


Abstract
Human monocarboxylate transporter 6 (MCT6) has recently been isolated, and its tissue distribution has been established at the mRNA level, but its functional properties remain unknown. The aim of this study is to investigate the transport properties of MCT6. When expressed in Xenopus laevis oocytes, MCT6 transported [3H]bumetanide in a pH- and membrane potential-sensitive but not proton gradient-dependent manner, with the Kt value of 84 μM. Furthermore, MCT6 transported various drugs such as probenecid and nateglinide. Neither [14C]l-lactic acid nor [3H]l-tryptophan, typical substrates of other MCT isoforms, was transported by MCT6. Four loop diuretics, i.e., furosemide, piretanide, azosemide, and torasemide, thiazides, probenecid, glibenclamide, and nateglinide inhibited the MCT6-mediated uptake of [3H]bumetanide. In contrast, short-chain carboxylic acids, such as l-lactic acid and succinic acid did not inhibit the MCT6-mediated uptake of bumetanide. These results suggest that the substrate specificity of MCT6 is distinct from those of other MCTs. Bumetanide would be a good tool for investigating the functional properties of MCT6. It is probable that MCT6 is involved in the disposition of various drugs, including bumetanide.
Human monocarboxylate transporter 6 (MCT6; SLC16A5) is a member of the MCT family, which comprises 14 members (Halestrap and Meredith, 2004) and was first cloned from a placenta cDNA library (Price et al., 1998). MCT6 shares 38% identity with MCT4 at the amino acid level. Northern blot analysis revealed that MCT6 mRNA is highly expressed in the kidney and placenta (Price et al., 1998), but expression at the protein level and the function of MCT6 are still unknown.
The first member of the MCT family was cloned from a mutated Chinese hamster ovary cell line with enhanced mevalonate uptake (Kim et al., 1992). Subsequently, the wild-type protein was demonstrated to catalyze proton-linked transport of l-lactic acid and pyruvic acid and was named monocarboxylate transporter 1 (MCT1) (Garcia et al., 1994a). Based on the nucleotide sequence of hamster MCT1, MCT1 orthologs have been cloned from human, rat, and mouse (Garcia et al., 1994b; Jackson et al., 1995; Takanaga et al., 1995; Carpenter et al., 1996). It has been reported that MCT1 cotransports H+ with l-lactic acid (Kim et al., 1992; Bröer et al., 1997, 1998). MCT1 (SLC16A1), MCT2 (SLC16A7), MCT3 (SLC16A8), and MCT4 (SLC16A3) all transport both l-lactic acid and pyruvic acid (Lin et al., 1998; Grollman et al., 2000; Manning Fox et al., 2000). T-type amino acid transporter 1 (TAT1, SLC16A10), another member of the MCT family, transports aromatic amino acids such as l-tryptophan (Kim et al., 2001, 2002). More recently, the rat ortholog of MCT8 (SLC16A2) has been shown to transport thyroid hormones (Friesema et al., 2003). The function of the other MCT family members remains unknown.
In the present study, we examined the substrate specificity of MCT6 using a Xenopus oocyte expression system. We also investigated the transport properties of MCT6 by using bumetanide, a newly identified substrate of MCT6.
Materials and Methods
Materials. [3H]Bumetanide (1.85 GBq/mmol) was purchased from American Radiolabeled Chemicals (St. Louis, MO). [3H]Probenecid was kindly provided by Fuji Biomedix Co., Ltd. (Saitama, Japan). [14C]Nateglinide and nateglinide were kindly supplied by Ajinomoto Co., Inc. (Tokyo, Japan). Nonradioactive bumetanide was purchased from MP Biomedicals (Irvine, CA). Azosemide, torasemide, and piretanide were kindly supplied by Sanwa Kagaku Kenkyusho Co. (Nagoya, Japan), Taisho Toyama Pharmaceutical Co. (Tokyo, Japan), and Nippon HEXAL Co. (Tokyo, Japan), respectively. Furosemide was purchased from Wako Pure Chemicals (Osaka, Japan). pSP64T-mKV1.3-EC plasmid was obtained from American Type Culture Collection (Manassas, VA). All other chemicals were of the highest grade commercially available.
