al., 2002). CBZ-IQ is readily interconverted with 2-OHIS via nonenzymatic oxidation/reduction (Fig. 1), and a two-step bioactivation pathway leading from 2-OHCBZ to 2-OHIS followed by oxidation to CBZ-IQ was originally proposed to explain CBZ-IQ formation (Ju and Uetrecht, 1999). The enzyme(s) responsible for converting 2-OHCBZ to 2-OHIS might be expected to be the most probable target(s) for covalent adduct formation, since further oxidation within the active site could generate the reactive iminoquinone, which could then subsequently bind to neighboring nucleophilic residues. Unfortunately, the identity of the enzyme(s) responsible for catalyzing 2-OHIS formation is unknown. Hence, in vitro studies were conducted to determine the role that human P450 enzymes play in the further metabolism of 2-OHCBZ to 2-OHIS and the ability of these compounds to inactivate the enzyme(s) catalyzing their formation.
Materials and Methods
Chemicals. CBZ, α-naphthoflavone, ketoconazole, omeprazole, quinidine, sulfafenazole, testosterone, 6β-hydroxytestosterone, troleandomycin, GSH, glucose 6-phospate, glucose-6-phosphate dehydrogenase, NAC, NADP, NADPH, EDTA, and sodium dithionite were purchased from Sigma-Aldrich (St. Louis, MO). The synthesis of 2-OHCBZ, 2-OHIS, and CBZ-IQ has been described elsewhere (Ju and Uetrecht, 1999; Pearce et al., 2002). All other reagents were of analytical grade.
Biological Reagents. Individual human liver microsomal preparations were obtained from BD Gentest (Woburn, MA; n = 10) and from XenoTech, LLC (Lenexa, KS; n = 24). Microsomes from baculovirus-infected insect cells (Supersomes) expressing human P450 enzymes (1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 3A4, 3A5, and 3A7) or control vector were purchased from BD Gentest). All recombinant enzymes were coexpressed with human NADPH-cytochrome P450 reductase; some enzymes (CYP2B6, CYP2C19, CYP2E1, CYP3A4, and CYP3A7) were also coexpressed with human cytochrome b5. The manufacturers provided protein concentrations, P450 contents, and P450 enzyme activities. Vials of microsomes were stored at –70°C until use. Microsomes were rapidly thawed in room temperature water and placed on ice prior to use. An inhibitory, polyclonal antibody against CYP3A4 and control IgG (both raised in rabbits) were purchased from XenoTech, LLC and used according to the manufacturer's recommendations.
In Vitro Incubation Conditions. In vitro enzyme assays were performed in 96-well microtiter plates. Standard incubation reactions (100 μl) contained human liver microsomes (50 μg of microsomal protein) or insect cell microsomes containing baculovirus-expressed cytochrome P450 enzymes (5 pmol) coexpressed with P450 reductase, potassium phosphate buffer (50 mM, pH 7.4), MgCl2 (3 mM), EDTA (1 mM), and 2-OHCBZ (5 to 500 μM) dissolved in methanol (≤1% v/v final concentration) at the final concentrations listed. After a 3-min preincubation, reactions were initiated by the addition of NADPH (1 mM) or an NADPH-generating system, consisting of NADP (1 mM), glucose-6-phosphate dehydrogenase (1 U/ml), and glucose 6-phosphate (5 mM), placed in a Thermo Electron Corporation (Waltham, MA) Benchtop Orbital Shaker incubator at 37 ± 0.1°C, and terminated after 0 to 60 min (typically 30 and 60 min for human liver microsomes and recombinant P450s, respectively) by the addition of 100 μl of ice-cold methanol containing the internal standard, CBZ (1 μM final concentration). Protein was precipitated by centrifugation at 10,000gmax for 10 min. An aliquot (25–75 μl) of the supernatant was analyzed by HPLC/MS via direct injection. Under these conditions, a preparation of human liver microsomes with high CYP3A activity (H042) catalyzed the conversion of 2-OHCBZ (60 μM) to 2-OHIS at rates that were proportional to incubation time (up to 60 min) and protein concentration (up to 1.0 mg/ml). With the exception of GSH- and NAC-trapping experiments, metabolism of 2-OHCBZ did not exceed 10%. Three replicate experiments were typically performed with two replicate samples per condition; exceptions to this generality are noted. Kinetic studies and correlation studies with human liver microsomes obtained from XenoTech, LLC were performed with duplicate and triplicate determinations, respectively.
Inhibition Experiments. Conversion of 2-OHCBZ (50 μM) to 2-OHIS by human liver microsomes was evaluated in the presence or absence (i.e., control) of known P450 isoform-selective inhibitors. The following chemical inhibitors were examined at the indicated concentrations: α-naphthoflavone (CYP1A2, 1 μM), sulfafenazole (CYP2C9, 10 μM), omeprazole (CYP2C19, 10 μM), quinidine (CYP2D6, 1 μM), ketoconazole (CYP3A4/5, 1 μM), and troleandomycin (CYP3A4/5, 100 μM). Inhibitors were dissolved in methanol and diluted in the incubation mixtures to a final solvent concentration of 1% (v/v). Control incubations contained an equal volume of methanol. Incubations containing the mechanism-based inhibitor troleandomycin were preincubated with human liver microsomes and an NADPH-generating system for 20 min before the reaction was started with substrate.
