


Abstract
Nebicapone (BIA 3-202; 1-[3,4-dihydroxy-5-nitrophenyl]-2-phenylethanone), a novel catechol-O-methyltransferase inhibitor, is mainly metabolized by glucuronidation. The purpose of this study was to characterize the major plasma metabolites of nebicapone following p.o. administration of nebicapone to healthy volunteers, and to determine the human UDP-glucuronosyltransferase (UGT) enzymes involved in nebicapone glucuronidation. Plasma samples were collected as part of a clinical trial at different time points postdose and were analyzed for nebicapone and its metabolites using a validated method consisting of a solid-phase extraction, followed by high-performance liquid chromatography/mass spectrometry detection. The primary metabolic pathways of nebicapone in humans involve mainly 3-O-glucuronidation, the major early metabolite, and 3-O-methylation, the predominant late metabolite. Of the nine commercially available recombinant UGT enzymes studied (UGT1A1, UGT1A3, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B7, and UGT2B15), only UGT1A9 exhibited high nebicapone glucuronosyltransferase specific activity (24.3 ± 1.3 nmol/mg protein/min). UGT1A6, UGT1A7, UGT1A8, UGT1A10, UGT2B7, and UGT2B15 exhibited low activity (0.1–1.1 nmol/mg protein/min), and UGT1A1 and UGT1A3 showed extremely low activities (less than 0.03 nmol/mg protein/min). The results show that nebicapone is mainly glucuronidated in humans and that multiple UGT enzymes are involved in this reaction.
l-3,4-dihydroxyphenylalanine (l-DOPA) therapy has revolutionized treatment of idiopathic Parkinson's disease (PD) by providing a p.o. administered source of dopamine precursor with ready access to the brain, and it remains the most widely used palliative drug for PD. However, l-DOPA is metabolized in the periphery by aromatic l-amino acid decarboxylase (AADC) and catechol-O-methyltransferase (COMT) enzymes, reducing brain availability. Therefore, an improvement in l-DOPA therapy is provided by a combination treatment of l-DOPA with an AADC inhibitor plus a COMT inhibitor, effectively increasing its availability to the brain (Backstrom et al., 1989).
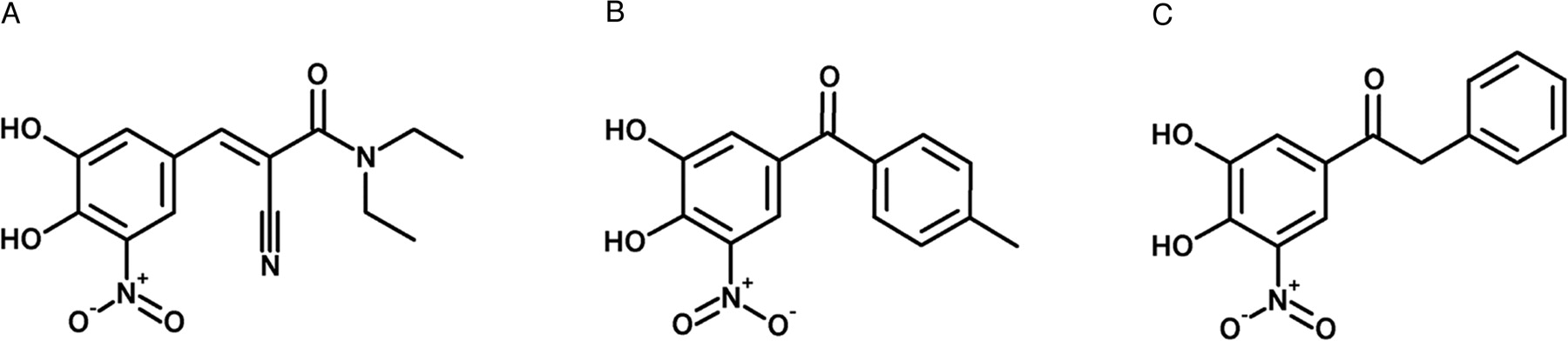
Second-generation COMT inhibitors entacapone and tolcapone (Fig. 1) have proven beneficial when used together with l-DOPA and an AADC inhibitor, such as carbidopa or benserazide, in the medical treatment of patients with PD (Dingemanse et al., 1995; Dingemanse, 1997; Heikkinen et al., 2001, 2002). When metabolized, these nitrocatechol derivatives are extensively conjugated, usually undergo direct glucuronidation catalyzed by UDP-glucuronosyltransferases (UGT), and are then rapidly excreted in the urine (Lautala et al., 1997). Additionally, both methylation and sulfation compete with glucuronidation for conjugation of the adjacent phenolic hydroxyls (Lautala et al., 1997). Major metabolites of tolcapone in human plasma are the 3-O-β-glucuronic acid (∼18.6%) and the 3-O-methyl conjugate (∼2.1%). In urine, the 3-O-β-glucuronic conjugates of tolcapone (∼13%) and its derivative N-acetyl amino (∼5.7%) are the predominant metabolites found (Jorga et al., 1999). Regarding entacapone, the only metabolite described in human plasma is the Z-isomer (∼5%) (Wikberg et al., 1993; Keranen et al., 1994); however, in human urine, besides the Z-isomer (∼25%), the 3-O-β-glucuronic acid derivative (∼70%) is the prevalent metabolite (Wikberg et al., 1993).