Methods.Construction of pSP64T-hMCT6. pSP64T vector containing a BglII restriction site was constructed from the pSP64T-mKV1.3-EC plasmid by digesting with BstE II, blunting with Klenow fragment, and ligating with a BglII linker. Next, pSP64T was digested with BglII and ligated with a NotI linker.
The plasmid containing MCT6 cDNA (clone KAT01693; Genbank accession no. AK000416) was obtained from New Energy and Industrial Technology Development Organization (NEDO) human cDNA project. MCT6 cDNA was amplified by polymerase chain reaction (PCR). PCR primers were designed to contain the 5′- and 3′-untranslated region sequences of MCT6. The forward primer was 5′-AAG ATT AAG CGG CCG CAA GAA AAG AGC ACT TCC CTG C-3′, and the reverse primer was 5′-TTA GTA TGC GGC CGC GGT CAA CAA TTT GCC CAA CTC-3′. Each primer contains a NotI restriction site (underlined). The PCR products were digested with NotI and ligated into the pSP64T-NotI vector. Finally, we obtained pSP64T-hMCT6. The identity of products was confirmed by means of sequencing.
cRNA Synthesis and Preparation of Xenopus oocytes. The capped cRNA of MCT6 was transcribed in vitro from BamHI-linearized pSP64T-hMCT6 with SP6 RNA polymerase. Oocytes were prepared from ovaries removed from female Xenopus laevis as previously described (Kobayashi et al., 2004). Oocytes (stages V–VI) were injected with cRNA and incubated in Barth's buffer [88 mM NaCl, 1 mM KCl, 0.33 mM Ca(NO3)2, 0.41 mM CaCl2 · 2H2O, 0.82 mM MgSO4 · 7H2O, 2.4 mM NaHCO3, 10 mM HEPES, and 50 ng/ml gentamicin sulfate, pH 7.6] at 18°C for 2 to 3 days.
Uptake Study. For uptake experiments, groups of 8 to 20 oocytes were transferred into 24-well multi-dishes and preincubated in uptake buffer (15 mM HEPES, 82.5 mM NaCl, 2.5 mM KCl, 1 mM Na2HPO4, and 1 mM MgCl2, pH 7.4 adjusted with Tris). The uptake buffer containing radiolabeled drugs (400 μl) was added to initiate the uptake after removal of preincubation buffer, and the mixture was incubated at room temperature (20–23°C) at pH 7.4 or pH 6.0, except in the pH dependence experiment. pH levels of uptake buffer were adjusted with 2-(N-morpholino)ethanesulfonic acid/Tris (pH 5.5, 6.0, and 6.5) or HEPES/Tris (pH 7.4 and 8.0). To examine the effects of Na+ and Cl–, NaCl in the buffer was replaced with N-methyl-d-glucamine, mannitol, or sodium gluconate. The high-potassium uptake buffer contained 15 mM NaCl, 120 mM KCl, 1 mM CaCl2, and 10 mM HEPES or 2-(N-morpholino)ethanesulfonic acid. The bicarbonate-containing uptake buffer was prepared by replacing 15 mM NaCl with 15 mM NaHCO3 and was gassed with 95% O2/5% CO2. To equilibrate intracellular pH (pHi) to external pH (pHo), we used the high-potassium uptake buffer containing 20 μM nigericin and 10 μM valinomycin (Sasaki et al., 1992; Emoto et al., 2002). The uptake was stopped by the addition of ice-cold uptake buffer, and oocytes were washed three times. The oocytes were divided into four to eight scintillation vials and dissolved in 250 μl of 10% sodium lauryl sulfate. The solution was slowly shaken for 1 h. A scintillation cocktail was added, and the radioactivity was counted using a liquid scintillation counter. Since uptake of all compounds in Table 1 by uninjected oocytes was essentially the same as that of oocytes injected with water both at pH 7.4 and 6.0, uninjected oocytes were used as the control instead of water-injected oocytes throughout this study. Values of uptake of [3H]bumetanide, [3H]probenecid, [3H]prostaglandin F2α, and [14C]nateglinide by uninjected oocytes at pH 7.4 were 0.06 ± 0.01, 0.51 ± 0.03, 0.19 ± 0.02, and 0.25 ± 0.02 (mean ± S.E., three separate experiments), respectively, and were not significantly different from those by water-injected oocytes: 0.10 ± 0.01, 0.58 ± 0.01, 0.26 ± 0.02, and 0.26 ± 0.01, respectively. The uptake by oocytes (uptake; μl/oocyte) is presented as the ratio of radioactivity in the sample (dpm/oocyte) to the initial concentration in the uptake buffer (dpm/μl).