Immunoinhibition studies were conducted with a polyclonal, inhibitory antibody against CYP3A4. An aliquot (12.5 μl) of the antibody or control (rabbit IgG) was incubated with a panel of human liver microsomes (0.05 mg) at room temperature for 20 min before addition of MgCl2, EDTA, phosphate buffer, and 2-OHCBZ (20 and 100 μM). Under these conditions, the antibody against CYP3A4 should inhibit ≥80% of CYP3A4 enzyme activity, according to the manufacturer's product information. Each 100-μl incubation (single determination for each condition) was initiated with NADPH-generating system and terminated with an equal volume of methanol after 30 min.
Preincubation-Dependent Inhibition of CYP3A4/5. Human liver microsomes (1 mg protein/ml) were preincubated with CBZ, 2-OHIS, CBZ-IQ (100 μM), or 2-OHCBZ (10, 100, or 500 μM) at 37 ± 1°C for 0 to 30 min in the presence of an NADPH-generating system. Controls contained microsomes preincubated in the presence or absence of the aforementioned compounds but lacked the NADPH-generating system. Preincubation mixtures were then diluted 10-fold with potassium phosphate buffer (50 mM, pH 7.4), MgCl2 (3 mM), EDTA (1 mM), and testosterone (250 mM). Reactions were initiated by the addition of the NADPH-generating system. Reactions were terminated with ice-cold methanol after 10 min. Protein was sedimented by centrifugation, and an aliquot of the supernatant was analyzed by reverse phase HPLC via direct injection, as described by Usmani et al. (2003). CYP3A4/5 activity was measured as testosterone 6β-hydroxylase (T6βH) activity. Incubations were performed in duplicate.
Formation of GSH and NAC Conjugates. 2-OHCBZ (10, 30, and 100 μM), 2-OHIS, or CBZ-IQ (100 μM) was incubated with or without GSH (0.01–5 mM) or NAC (0.5 mM) in buffer containing potassium phosphate (50 mM, pH 7.4), MgCl2 (3 mM), and EDTA (1 mM) ± human liver microsomes (0.5 mg/ml), ± NADPH (1 mM), or an NADPH-generating system. In general, reactions were conducted at 37 ± 0.1°C and terminated after the desired time interval (0.5–3.5 h) by the addition of an equal volume of ice-cold methanol. Some reactions containing 2-OHIS or CBZ-IQ were performed at room temperature in the absence of human liver microsomes and NADPH. Samples were subjected to centrifugation (10,000gmax for 10 min), and an aliquot of the supernatant was analyzed by HPLC/MS via direct injection. Reactions were performed as single determinations; reaction conditions found to yield GSH or NAC conjugates were confirmed with a replicate incubation.
HPLC/MS Analysis. 2-OHCBZ and its metabolites were resolved on a reversed-phase Phenomenex (Torrance, CA) Luna C-8 (2) column (4.6 mm × 25 cm, 5-μm particle size) preceded by a Phenomenex C-8 guard column (4 mm × 3 mm i.d., 5-μm particle size) using a Hewlett Packard (Palo Alto, CA) HP1100 HPLC system equipped with an HP1100 degasser, binary pump, autosampler, column heater, diode array detector, and mass spectral detector. The mobile phase consisted of 0.1% aqueous acetic acid (solvent A) and methanol (solvent B) and was delivered at a constant flow of 0.6 ml/min. The solvent program was as follows: 0 to 5 min, 40% B; 5 to 15 min, a linear gradient from 40 to 50% B; 15 to 20 min, a linear gradient from 50 to 70% B; 20 to 40 min, 70% B; 40 to 41 min, a linear gradient from 70 to 40% B; 41 to 45 min, 40% B. The column temperature was maintained at 40°C. Under these conditions, 2-OHCBZ, 2-OHIS, and the internal standard (CBZ) eluted at ∼27.0, 29.5, and 30.5 min, respectively. The column effluent was monitored by UV detection (290 nm; for verification of metabolite identity) and by atmospheric pressure chemical ionization detection with a mass spectrometer operating in a selective positive ion-monitoring mode. Ion detection was optimized for 2-OHIS detection. The drying gas temperature and flow were maintained at 200°C and 5 l/min, respectively, and the nebulizer pressure was set at 20 psig. The vaporizer temperature was maintained at 400°C. The capillary voltage was set at 3 kV and the corona current was set at 4.5 μA. Under these conditions, 2-OHIS yielded [MH]+ ions at m/z 208 and 210; other ions of interest were monitored as [MH]+ ions. Data were collected and integrated with Hewlett Packard Chemstation V A.0401 software. 2-OHCBZ and 2-OHIS were quantified by comparison of their peak areas (determined by mass spectral analysis) with those of analytical standards. The lower limit of quantification for the assay was 1.2 pmol for 2-hydroxyiminostilbene. The analytical method was linear over a standard concentration range of 30 nM to 10 μM (r2 > 0.99).