No methylation products of entacapone were detected in human plasma or urine, possibly because the nitro group of entacapone hinders methylation of the catechol (Wikberg et al., 1993). As an alternative to molecular conjugation with endogenous species like glucuronidation, sulfation, methylation, glutathione conjugation, and acetylation (phase II drug metabolism reactions), these drugs could undergo oxidation, reduction, and hydroxylation (phase I drug metabolism reactions); however, such phase I metabolites are minor (Wikberg et al., 1993; Jorga et al., 1999).

Structures of entacapone (A), tolcapone (B), and nebicapone (C).
The human UGT family presently comprises 16 individual distinctly expressed enzymes, not including variant enzymes (Taskinen et al., 2003). UGT are lumenally facing in the endoplasmatic reticulum and are expressed not only in liver but also in extrahepatic tissues, where the extent of glucuronidation can be substantial. Some UGT enzymes (e.g., UGT1A7, UGT1A8, UGT2A1, and UGT1A10) are expressed only in extrahepatic tissues (Kiang et al., 2005). Hepatic expression of UGT shows little interindividual variation, as opposed to the high interindividual variability found in the gastrointestinal tract (Strassburg et al., 1998, 2000), further accentuated by the differential enzyme expression within different segments of the gastrointestinal tract (Strassburg et al., 1998, 2000; Antonio et al., 2003; Basu et al., 2004). Genetic polymorphisms have been described for several UGT, and some of them have been associated with interindividual variations in drug effect (Chung et al., 2005) and toxicity (Marsh and McLeod, 2004; Massacesi et al., 2006). Therefore, the identification of specific enzymes involved in the metabolism of a drug is important.
There have been some studies identifying UGT able of conjugating entacapone and tolcapone with glucuronic acid. Entacapone was described to be a substrate for UGT1A1, UGT1A3, UGT1A7, UGT1A8, UGT1A10, UGT2B7, and UGT2B15 and a particularly good one for UGT1A9 (Lautala et al., 2000). Tolcapone, on the other hand, was tested on a limited number of enzymes, and it was described to be efficiently conjugated by UGT1A9 and to a lesser extent by UGT2B7 and UGT2B15 (Lautala et al., 2000).
Nebicapone (BIA 3-202, Fig. 1) is a new COMT inhibitor currently being developed for use as an adjunct to l-DOPA/AADC inhibitor therapy in the treatment of PD. It is a reversible and mainly peripherally acting inhibitor that was shown to decrease the biotransformation of l-DOPA to 3-O-methyl-DOPA by inhibition of COMT in clinical trials (Almeida and Soares-Da-Silva, 2003; Silveira et al., 2003; Almeida et al., 2004).
The purpose of the study presented herein was to quantify major metabolites of nebicapone in human plasma using a sensitive and specific HPLC/MS assay, and to investigate the UGT enzymes involved in nebicapone glucuronidation in vitro.
Materials and Methods
Chemicals. Nebicapone (BIA 3-202; 1-[3,4-dihydroxy-5-nitrophenyl]-2-phenyl-ethanone), its metabolites BIA 3-467 [1-(5-acetamido-3,4-dihydroxyphenyl)-2-phenyl-ethanone], BIA 3-476 [1-(3-O-β-d-glucopyranuronosido-4hydroxy-5-nitrophenyl)-2-phenyl-ethanone], BIA 3-465 prepared as a pyridinium salt [pyridinium-1-(4-hydroxy-5-nitro-3-O-sulfatophenyl)-2-phenyl-ethanone], BIA 3-270 [1-(4-hydroxy-3-methoxy-5-nitrophenyl)-2-phenyl-ethanone], and tolcapone as internal standard were synthesized in the Laboratory of Chemistry, BIAL (S. Mamede Coronado, Portugal), with purities >99.5%. All the other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Recombinant human UGT expressed in baculovirus-infected insect cells were purchased from Invitrogen (Carlsbad, CA) (UGT1A1, UGT1A3, UGT1A6, UGT1A7, UGT1A10, and UGT2B7) and from BD Biosciences (San Jose, CA) (UGT1A8, UGT1A9, and UGT2B15). Pooled human liver microsomes (HLM) (from 48 donors) and intestinal microsomes (HIM) (prepared from the duodenum and jejunum sections of each of five donors) were purchased from BD Gentest. The protein contents were used as described in the data sheets provided by the manufacturers.