Uptake of various compounds by MCT6
Uptake of each compound in MCT6-expressing oocytes or uninjected control oocytes was measured at room temperature and pH 7.4 or 6.0 for 30 min. Each value represents the mean ± S.E.M. of three to five separate experiments. Significance of differences between the uptake in uninjected oocytes and MCT6-expressing oocytes was determined by the unpaired Student's t test (* p < 0.05).
MCT6-specific uptake was calculated by subtraction of the uptake by uninjected oocytes from that by MCT6 cRNA-injected oocytes. To estimate the kinetic parameters of the saturable uptake via MCT6, the Michaelis-Menten eq. 1 was fitted to the experimental data by means of nonlinear least-squares regression analysis (Yamaoka et al., 1981).  where J and C represent the uptake rate (pmol/oocyte/30 min) and the concentration of substrate (μM), respectively. Jmax and Kt represent the maximum uptake rate for the saturable component and the half-maximal concentration.
where J and C represent the uptake rate (pmol/oocyte/30 min) and the concentration of substrate (μM), respectively. Jmax and Kt represent the maximum uptake rate for the saturable component and the half-maximal concentration.
For inhibition curves, the following sigmoidal inhibition eq. 2 was fitted to the data by means of nonlinear least-squares regression analysis.  where E, IC50, and C represent the relative uptake rate of substrate (percentage of control), the concentration of inhibitor which gives 50% inhibition, and the concentration of inhibitor, respectively.
where E, IC50, and C represent the relative uptake rate of substrate (percentage of control), the concentration of inhibitor which gives 50% inhibition, and the concentration of inhibitor, respectively.
Statistical Analysis. Statistical analysis was performed using the unpaired Student's t test or analysis of variance followed by Dunnett's test. A difference between means was considered statistically significant when the P value was less than 0.05.
Results
Transport of Organic Compounds via MCT6. [14C]Bumetanide was found to be taken up specifically by oocytes injected with MCT6 cRNA (MCT6-expressing oocytes) in comparison with uninjected control oocytes, showing that bumetanide is a substrate of MCT6. We therefore examined the uptake of various compounds via MCT6 using bumetanide as positive control. Uptake of [3H]estrone-3-sulfate [ES; a substrate of organic anion transporter (OAT) 3, OAT4, and organic anion transporting polypeptide (OATP) 1A2, OATP1B1, OATP1B3, OATP2B1, and OATP3A1] (Kullak-Ublick et al., 2001; Hagenbuch and Meier, 2004), [14C]p-aminohippuric acid (PAH; a typical substrate of OAT1, OAT3, and OAT4) (Koepsell and Endou, 2004), [3H]valproic acid, [14C]salicylic acid, [3H]hexanoic acid, and [14C]l-leucine by MCT6-expressing oocytes was similar to that by control oocytes at both pH 7.4 and 6.0 (Table 1). The uptake of [3H]valproic acid was lower than that by control oocytes. However, uptake of [14C]nateglinide or [3H]prostaglandin F2α by MCT6-expressing oocytes was significantly higher than that by control oocytes (Table 1). The uptake rate of [14C]nateglinide at 8 μM was higher than that at 57 μM but still lower than that of [3H]bumetanide (Table 1). The values of uptake of [3H]probenecid by MCT6-expressing oocytes at pH 7.4 and 6.0 were significantly higher and lower, respectively, than those by control oocytes (Table 1). MCT6 did not mediate the uptake of [14C]l-lactic acid, a typical substrate of MCT1-MCT4, or [3H]tryptophan, a typical substrate of TAT1 (data not shown). Uptake of [3H]bumetanide by MCT1- and MCT4-expressing oocytes were similar to that by control oocytes (data not shown).