Data Analysis. Coefficients of determination (r2) between the rates of 2-OHIS formation and the activities of cytochrome P450 enzymes were determined using least-squares regression analysis. Significance was determined by Pearson's regression analysis using a two-tailed Student's t test (SPSS 11.0; SPSS Inc., Chicago, IL). Kinetic data were analyzed by nonlinear regression without weighting (GraFit 5; Erithacus Software, Horley, Surrey, UK).

Representative HPLC/MS chromatograms obtained by atmospheric pressure chemical ionization at selected ion currents of [MH]+ ions of 2-OHCBZ and its metabolites formed by human liver microsomes. Total ion count (T.I.C.): 1, 2-OHCBZ (26.5 min); 2, 2-OHIS (29.5 min); 3, CBZ (internal standard, 30.5 min).
Results
Metabolism of 2-OHCBZ by Human Liver Microsomes. A representative chromatogram from an incubation of 2-OHCBZ with NADPH-fortified human liver microsomes is shown in Fig. 2. Human liver microsomes converted 2-OHCBZ to 2-OHIS and to at least two other unidentified metabolites. The latter two metabolites each had an apparent mass of 268 (MH+ ions at m/z 269), consistent with that of a di-hydroxycarbamazepine metabolite. No CBZ-IQ formation was observed under these incubation conditions. In the absence of NADPH or human liver microsomes, no 2-OHCBZ metabolites were detected.
The kinetics of 2-OHIS formation were investigated in a pooled sample of human liver microsomes and in microsomal preparations from three individuals (H042, H030, and H056). Visual inspection of Eadie-Hofstee plots (rate versus rate/[substrate]) of the data for each of the microsomal preparations suggested that 2-OHIS formation appeared to conform to monophasic, Michaelis-Menten kinetics. Unfortunately, saturating concentrations of substrate could not be achieved in these experiments (2-OHCBZ solubility was limited to ∼500 μM in incubation mixtures), precluding an accurate determination of apparent kinetic parameters. However, estimates of these parameters were generated by fitting kinetic data to a one-enzyme model using nonlinear regression software (GraFit 5). Estimated kinetic parameters are shown in Table 1, and a representative substrate-velocity plot and the corresponding Eadie-Hofstee plot obtained with pooled human liver microsomes are shown in Fig. 3.
Apparent kinetic parameters for the formation of 2-hydroxyiminostilbene in human liver microsomes and insect cell microsomes containing recombinant CYP3A4
Values are presented ± standard error.
Intersubject Variability and Correlation Experiments. Human liver microsomes prepared from 34 donors were examined for their ability to convert 2-OHCBZ to 2-OHIS at three substrate concentrations (20, 100, and 500 μM). The rate of 2-OHIS formation varied >45-fold among the microsomal samples [range (rates in pmol/mg protein/min ± S.D.), at respective substrate concentrations: 0.4 ± 0.9 to 22.8 ± 5.6, 20 μM; 3.0 ± 0.5 to 213 ± 43, 100 μM; and 10.9 ± 1.8 to 496 ± 38, 500 μM)].
Initially, correlation studies were conducted with a relatively small number of liver microsomal samples (n = 10; obtained from BD Gentest), whose P450 activities were characterized with one set of isoform-selective reactions. The sample-to-sample variation in the rates of 2-OHIS formation (at substrate concentrations of 20, 100, and 500 μM) correlated significantly with CYP3A4/5 activity (r2 ≥ 0.784, P < 0.05) and with CYP2B6 activity (r2 ≥ 0.706, P < 0.05), but not with any other cytochrome P450 activities in this set of samples (Table 2). Visual inspection of the plots of 2-OHIS formation against CYP3A4/5 or CYP2B6 activities suggested that the observed correlations might be unduly influenced by data points associated with two microsomal samples, each of which possessed high CYP2B6 and CYP3A4/5 activities (data not shown).
Regression analysis (r2) of the relationship between the rates of 2-hydroxycarbamazepine conversion to 2-hydroxyiminostilbene with the sample-to-sample variation in cytochrome P450 activity in human liver microsomes
Values listed are expressed as r2 (P value). Values in bold indicate significant coefficients of determination between P450 activity and rate of 2-hydroxyiminostilbene formation at P < 0.05. HLM Set 1 was obtained from BD Gentest, HLM Set 2 was obtained from XenoTech, LLC.

Effect of substrate concentration on the rate of 2-OHIS formation by pooled human liver microsomes and recombinant CYP3A4. Representative plots of the rates of 2-OHIS formation versus substrate concentration by pooled human liver microsomes (A) and recombinant CYP3A4 (B). Insets, Eadie-Hofstee plots. 2-OHCBZ (5–500 μM) was incubated with pooled human liver microsomes (0.5 mg of microsomal protein/ml) or insect cell microsomes containing baculovirus-expressed CYP3A4 (0.5 pmol/ml) coexpressed with P450 reductase in 100-μl reaction mixtures at 37 ± 0.1°C, and terminated after 30 or 60 min for human liver microsomes and recombinant CYP3A4, respectively. After precipitation of microsomal protein, an aliquot (75 μl) of the supernatant was analyzed by HPLC/MS via direct injection, respectively, as described under Materials and Methods.