Human Administration. The assay of nebicapone and its metabolites was performed on plasma samples collected from six young healthy volunteers (Caucasians, male and female) who received a 400-mg single dose of nebicapone while participating in a clinical trial (Almeida et al., 2004). Blood samples were taken at the following times: predose and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 18, and 24 h postdose. Blood samples were taken into potassium EDTA and centrifuged immediately at 1500g for 10 min at approximately 4°C. The resulting plasma was stored below –20°C until required for analysis.
Extraction of Nebicapone and Its Metabolites from Plasma. A 500-μl volume of plasma specimen was added to 500 μl of phosphate buffer (0.1 M, pH 2) containing 400 ng/ml tolcapone as internal standard. The samples were placed on an automatic liquid handler (ASPEC-XL4, Gilson, Villier Le Bel, France) for solid-phase extraction. The solid-phase extraction cartridges (Oasis, HLB, 30 mg, 1 ml, Waters) were conditioned with 1 ml of acetonitrile and then washed twice with 1 ml of phosphate buffer (0.1 M, pH 2). Specimens (900 μl) were loaded onto the cartridges, and the cartridges were washed twice with 1 ml of phosphate buffer (0.1 M, pH 2.0). After the second wash, the cartridges were flushed with air push of 10 ml at 6 ml/min. The cartridges were eluted twice with 250 μl of acetonitrile containing 1% formic acid with an air push of 2 ml at 6 ml/min. Four hundred microliters of water containing 1% formic acid was added to the eluted sample and mixed twice with aspiring dispensing cycles. The eluted samples were injected (5 μl) into an HPLC/MS.
Nebicapone Glucuronidation Screening by Recombinant UGT. Glucuronidation activity by UGT1A1, UGT1A3, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B7, and UGT2B15 was measured using the following assay conditions: the incubation mixture (100 μl total volume) contained 0.4 mg/ml total protein, 10 mM MgCl2, 2 mM uridine 5′-diphosphoglucuronic acid, 25 μg/ml alamethicin, 5 mM saccharolactone in 50 mM phosphate buffer, pH 7.5, and 20 μM nebicapone. Drug was dissolved in dimethyl sulfoxide, and the final concentration of dimethyl sulfoxide in the reaction was less than 0.5% (v/v). Reactions were preincubated 5 min and were initiated with the drug. Reaction mixtures were incubated for up to 60 min and stopped with 100 μl of 1% formic acid in acetonitrile. All the incubations were performed in a water bath shaking at 37°C. After removal of the protein by centrifugation for 3 min at 15,000g, supernatant was filtered through 0.20-μm Spin-X filters (Corning, NY) and injected on a HPLC/MS.
Kinetics of Nebicapone Glucuronidation by Selected UGT and in Pooled HLM and HIM. Rates of glucuronidation were determined, as described above for UGT screening, with nebicapone concentrations ranging from 5 to 1000 μM and with 15-min incubation time for all the recombinant UGT except UGT1A9. With UGT1A9, HLM and HIM incubation time was 5 min, and total protein concentration was 0.1 mg/ml.
All the preparations were evaluated for linearity of product formation with respect to incubation time (0–60 min). HLM, HIM, and UGT1A8, UGT1A9, and UGT2B7 were also evaluated for linearity of product formation with respect to protein concentration (0.05–0.6 mg/ml). All the experiments were performed with samples in duplicate.
HPLC/MS Analysis. The analysis of plasma sample extracts was performed using HPLC/MS (Agilent, atmospheric pressure/electrospray ionization, 1100 Series, Agilent Technologies) with negative ion detection. Separation was performed on a Zorbax SB-C18, 3-μm, 30 × 4.6 mm column (Agilent) using mobile phase A, water containing 1% formic acid (v/v), and B, acetonitrile containing 1% formic acid (v/v), with gradient conditions of 80% A and 20% B at 0 min and 50% A/50% B at 20 min. Selected ion monitoring with the detection of each compound of interest was used for quantification, namely, m/z 284.3 (BIA 3-467), 352.2 (BIA 3-465), 448.4 (BIA 3-476), 272.2 (nebicapone and tolcapone), and 286.3 (BIA 3-270). A time-programmed detection was used for the following ions: group 1, BIA 3-467, BIA 3-465, and BIA 3-476; group 2, nebicapone, tolcapone, and BIA 3-270. For maximal sensitivity, the fragment energy was set to 120 V, and further settings were 3500 eV for the capillary voltage, 350°C nebulizer gas temperature, and 40 psi nebulizer pressure. The method was validated in accordance with Food and Drug Administration guidance for industry (http://www.fda.gov/cder/guidance/4252fnl.pdf). The limits of quantification were defined as the lowest concentration of the range examined that has acceptable precision and accuracy [accuracy within 20% of the nominal value, and the coefficient of variation (%) did not deviate more than 20%]. Therefore, the limits of quantification were 0.22 μM for BIA 3-467, 0.14 μM for BIA 3-476, 0.12 μM for BIA 3-465, 0.15 μM for nebicapone, and 0.14 μM for BIA 3-270. The intrabatch and interbatch coefficient of variation and accuracy were within 15% of the actual value for all the analytes and in all the concentrations checked.