Time course (A), concentration dependence (B), and pH dependence (C) of the uptake of [3H]bumetanide by MCT6. Uptake of 0.1 μM [3H]bumetanide into MCT6-expressing (closed circles; A and C) or uninjected control oocytes (open circles) was measured at room temperature and pH 7.4 (A and B) for 30 min (C). MCT6-mediated uptake (open squares) was calculated by subtracting the uptake amount of control oocytes from that of MCT6-expressing oocytes. Each point represents the mean ± S.E.M. (n = 3–5; *, p < 0.05 versus uninjected). The solid line in B represented the fitting line obtained from the data fitted with eq. 1 using nonlinear least-squares regression analysis.
Functional Characteristics of MCT6. The uptake of [3H]bumetanide via MCT6 linearly increased up to 60 min and reached a steady state at 90 to 120 min (Fig. 1A). In the following experiments, the uptake was examined at 30 min. When the concentration dependence of [3H]bumetanide uptake by MCT6-expressing oocytes was examined, the uptake was saturable with regard to substrate concentration (Fig. 1B). Kinetic analysis with the Michaelis-Menten eq. 1 yielded a Kt value of 84.2 (56.3, 125.9; mean – S.D., mean + S.D.) μM and a Jmax value of 45.2 ± 21.88 pmol/oocyte/30 min (mean ± S.D. of ten separate experiments). Figure 1C shows the effect of extracellular pH on the uptake of [3H]bumetanide via MCT6. At each pH between 8.0 to 5.5, the uptake by the MCT6-expressing oocytes was higher than that by control oocytes. The uptake rate of [3H]bumetanide via MCT6 increased with decreasing extracellular pH.
The Kt and Jmax values at pH 6.0 were 83.4 (64.9, 107.2; mean – S.D., mean + S.D.) μM and 83.1 ± 27.0 pmol/oocyte/30 min (mean ± S.D. of four separate experiments), respectively. Although the Kt values were comparable at pH 7.4 and 6.0, the Jmax value at pH 6.0 was about twice that at pH 7.4. Therefore, the increase of uptake activity of MCT6 for [3H]bumetanide at acidic extracellular pH can be ascribed to an increased maximum transport rate, rather than to increased affinity for MCT6.
When NaCl in uptake buffer was replaced with N-methyl-d-glucamine, mannitol, or sodium gluconate to investigate the involvement of sodium ion and chloride ion, there was no significant change of uptake of [3H]bumetanide by MCT6 at pH 7.4 (Table 2). We also examined the effect of membrane potential on the uptake of [3H]bumetanide by MCT6. Under the condition of intracellular K+ concentration ([K+]in) = extracellular K+ concentration ([K+]out), attained by preincubation with the high-potassium buffer containing 10 μM valinomycin, the uptake of [3H]bumetanide by MCT6 was significantly higher than that in standard uptake buffer (Fig. 2A), suggesting that MCT6-mediated transport of bumetanide is dependent on membrane voltage. As shown in Fig. 2B, extracellular acidic pH also increased the uptake of [3H]bumetanide by MCT6 under the condition of [K+]in = [K+]out. These results indicates that the uptake of [3H]bumetanide by MCT6 is pH- and membrane potential-sensitive.
Effects of extracellular Na+, and K+, and Cl- on the uptake of [3H]bumetanide by MCT6
The uptake of [3H]bumetanide into MCT6-expressing oocytes or uninjected control oocytes was performed at room temperature and pH 7.4 for 30 min in the standard uptake buffer (NaCl) or Na+ - and/or Cl–-free buffer. Each value represents the mean ± S.E.M. of three separate experiments.