To establish that the relationships observed between 2-OHIS formation and CYP3A4 and CYP2B6 activities were valid, additional correlation studies were performed with a second, larger panel of human liver microsomes (n = 24, obtained from XenoTech, LLC). The P450 activities in this panel of microsomal samples were determined with a second set of isoform-selective reactions. Although some P450 activities were determined with different substrates or concentrations of substrate, certain P450 activities (CYP2A6, 2C8, 2C9, 2E1, and 3A4/5) were characterized under conditions that permitted direct comparison between the two panels of microsomes. As shown in Table 2, formation of 2-OHIS in the panel of 24 microsomal samples was significantly correlated with several P450 activities. This finding was not unexpected because a number of isoform activities were significantly correlated with one another (data not shown). For example, CYP3A4/5 activity was significantly correlated with all other measured P450 activities except those of CYP2C9, CYP2E1, and CYP4A. It should be noted, however, that the highest coefficients of determination (r2 > 0.780) were found between the rate of 2-OHIS formation and CYP3A4/5 or CYP2B6 activity. An example of the relationship between the rate of 2-OHIS formation and T6βH activity (CYP3A4/5) in the two commercial panels of human liver microsomes examined is shown in Fig. 4.
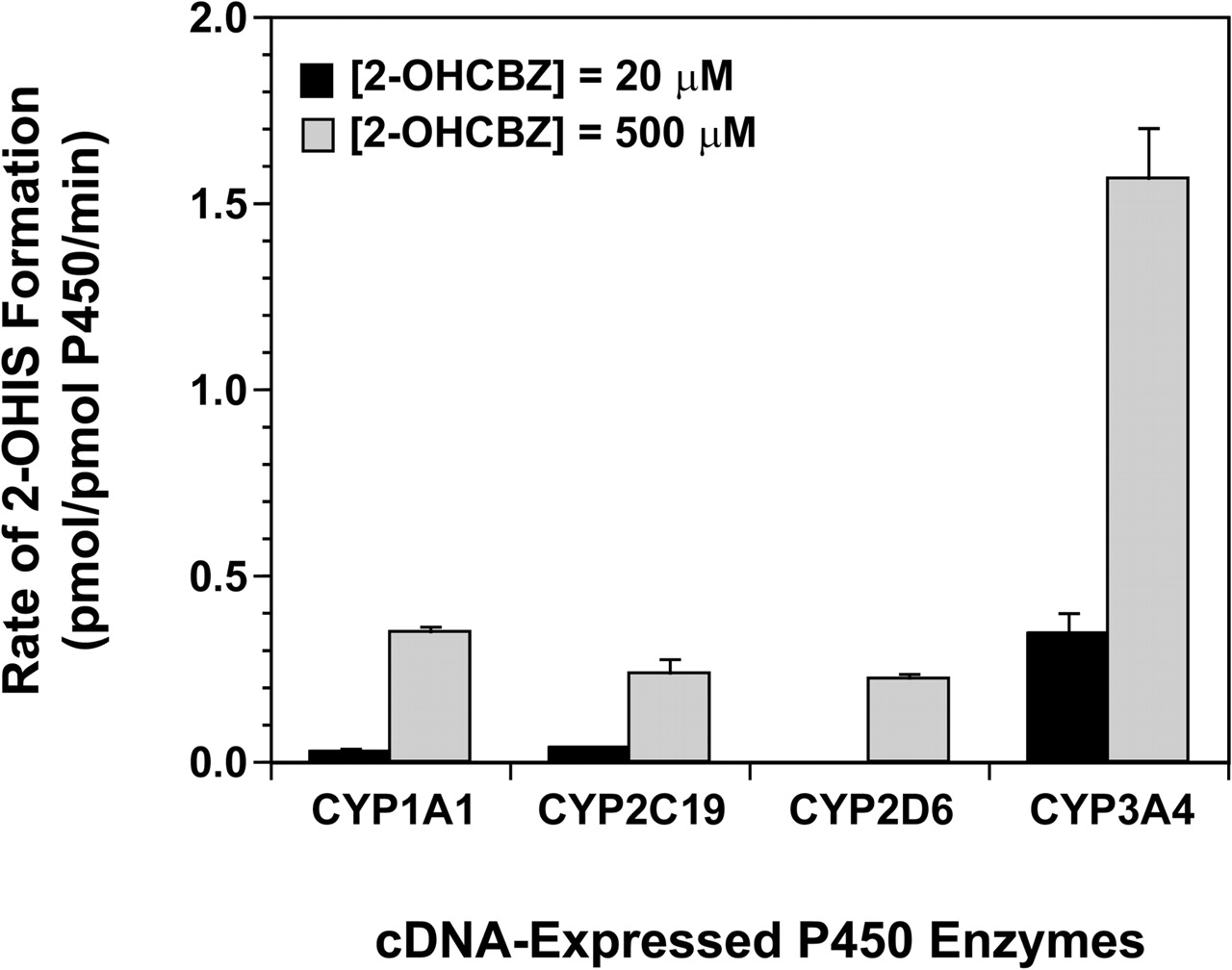
Formation of 2-OHIS by cDNA-Expressed Human P450 Enzymes. Microsomes containing one of 14 cDNA-expressed human P450 enzymes (CYP1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 3A4, 3A5, and 3A7) or a vector control were screened for their ability to catalyze the formation of 2-OHIS from a relatively high concentration of 2-OHCBZ (500 μM). Only four of the recombinant enzymes examined, namely CYP1A1, CYP2C19, CYP2D6, and CYP3A4, produced detectable quantities of 2-OHIS. When these four P450 enzymes were incubated with 20 μM 2-OHCBZ, CYP3A4 catalyzed the formation of 2-OHIS at a rate of 0.344 pmol/pmol P450/min, which was at least 10 times the rates catalyzed by either CYP1A1 or CYP2C19 (Fig. 5). CYP2D6 did not form detectable amounts of 2-OHIS at this substrate concentration. Normalization of the rates of 2-OHIS formation (at a substrate concentration of 20 μM) with respect to the relative abundance of each P450 isoform present in native human liver microsomes (Rodrigues, 1999) suggested that ∼98% of P450-mediated 2-OHIS formation can be attributed to CYP3A4.