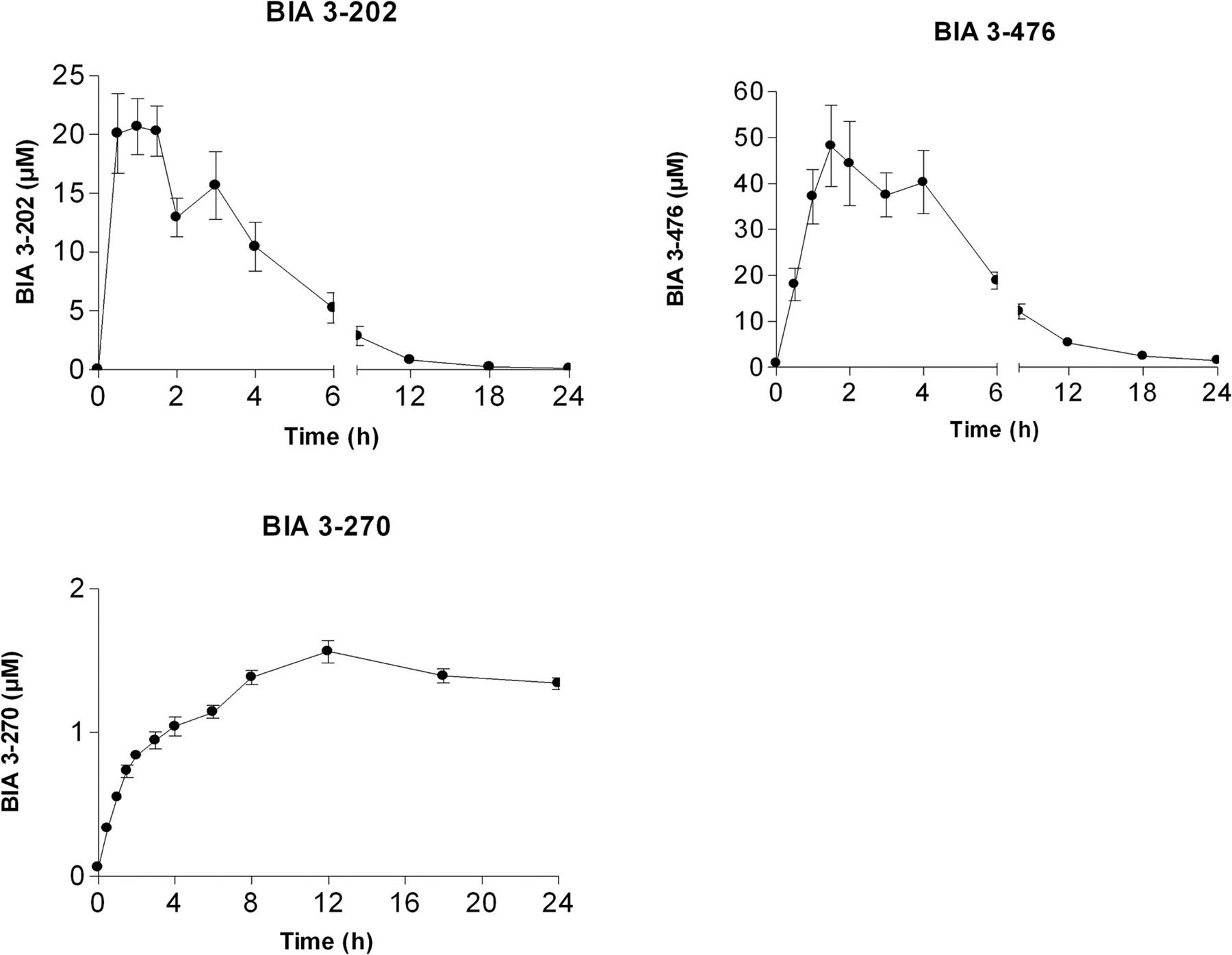
Mean plasma nebicapone (BIA 3-202) and its metabolites, 3-O-glucuronide (BIA 3-476) and 3-O-methylated (BIA 3-270) concentration-time profile following p.o. administration of 400 mg of nebicapone to humans. Symbols are mean ± S.E.M.; n = 6 humans.
For the determination of nebicapone glucuronide in in vitro studies, the same analytical column and mobile phases were used with gradient conditions of 70% A and 30% B at 0 to 5 min and 50% A/50% B at 10 min. Selected ion monitoring with the detection of m/z 448.4 was used for quantification of nebicapone glucuronide.
Data Analysis. The pharmacokinetic parameters of nebicapone and its metabolites were calculated from plasma concentration-time profiles using GraphPad Prism v4 (GraphPad Software Inc., San Diego, CA).