Membrane potential-sensitive and pH-sensitive [3H]bumetanide uptake via MCT6. A, after preincubation in the standard uptake buffer (2.5 mM K+) or the high-potassium uptake buffer (120 mM K+) containing 10 μM valinomycin at pH 7.4 for 30 min, uptake of [3H]bumetanide into MCT6-expressing or uninjected control oocytes for 30 min was measured in the same buffer. MCT6-mediated uptake is shown (mean ± S.E.M., three separate experiments; *, p < 0.05 versus control). B, after preincubation in the high-potassium uptake buffer (120 mM K+) containing 10 μM valinomycin at pH 7.4 for 30 min, uptake of [3H]bumetanide into MCT6-expressing or uninjected control oocytes for 10 min was measured at pH 7.4, 6.0, or 5.5. MCT6-mediated uptake is shown (mean ± S.E.M., n = 5). C and D, after preincubation, uptake of [14C]l-lactic acid by MCT1-expressing oocytes (C) and uptake of [3H]bumetanide by MCT6-expressing oocytes was measured under each condition as follows: open triangles, preincubation and uptake buffers were the high-potassium buffer containing 10 μM valinomycin at pH 7.4 ([K+]in = [K+]out, pHi ≈ pHo); closed triangles, preincubation and uptake buffers were the high-potassium buffer containing 10 μM valinomycin and 20 μM nigericin at pH 7.4 ([K+]in = [K+]out, pHi = pHo); open circles, after preincubation in the high-potassium buffer containing 10 μM valinomycin at pH 7.4, uptake was measured in the high-potassium buffer containing 10 μM valinomycin at pH 6.0 ([K+]in = [K+]out, pHi > pHo); closed circles, preincubation and uptake buffers were the high-potassium buffer containing 10 μM valinomycin and 20 μM nigericin at pH 6.0 ([K+]in = [K+]out, pHi = pHo). MCT1- or MCT6-mediated uptake is shown (mean ± S.E.M., n = 5; *, p < 0.05 versus the value in the absence of nigericin).
Furthermore, we investigated the mechanism of the pH effect on MCT6 activity. Under the condition of [K+]in = [K+]out, the uptake of [14C]l-lactic acid by MCT1, which is proton-coupled, was significantly reduced by removing the proton gradient with nigericin (Fig. 2C), but the uptake of [3H]bumetanide by MCT6 was not (Fig. 2D). These results imply that a proton gradient is not the driving force for MCT6.
We also examined the effect of  on the MCT6-mediated [3H]bumetanide uptake under the condition of high extracellular K+ concentration. The uptake of [3H]bumetanide via MCT6 for 30 min after preincubation in high K+ and
on the MCT6-mediated [3H]bumetanide uptake under the condition of high extracellular K+ concentration. The uptake of [3H]bumetanide via MCT6 for 30 min after preincubation in high K+ and  -containing buffer for 30 min was 0.91 ± 0.11 μl/oocyte (mean ± S.E., n = 5) and was not significantly different from that after preincubation in high K+ but not
-containing buffer for 30 min was 0.91 ± 0.11 μl/oocyte (mean ± S.E., n = 5) and was not significantly different from that after preincubation in high K+ but not  -containing buffer, 0.95 ± 0.06 μl/oocyte.
-containing buffer, 0.95 ± 0.06 μl/oocyte.
Inhibitory Effect of Various Compounds on the Uptake of Bumetanide Mediated by MCT6.Figure 3 shows the inhibitory effects of several loop diuretics (furosemide, piretanide, azosemide, and torasemide) on the uptake of [3H]bumetanide. All of these drugs concentration-dependently inhibited the MCT6-mediated uptake of [3H]bumetanide. The IC50 values of furosemide, piretanide, azosemide, and torasemide were 46.1 ± 16.4, 163.2 ± 74.7, 20.8 ± 5.7, and 13.0 ± 9.4 μM, respectively (estimate ± S.E.). Table 3 shows the inhibitory effects of various compounds other than loop diuretics on the MCT6-mediated uptake of [3H]bumetanide. l-Lactic acid (a typical substrate of MCT1-MCT4), pyruvic acid (a substrate of MCT1, MCT2, and MCT4), succinic acid, and citric acid did not affect the MCT6-mediated uptake of [3H]bumetanide. The effects of cationic compounds were subsequently examined. The MCT6-mediated uptake of [3H]bumetanide was inhibited by cimetidine but not by choline. In addition, PAH and ES inhibited the MCT6-mediated uptake of [3H]bumetanide. Thiazides, probenecid, glibenclamide, and nateglinide also inhibited the uptake of [3H]bumetanide.