Similar to the results obtained with human liver microsomes, formation of 2-OHIS by recombinant CYP3A4 appeared to conform to traditional Michaelis-Menten kinetics, based on visual examination of the Eadie-Hofstee plot of the data (Fig. 3). Nonlinear regression of the data from kinetic studies yielded an estimated apparent Km of 124 μM for CYP3A4, a Km value comparable to those generated for the human liver microsomal preparations (Table 1).

Relationship between the rate of 2-OHIS formation (100 μM) and testosterone 6β-hydroxylase activity (CYP3A4/5) in human liver microsomes obtained from BD Gentest (solid circles) and XenoTech LLC (open circles).
Inhibition of 2-OHIS Formation. Based on the correlation and recombinant human P450 data, selective chemical inhibitors were chosen to assess further the contribution of CYP3A4 and other selected P450 enzymes (CYP1A1/2, CYP2C19, and CYP2D6) in the conversion of 2-OHCBZ (100 μM) to 2-OHIS. Since these studies implicated CYP3A4 as the major enzyme responsible for catalyzing 2-OHIS formation, chemical inhibitors were incubated with human liver microsomes from two donors, one with high CYP3A4/5 activity (H112), and one with low CYP3A4/5 activity (H066). As illustrated in Fig. 6, the CYP3A inhibitors ketoconazole and troleandomycin markedly inhibited the formation of 2-OHIS (>80%) in both microsomal preparations. α-Naphthoflavone, an inhibitor of CYP1A2 with the ability to stimulate the activity of CYP3A enzymes (Shou et al., 1994), stimulated the formation of 2-OHIS by 2- to 3-fold. The other chemicals examined caused little or no inhibition of 2-OHIS formation (<15%).

2-OHIS formation by human recombinant cytochrome P450 enzymes. 2-OHCBZ (20 and 500 μM) was incubated with heterologously expressed human P450 enzymes as described under Materials and Methods. Each bar represents the mean ± S.D. of six determinations.

Effects of various P450 isoform-selective inhibitors on the formation of 2-OHIS by human liver microsomes. Human liver microsomes were incubated with 2-OHCBZ (50 μM) in the presence or absence of various chemicals, as described under Materials and Methods. Final inhibitor concentrations are indicated in parentheses. Each bar represents the mean ± S.D. of six determinations. The uninhibited rates of 2-OHIS formation were 15.7 ± 1.1 and 87.6 ± 11.5 pmol/mg/min in human liver microsomes with low (H066) and high (H112) CYP3A activity (H112), respectively.
Consistent with these results, polyclonal antibody against CYP3A4 virtually eliminated the formation of 2-OHIS in human liver microsomes (BD Gentest panel of 10); no detectable metabolite was produced in any of the microsomal samples in the presence of CYP3A4 antibody at substrate concentrations of 20 or 100 μM (data not shown). In general, rabbit preimmune IgG had little or no effect on the formation of 2-OHIS in human liver microsomes.
Preincubation-Dependent Inhibition of CYP3A4/5. Preincubation time-dependent inhibition of CYP3A4 by 2-OHCBZ (10, 100, or 500 μM) or by CBZ, 2-OHIS, or CBZ-IQ (100 μM) was investigated in human liver microsomes. To minimize any competitive enzyme inhibition that might be present, human liver microsomes were preincubated with one of the aforementioned compounds in the presence of an NADPH-generating system for 0 to 30 min, followed by a 10-fold dilution of the incubation volume before the determination of CYP3A activity, as measured by T6βH activity. Using this approach, none of the compounds examined caused a significant decrease in CYP3A activity (data not shown).