Kinetic parameters of nebicapone glucuronidation were obtained by fitting velocity data to the following models (eqs. 1–3) with GraphPad Prism (GraphPad Software Inc.): Michaelis-Menten equation,  where v is the rate of the reaction, Vmax is the maximum velocity, Km is the Michaelis constant, and S is the substrate concentration; Hill equation, which describes sigmoidal kinetics (Houston and Kenworthy, 2000),
where v is the rate of the reaction, Vmax is the maximum velocity, Km is the Michaelis constant, and S is the substrate concentration; Hill equation, which describes sigmoidal kinetics (Houston and Kenworthy, 2000),  where S50 is analogous to Km, and n is the Hill coefficient; and substrate inhibition model (Houston and Kenworthy, 2000),
where S50 is analogous to Km, and n is the Hill coefficient; and substrate inhibition model (Houston and Kenworthy, 2000),  where Ksi is the constant describing the substrate inhibition interaction. All the data are reported as mean ± S.E.M.
where Ksi is the constant describing the substrate inhibition interaction. All the data are reported as mean ± S.E.M.
Results
Metabolism and Elimination of Nebicapone in Human Plasma. Nebicapone and its major metabolites were quantified for an administration dose of 400 mg. The concentration-time profiles are presented in Fig. 2, and the pharmacokinetic parameters derived from these curves are summarized in Table 1. Nebicapone was rapidly absorbed, reaching a maximum plasma concentration (Cmax) of 28.3 ± 1.4 μM within 1.3 h before falling back to low levels over the next 8 h and returning to baseline at 12 h. Nebicapone presented an area under the curve (AUC)0–24 of 99.1 ± 17.2 h/μM and was mainly conjugated to the 3-O-β-glucuronic acid derivative (BIA 3-476) representing ∼70% of the total nebicapone and metabolites AUC. BIA 3-476 reached Cmax of 60.8 ± 7.8 μM within 2.5 h after administration and remained the major circulating metabolite of nebicapone at 8 h after administration. The mean AUC0–24 of the fraction of nebicapone glucuronide was 303.1 ± 96.3 h/μM. The lower limit of quantification for BIA 3-476 was approached by 12 h. After this time, the 3-O-methyl derivative of nebicapone, BIA 3-270, became the predominant metabolite in plasma with a Tmax of 12 h, an AUC0–24 of 30.6 ± 9.1 h/μM, and Cmax of 1.7 ± 0.1 μM, representing ∼7% of the total nebicapone and metabolites AUC. The 3-O-sulfate and acetamino derivatives of nebicapone (BIA 3-465 and BIA 3-467, respectively) were very minor metabolites (representing less than ∼1% of the total nebicapone and metabolites AUC) and were not quantified.
Mean (S.D.) pharmacokinetic parameters of nebicapone and its metabolites BIA 3-476 and BIA 3-270 in humans after p.o. administration of nebicapone (n = 6)
Tmax values are the median with range values in parentheses.
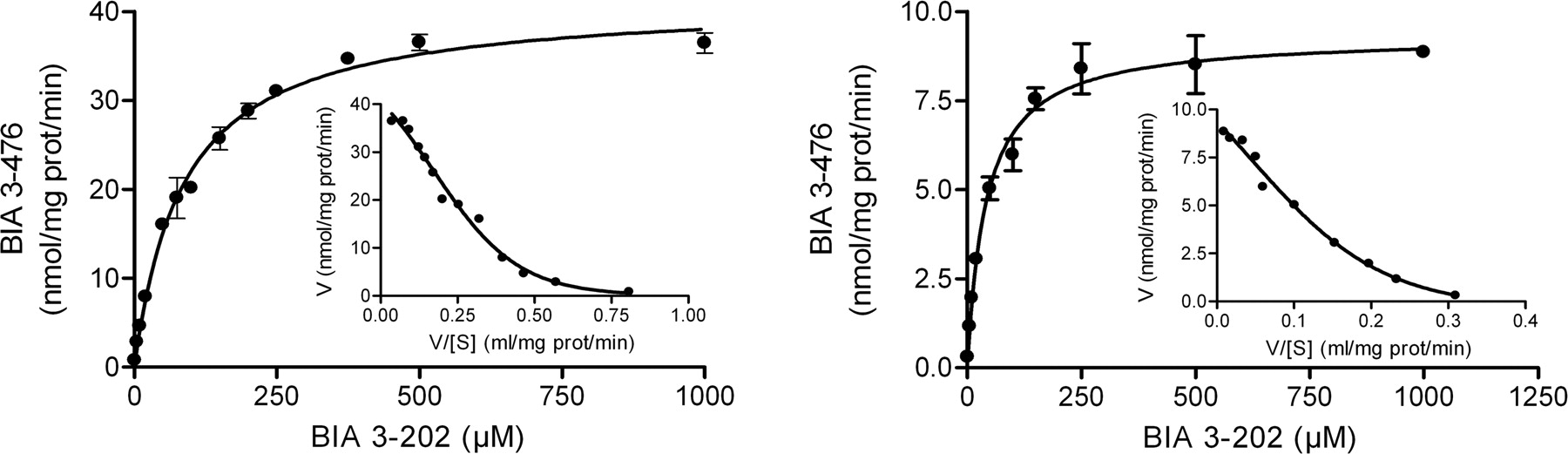
Kinetics of nebicapone (BIA 3-202) glucuronidation in human liver (A) and intestine (B) microsomal pools. Nebicapone concentrations ranged from 1 to 1000 μM. Each inset shows the Eadie-Hofstee representation of the experimental data with computer-generated best fit. Values represent mean ± S.E.M. of duplicates.