Inhibitory effects of anions and cations on the uptake of [3H]bumetanide by MCT6
Uptake rate of 0.1 μM [3H]bumetanide inMCT6-expressing oocytes or uninjected oocytes was measured at room temperature and pH 7.4 or pH 6.0 for 30 min in the presence or absence of inhibitor. MCT6-specific uptake was obtained by subtracting the uptake amount of uninjected oocytes from that of MCT6-expressing oocytes. The results are shown as percentages of control uptake measured in the absence of inhibitor. Each value represents the mean ± S.E.M. (n = 4–10).
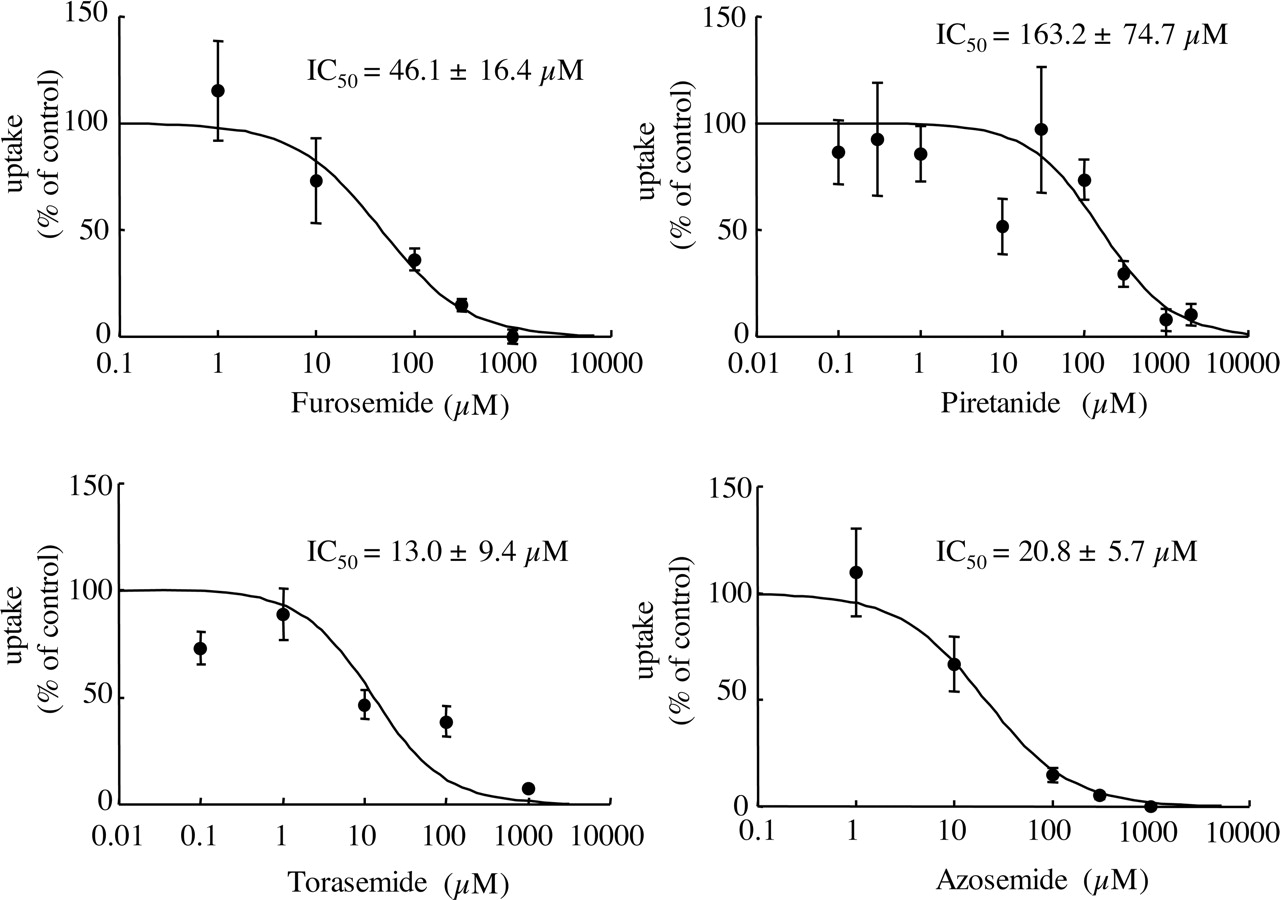
Inhibitory effects of loop diuretics on the uptake of [3H]bumetanide by MCT6. MCT6-expressing and control oocytes were incubated with uptake buffer containing 0.1 to 0.2 μM[3H]bumetanide and the indicated concentrations of inhibitors at room temperature, pH 7.4, for 30 min. MCT6-mediated uptake was calculated by subtracting the uptake amount of uninjected control oocytes from that of MCT6-expressing oocytes (mean ± S.E.M., n = 3–5). Equation 2 was fitted to the data using nonlinear least-squares regression analysis (estimate ± S.E.), and the result is represented by the solid line.