Formation of GSH- and NAC-Conjugates. In the presence of either NADPH (1 mM) or an NADPH-generating system (as described in sections pertaining to incubation conditions under Materials and Methods), human liver microsomes failed to form GSH- or NAC-conjugates when incubated with 2-OHCBZ as substrate (up to 3.5 h incubation time). These incubations, however, did contain substantial amounts of 2-OHIS (concentrations approximating 7–10% of the initial substrate concentration). Similarly, when either 2-OHIS or CBZ-IQ was incubated with NADPH or an NADPH-generating system, in the presence or absence of human liver microsomes, neither GSH- nor NAC-conjugates of these compounds were detected. It was noted, however, that in those incubations containing CBZ-IQ as substrate, the majority (>95%) of the CBZ-IQ present had been converted to 2-OHIS. Additional experiments demonstrated that CBZ-IQ could be reduced to 2-OHIS in an aqueous medium in the presence of NADPH, an NADPH-generating system, or sodium dithionite.

Representative HPLC/MS chromatograms obtained by atmospheric pressure chemical ionization at selected ion currents of [MH]+ ions present in 60-min incubations containing glutathione (5 mM, upper panel) or N-acetyl cysteine (0.5 mM, lower panel) in the presence (B) or absence (A) of CBZ-IQ. The selected ion current for each ion is provided in the upper left corner of each panel.
In contrast, CBZ-IQ alone readily formed GSH- and NAC-conjugates when incubated with GSH or NAC in a buffer consisting of potassium phosphate (50 mM, pH 7.4), MgCl2 (3 mM), and EDTA (1 mM), but lacking NADPH and human liver microsomes, as shown in Fig. 7. GSH- and NAC-conjugates of CBZ-IQ were monitored as [MH]+ ions at m/z 515 and 371, respectively. GSH and NAC conjugates, with spectral properties identical to those observed in incubations containing CBZ-IQ, were also detected in incubations that contained 2-OHIS as substrate. However, GSH- and NAC-conjugates formed only under conditions that permitted oxidation of 2-OHIS to CBZ-IQ, such as allowing incubations to sit at room temperature on a laboratory bench for 2 h before the addition of conjugating agent. When GSH was added to incubation mixtures 10 min after addition of 2-OHIS, no GSH-conjugate was detected.
Discussion
Although the mechanisms of many idiosyncratic hypersensitivity reactions are poorly understood, bioactivation of the offending parent drug to chemically reactive metabolites is considered to be the critical initiating event. Almost without exception, drugs associated with idiosyncratic hypersensitivity have been demonstrated to form cytotoxic or protein-reactive metabolites in a variety of human or rodent in vitro and in vivo assay systems (Naisbitt et al., 2001; Ju and Uetrecht, 2002; Peggs et al., 2002). In general, the enzymes responsible for catalyzing reactive metabolite formation also serve as major targets and become covalently modified by the reactive species. The cytochrome P450 enzymes responsible for bioactivating the drugs halothane, tienilic acid, and dihydralazine (Bourdi et al., 1994; Eliasson and Kenna, 1996; Lecoeur et al., 1996), all of which are associated with drug-induced hypersensitivity reactions, form covalent adducts with the respective reactive species produced, potentially suffering inactivation in the process. Moreover, patients who have experienced a drug-induced hypersensitivity reaction with one of these compounds have also been found to have circulating autoantibodies that recognize the P450 enzyme responsible for each drug's bioactivation.
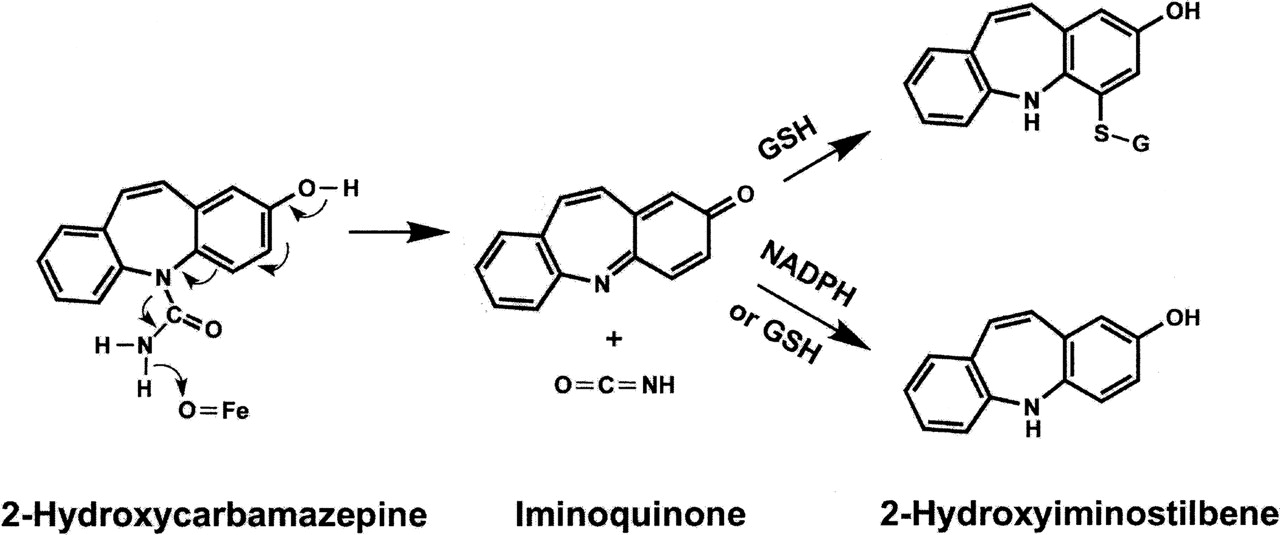
Proposed mechanism for the oxidation of 2-OHCBZ to CBZ-IQ and isocyanic acid, followed by reactions with NADPH and GSH. O=Fe represents the active site of cytochrome P450. CBZ-IQ can act as an electrophil and react with GSH to form an adduct. CBZ-IQ can also be reduced by NADPH or GSH to 2-OHIS. The reduction by GSH presumably occurs when attack is on the nitrogen rather than carbon, and the unstable product reacts with another molecule of GSH to form glutathione disulfide and 2-OHIS.