Nebicapone Glucuronidation by Human Microsomes. Kinetic analysis of nebicapone was performed in human liver and intestine pools of microsomes. As shown in Fig. 3, both preparations displayed typical hyperbolic kinetics, although the Eadie-Hofstee plots of the data were clearly biphasic (Hutzler and Tracy, 2002), which is indicative of the involvement of more than one enzyme in the reaction. The apparent kinetic parameters derived from these curves fitted to the Michaelis-Menten equation (eq. 1) are listed in Table 2. Both apparent Km and Vmax values were higher for liver than for intestine microsomes (about 2- and 4-fold higher, respectively). The intrinsic clearance (Clint = Vmax/Km) calculated for intestine was 222 μl/mg protein/min, and the one calculated for liver was 475 μl/mg protein/min, 2.1-fold higher than the one for intestine.
Apparent kinetic parameters of nebicapone glucuronidation in HLM, HIM, and recombinant UGT enzymes.
Nebicapone Glucuronidation by Recombinant UGT. Nine commercially available UGT enzymes were used to evaluate their ability to conjugate nebicapone to BIA 3-476 (Fig. 4). From the tested UGT, only UGT1A9 produced significant amounts of BIA 3-476 (24.3 ± 1.3 nmol/mg protein/min). UGT1A6, UGT1A7, UGT1A8, UGT2B7, and UGT2B15 produced small amounts of BIA 3-476 (between 0.1 and 1.1 nmol/mg protein/min). UGT1A1 and UGT1A3 also conjugated nebicapone, although only at longer incubation times (more than 30 min) and at extremely low levels, 0.028 ± 0.002 and 0.016 ± 0.002 nmol/mg protein/min, respectively. No metabolite formation was detected with control baculosomes/Supersomes over an incubation period of 60 min.
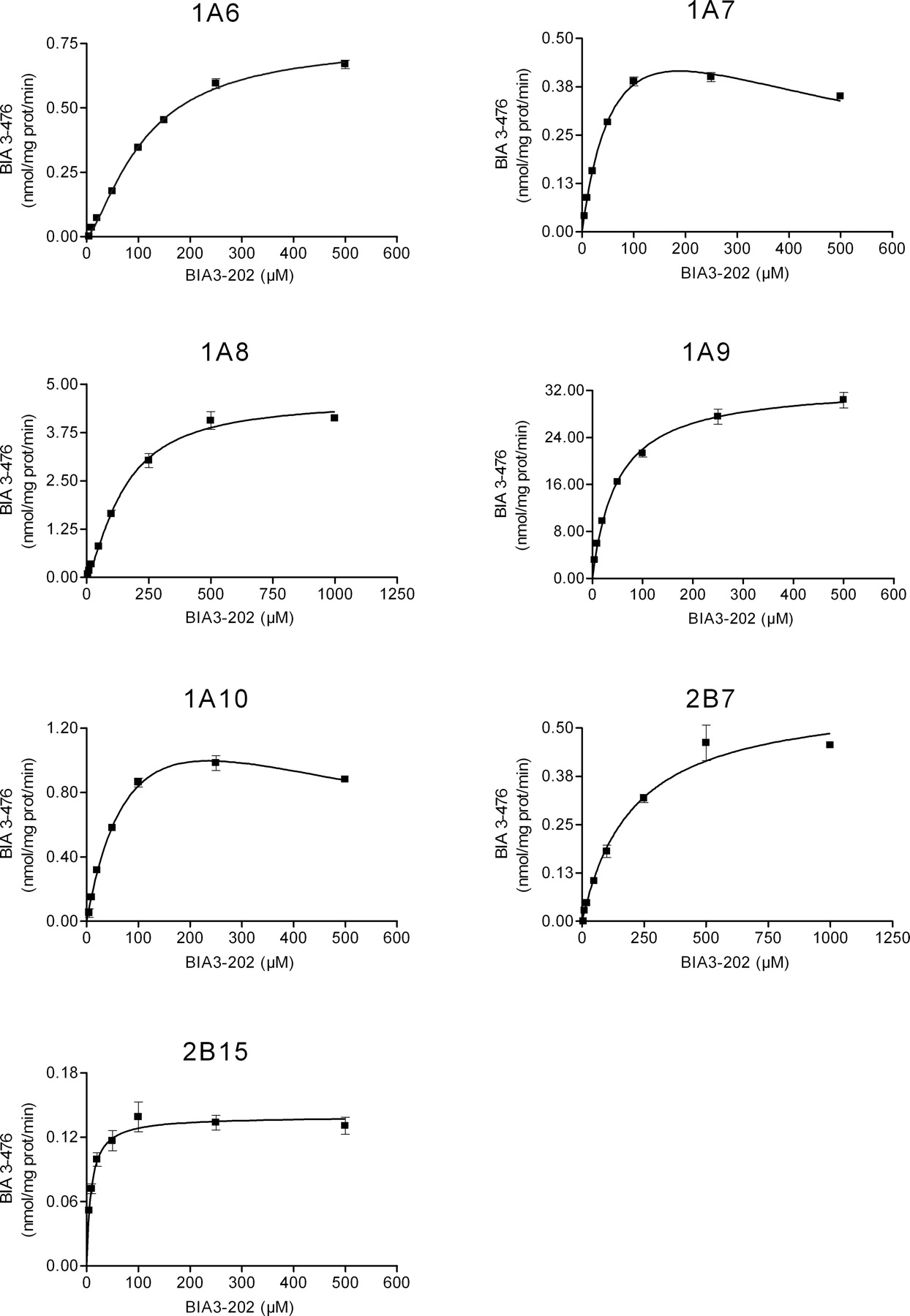
Kinetics of Nebicapone Glucuronidation by Recombinant UGT. The characterization of nebicapone glucuronidation kinetics was performed for all the UGT with activities higher than 0.1 nmol/mg protein/min, namely, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B7, and UGT2B15. Each enzyme was incubated with different concentrations of nebicapone (1–1000 μM), and the initial rates were determined. Rates were fitted to the most suitable model using Eadie-Hofstee plots (not shown) as diagnostic tool. Accordingly, the