Discussion
Price et al. (1998) identified human MCT6 cDNA and examined the tissue distribution of MCT6 mRNA by Northern blot analysis, but the present study is the first to describe the functional characteristics of MCT6. We found that MCT6 transports bumetanide, but not l-lactic acid, a typical substrate of MCT1-MCT4, or l-tryptophan, a typical substrate of TAT1. In contrast, MCT1 and MCT4 transported l-lactic acid but not bumetanide. These results suggest that the substrate specificity of MCT6 is different from those of other MCTs, such as MCT1-MCT4 and TAT1. The substrates of other MCTs are endogenous compounds such as l-lactic acid, thyroid hormone, and aromatic amino acids (Halestrap and Meredith, 2004), and there is no evidence that the other MCTs mediate the transport of therapeutic drugs. In contrast, MCT6 mediated the transport of bumetanide, probenecid, and nateglinide and was inhibited by various drugs (Tables 1 and 3). Therefore, it seems likely that MCT6 participates in the membrane transport of various drugs. There are similar peptide sequences of rat (ex. XM_346905) and mouse (ex. XM_126601) in the GenBank database (http://www.ncbi.nlm.nih.gov). Further studies will be required to elucidate the difference in MCT6-mediated drug transport between humans and rodents. In addition, the physiological and pharmacokinetic roles of human MCT6 remain to be elucidated.
The uptake of [3H]probenecid by MCT6-expressing oocytes at pH 6.0 was lower than that by control oocytes. Further investigation is necessary to compare the properties of probenecid uptake by MCT6 with those of bumetanide uptake and to understand the action of pH on the transport activity of MCT6.
MCT6 did not transport PAH and ES, typical substrates of organic anion transporters such as OATs and OATPs, which are involved in the transport of various anionic drugs (Table 1). These results suggest that the substrate specificity of MCT6 is different from those of OATs and OATPs. The MCT6-mediated uptake of [3H]bumetanide was inhibited by PAH and ES (Table 3). Kinetic analysis of the inhibitory effects of PAH and ES remains to be performed.
Four loop diuretics structurally related to bumetanide, i.e., furosemide, piretanide, azosemide, and torasemide, inhibited the MCT6-mediated uptake of [3H]bumetanide (Fig. 3), as did several thiazides, probenecid, glibenclamide, and nateglinide. In contrast, short-chain carboxylic acids, such as l-lactic acid and succinic acid, did not inhibit the MCT6-mediated uptake of bumetanide (Table 3). All known substrates of other MCT isoforms have a carboxylate moiety. However, MCT6 was inhibited by azosemide and torasemide, which have an anionic sulfonamide moiety but not a carboxylate moiety, and cimetidine, an organic cation but not by short-chain carboxylic acids, suggesting that a carboxylate group may not be essential for affinity to MCT6.