Similarly, CBZ can be bioactivated by human hepatic P450s to cytotoxic (Riley et al., 1988; Pirmohamed et al., 1993) and protein-reactive (Pirmohamed et al., 1992; Lillibridge et al., 1996) metabolites. Studies with cDNA-expressed human P450s suggest that CYP3A4 contributes approximately 65% to [14C]CBZ covalent adduct formation in human liver microsomes (Wolkenstein et al., 1998). Furthermore, antibodies that recognize a linear amino acid sequence localized to the CYP3A K-helix have been observed in the sera of a subset of patients with hypersensitivity reactions to phenytoin or carbamazepine (Leeder et al., 1996). Although the events linking drug bioactivation and subsequent formation of autoantibodies have remained elusive, the observation that suicide inactivation of CYP3A4 by cumene hydroperoxide results in adduct formation in the vicinity of the K-helix as well as accelerated proteasomal degradation by ubiquitin-dependent processes (He et al., 1998) provides some intriguing possibilities. Therefore, we have focused our attention on reactive metabolites of CBZ that may function in this manner.
Based on its demonstrated capability to react with sulfhydryl-containing nucleophils, such as GSH and NAC, CBZ-IQ was identified as a reactive species with the potential to function as a suicide-inactivator. Originally it was assumed that CBZ-IQ was formed by hydrolysis of the urea followed by oxidation of 2-OHIS, although in retrospect the hydrolysis of a urea is difficult. However, the data provide strong evidence for an oxidative mechanism rather than hydrolysis because the formation of 2-OHIS is mediated by P450 and requires NADPH. The P450-mediated oxidation of a phenol with a para substituent involving loss of two hydrogens to form a quinone-type product is well known, e.g., the oxidation of acetaminophen to an imidoquinone. What is different about this oxidation is that there is no hydrogen on the para nitrogen; however, there is a hydrogen on the other urea nitrogen. It is also known that N-methylformamide is oxidized by P450 with loss of two hydrogens to form methyl isocyanate (Guengerich, 2001). By combining these two mechanisms, CBZ-IQ can be formed by direct oxidation of 2-OHCBZ; specifically, P450 abstracts a hydrogen atom from the urea nitrogen followed by the abstraction of another electron leading to CBZ-IQ and isocyanic acid, as shown in Fig. 8. Alternatively, the hydrogen could be abstracted from the phenol with electrons flowing in the opposite direction; the site of hydrogen abstraction would likely depend on how the molecule orients in the enzyme pocket.
Delineation of the pathway leading to CBZ-IQ will require more extensive, mechanistic studies than those conducted in the current study, which was undertaken to identify the human P450 enzymes involved in the conversion of 2-OHCBZ to 2-OHIS (or to CBZ-IQ, followed by reduction to 2-OHIS) and to determine the ability of 2-OHIS and CBZ-IQ to inactivate the enzymes involved in this process. Results from correlation analyses, inhibition experiments, and studies with recombinant cDNA-expressed enzymes clearly indicate that CYP3A4 is the principal P450 enzyme responsible for the conversion of 2-OHCBZ to 2-OHIS in human liver microsomes. These results are consistent with the data of Wolkenstein et al. (1998), which demonstrated that CYP3A4 was the P450 enzyme primarily responsible for the bioactivation of [14C]carbamazepine, and with previous work from our group related to the existence of serum antibodies capable of recognizing a linear amino acid sequence localized to the CYP3A K-helix (Leeder et al., 1996). However, the capacity for CBZ-IQ to adduct or inactivate CYP3A4 is unclear. Time-dependent inhibition experiments conducted with CBZ, 2-OHCBZ, 2-OHIS, and CBZ-IQ failed to inhibit human liver microsomal CYP3A activity, suggesting that CYP3A4 is not selectively inactivated by any metabolite species in the pathway leading to formation of CBZ-IQ (including, presumably, isocyanic acid). Furthermore, experiments conducted to determine whether GSH- or NAC-adducts could be generated in vitro using 2-OHCBZ as substrate failed to demonstrate adduct formation. In addition, GSH- and NAC-adducts were formed with CBZ-IQ or 2-OHIS only under oxidative conditions, i.e., in the absence of NADPH. Even though CBZ-IQ was not observed, the data imply its direct formation by P450, yet no inactivation of P450 was observed. Possible explanations include the following: 1) it was reduced to 2-OHIS before it had time to react, 2) it reacted with P450, but this did not lead to inactivation of P450, or 3) iminoquinones are selective for free sulfhydryl groups and there is no free sulfhydryl group close to the active site with which CBZ-IQ could react.