experimental data from UGT1A9, UGT2B7, and UGT2B15 were fitted with Michaelis-Menten equation (eq. 1); data from UGT1A6 and UGT1A8 were fitted with the Hill model (eq. 2); and data from UGT1A7 and UGT1A10, with an obvious substrate inhibition profile, were fitted with eq. 3. The resulting curves are represented in Fig. 5, and the apparent kinetic parameters, Km and Vmax, derived from these curves are shown in Table 2. There is a rather considerable range of affinities for the conjugation of nebicapone as shown by the apparent Km values determined. The enzyme with the highest affinity was UGT2B15, with a Km of 9 μM, followed by UGT1A9 and UGT1A7 with Km values of 50 and 75 μM, respectively. UGT1A6 and UGT1A10 had apparent affinities close to 100 μM, and UGT1A8 and UGT2B7 were the ones with the lower affinities, Km of 149 and 206 μM, respectively.

Apparent glucuronidation rates catalyzed by recombinant human UGT enzymes. Rates were determined at 20 μM nebicapone. Values represent mean ± S.E.M. of three or four determinations.
When comparing the Km values for the HLM (87 μM) and the liver-expressed UGT, specifically UGT1A6, UGT1A9, UGT2B7, and UGT2B15, it is not possible to establish which is the most important enzyme(s) involved in the conjugation of nebicapone in the liver because two of the enzymes (UGT2B15 and UGT1A9) have higher affinities than the HLM, and the other two (UGT1A6 and UGT2B7) have lower affinities.
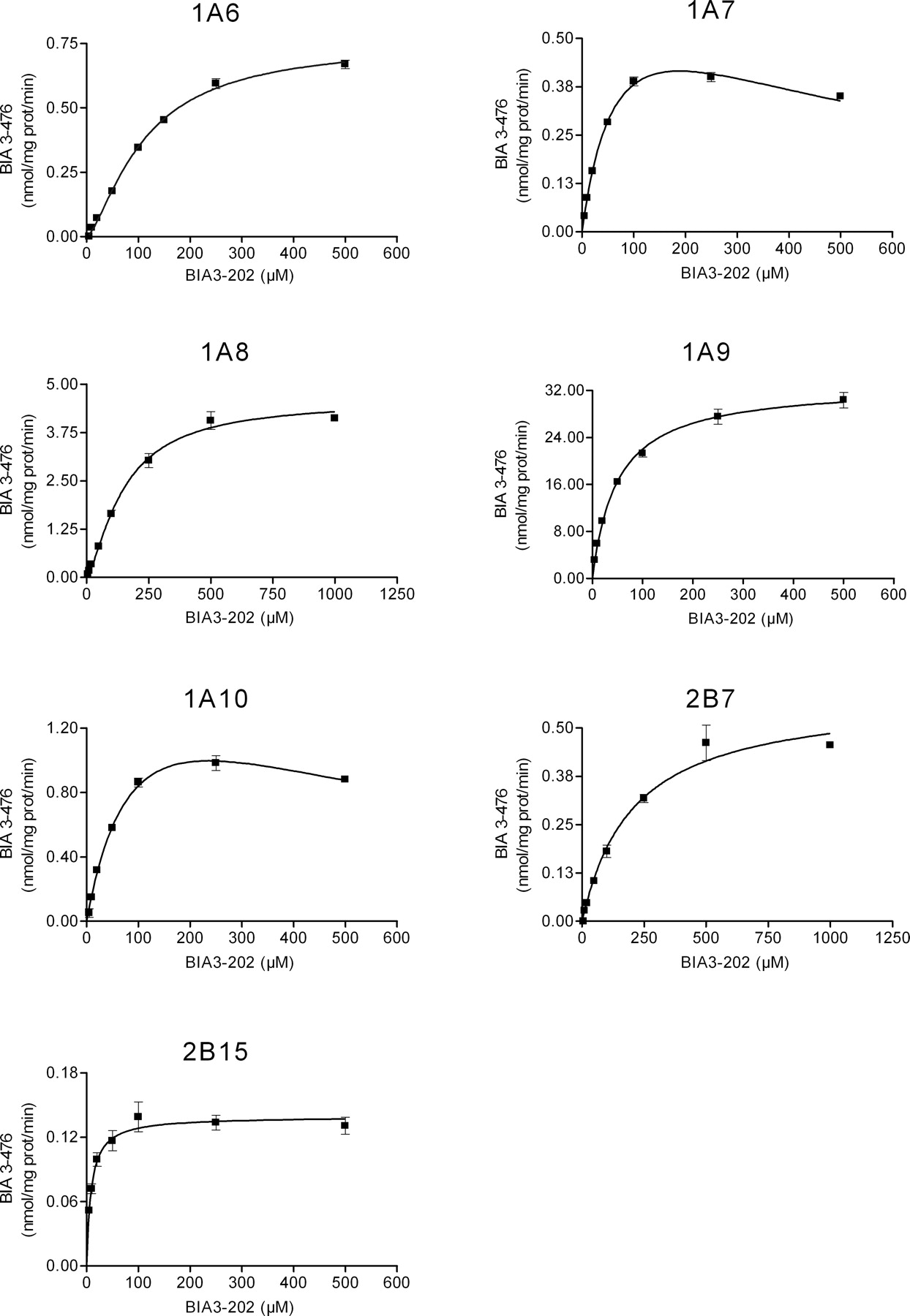
Kinetics of nebicapone glucuronidation by recombinant human UGT enzymes (UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B7, and UGT2B15). Nebicapone concentrations ranged from 1 to 1000 μM. Values represent mean ± S.E.M. of duplicates. Lines represent the fitting curves to the Michaelis-Menten equation (UGT1A9, UGT2B7, UGT2B15), Hill equation (UGT1A6, UGT1A8), or substrate inhibition equation (UGT1A7, UGT1A10) as described under Materials and Methods.
The comparison of the affinities of the intestinal-expressed enzymes (namely, UGT1A6, UGT1A8, UGT1A9, UGT1A10, UGT2B7, and UGT2B15) with the affinity of HIM (Km = 42 μM) suggests an important contribution of UGT1A9 in the intestinal conjugation of nebicapone because it is the enzyme with similar affinity to the one of HIM; however, involvement of the high affinity UGT2B15 together with one or more of the lower affinity enzymes (UGT1A6, UGT1A8, UGT1A10, and UGT2B7) cannot be excluded.