It has been demonstrated that the transport of l-lactic acid by MCT1 is proton-coupled (Poole and Halestrap, 1993). MCT2, MCT3, and MCT4 transport monocarboxylates in a pH-dependent manner. On the other hand, transport mediated by MCT8 and TAT1 has been shown to be pH-insensitive (Kim et al., 2002; Halestrap and Meredith, 2004). The uptake of bumetanide via MCT6 was shown to be sodium- and chloride-independent, and pH- and membrane potential-sensitive, but not proton gradient-dependent. Under the condition of membrane depolarization, the presence of bicarbonate or diminution of the inward proton gradient did not affect the uptake of bumetanide via MCT6, suggesting that MCT6 is neither a proton-coupled transporter nor an OH– or  exchanger. When the membrane potential was depolarized, the uptake of bumetanide via MCT6 was increased. Our results suggest that MCT6-mediated transport of bumetanide will be the movement with net charge(s). Therefore, under physiological conditions, MCT6 may be involved in the facilitated diffusion of organic anions along the electrochemical potential gradient. To test this hypothesis, further electrophysiological studies are needed.
exchanger. When the membrane potential was depolarized, the uptake of bumetanide via MCT6 was increased. Our results suggest that MCT6-mediated transport of bumetanide will be the movement with net charge(s). Therefore, under physiological conditions, MCT6 may be involved in the facilitated diffusion of organic anions along the electrochemical potential gradient. To test this hypothesis, further electrophysiological studies are needed.
The transepithelial transport of bumetanide in the kidney has been suggested to involve hOAT1 to hOAT3 localized on the basolateral side and hOAT4 localized on the apical side of the proximal tubules (Hasannejad et al., 2004; Kobayashi et al., 2005). Since MCT6 mRNA is expressed predominantly in the kidney (Price et al., 1998), MCT6 may be involved in the renal secretion or reabsorption of bumetanide. The Kt values of bumetanide for hOAT2, hOAT3, and hOAT4 were 7.52, 1.59, and 0.31 μM, respectively (Hasannejad et al., 2004; Kobayashi et al., 2005), but the affinity of bumetanide for MCT6 (Kt value at pH 7.4; 84 μM) was lower. The clinical plasma concentration and plasma protein binding of bumetanide were reported to be about 0.27 μM and 95%, respectively (Davies et al., 1974; Pentikainen et al., 1977). Consequently, the estimated plasma unbound concentration of bumetanide is 0.014 μM, which is lower than Kt values of MCT6, hOAT2, hOAT3, and hOAT4. Therefore, none of these transporters should be saturated, and they are all possibly involved in the renal secretion and/or reabsorption of bumetanide. The renal localization of MCT6 and its quantitative contribution to the renal secretion and/or reabsorption of bumetanide needs further study.
Even under the condition of membrane depolarization, the uptake of bumetanide by MCT6 was elevated by extracellular acidic pH. However, our results indicate that the pH gradient across the cell membrane is not the driving force of MCT6. The mRNA of MCT6 is highly expressed in placenta and kidney. In kidney, the intracellular pH of tubular epithelial cells is lower than the pH of the extracellular fluid, and urine is acidic under normal conditions. In placenta, fetal blood is slightly more acidic (pH 7.3) than maternal blood (pH 7.4). In addition, Na+/H+ exchangers on the luminal and basolateral membrane of tubular epithelium and brush-border membrane of trophoblast cells produce a microacidic environment. Although the physiological meaning of the pH sensitivity of MCT6 activity is still unknown, MCT6 could be regulated by the acidic environment in these tissues.
In conclusion, we identified bumetanide for the first time as an MCT6 ligand. MCT6 transports bumetanide in a pH- and membrane potential-sensitive but not proton gradient-dependent manner, and the substrate specificity of MCT6 is different from those of the other MCTs. It is probable that MCT6 is involved in the disposition of various drugs, including bumetanide. Bumetanide should be a good tool for investigating the functional properties of MCT6 and the differences of substrate recognition sites among MCT isoforms.
Footnotes
-
This work was supported in part a grant-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.005264.
-
ABBREVIATIONS: MCT, monocarboxylate transporter; TAT1, T-type amino acid transporter 1; PCR, polymerase chain reaction; pHi, intracellular pH; pHo, external pH; ES, estrone-3-sulfate; OAT, organic anion transporter; OATP, organic anion transporting polypeptide; PAH, p-aminohippuric acid.
- Received April 21, 2005.
- Accepted September 16, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}