The role of this bioactivation pathway in the pathogenesis of CBZ idiosyncratic toxicity is speculative at this time. It is possible that in situations where there is little or no reductive capacity (e.g., in GSH-depleted states, or in cells with high oxidative potential, such as macrophages) and perhaps distal to the site of formation, CBZ-IQ may potentially function as a reactive species in vivo. Such a situation is not without precedent. Felbamate, another antiepileptic drug whose use is associated with idiosyncratic aplastic anemia, undergoes biotransformation in the liver to form the unstable intermediate, 3-carbamoyl-2-phenylproprionaldehyde (Thompson et al., 1997). 3-Carbamoyl-2-phenylproprionaldehyde can undergo 1) reversible cyclization to form a stable compound that may be transported to tissues distant from its site of formation (Dieckhaus et al., 2001), 2) further oxidation to form the “nontoxic” 3-carbamoyl-2-phenylpropionic acid (the major metabolite formed in humans in vivo), or 3) β-elimination to form 2-phenylpropenal (commonly called atropaldehyde), a potent electrophil that is toxic to cells in culture (Thompson et al., 1996). Atropaldehyde undergoes rapid conjugation with GSH and can be observed in the urine of felbamate-treated patients as mercapturate derivatives (Thompson et al., 1997). Thus, formation of 3-carbamoyl-2-phenylproprionaldehyde represents a “commitment step” whereby the molecule commits to either a detoxication pathway (leading to 3-carbamoyl-2-phenylpropionic acid) or to a toxic pathway (leading to atropaldehyde).
Certain similarities exist between the paradigm presented for atropaldehyde formation and the generation of the reactive metabolite, CBZ-IQ. Both 2-OHCBZ and 2-OHIS are present in measurable concentrations in the plasma (Lertratanangkoon and Horning, 1982), which could represent a “metabolic depot” for transport to tissues, e.g., skin or bone marrow, which might favor formation of CBZ-IQ. Furthermore, formation of 2-OHIS may represent a “commitment step” analogous to that of aldehyde carbamate formation. Although 2-OHIS is readily oxidized/reoxidized to CBZ-IQ under the appropriate conditions, 2-OHIS is also capable of undergoing conjugation with glucuronic acid. Whereas oxidation to CBZ-IQ represents a bioactivation pathway, glucuronidation of 2-OHIS may well represent a detoxification pathway. It is also important to note that both felbamate and carbamazepine are typically administered in doses of hundreds of milligrams to grams per day (Sifton, 1999), suggesting that a substantial burden of protein- or thiol-reactive metabolites may be formed on a daily basis. Therefore, it is conceivable that individuals with a low capacity to detoxify reactive metabolites or individuals who possess a normal constitutive capacity for detoxication, but are unable to respond to an electrophillic or oxidative challenge, may represent individuals at risk for idiosyncratic toxicity.
In summary, we have demonstrated that CYP3A4 is largely, if not solely, responsible for the conversion of 2-OHCBZ to 2-OHIS in human liver microsomes. Furthermore, time-dependent inhibition experiments conducted with CBZ, 2-OHCBZ, 2-OHIS, and CBZ-IQ failed to inhibit CYP3A activity in human liver microsomes, which suggests that CYP3A4 is not selectively inactivated by any of these metabolites. GSH- and NAC-conjugates can be formed from CBZ-IQ and 2-OHIS, but only under conditions where local redox status favors net oxidation. Cumulatively, these results suggest that CBZ-IQ may not form CYP3A4-adducts that could potentially elicit hypersensitivity responses. However, the possibility remains that CBZ-IQ is capable of functioning as a reactive metabolite in tissues with little reductive capacity, which could lead to localized, idiosyncratic toxicities. Clearly, this issue warrants further investigation.
Footnotes
-
Supported by Grant R01GM58883-04, National Institute of General Medical Sciences (J.S.L.).
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.004861.
-
ABBREVIATIONS: CBZ, carbamazepine, 5H-dibenzo[b,f]azepine-5-carboxamide; P450, cytochrome P450; 2-OHIS, 2-hydroxyiminostilbene; CBZ-IQ, carbamazepine iminoquinone; GSH, glutathione; NAC, N-acetylcysteine; 2-OHCBZ, 2-hydroxycarbamazepine; HPLC/MS, high-performance liquid chromatography-mass spectrometry; T6βH, testosterone 6β-hydroxylase; ketoconazole, cis-1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-(1H-imidazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]piperazine; α-naphthoflavone, 7,8-benzoflavone; omeprazole, 5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridinyl)methyl]sulfinyl]-1H-benzimadazole; quinidine, dextro-6′-methoxycinchonan-9-ol; sulfaphenazole, 4-amino-N-(1-phenyl-1H-pyrazol-5-yl)benzenesulfonamide.
- Received March 25, 2005.
- Accepted August 31, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}