Discussion
In the work here presented, the major metabolites of nebicapone were quantified in human plasma (Fig. 6), and the kinetic parameters from UGT enzymes able to metabolize nebicapone were determined. The UGT kinetic parameters for HLM and HIM were compared in an attempt to elucidate which enzymes contribute to the nebicapone glucuronidation in vivo.
Nebicapone 3-O-glucuronide was rapidly formed and accounted for most of the nebicapone-related material in human plasma, whereas the 3-O-methylated derivative was responsible for low but sustained nebicapone-related material in plasma over 24 h. This profile is similar to the one observed for tolcapone upon human administration (Jorga et al., 1999). The 4-O-glucuronide of nebicapone was not found in human plasma, similarly to what was observed for other nitrocatechols, such as tolcapone (Jorga et al., 1999), thus providing additional evidence for the hypothesis that nitrocatechol glucuronidation in humans is highly regioselective (Wikberg et al., 1993).

Nebicapone and metabolites detected.
Glucuronidation and sulfation are often competing reactions for phenolic substrates; however, in the case of nebicapone, glucuronidation is preferred over sulfation, which is the case of tolcapone (Jorga et al., 1999) and entacapone (Wikberg et al., 1993).
N-Acetylation of nebicapone resulting from the 5-nitro group reduction was detected in some samples; however, the levels were below the limit of quantification. Limited amounts of N-acetylamino derivative of tolcapone were also reported in human plasma (Jorga et al., 1999); on the other hand, no nitro reduction was observed for entacapone (Wikberg et al., 1993).
The small amount of the nebicapone O-methylated derivative observed in this study is in agreement with the fact that nitrocatechols are poor substrates for COMT because of the strong electronegative nitro substituent (Ovaska and Yliniemela, 1998). The 3-O-methylation of nebicapone reduces the polarity of the molecule and yields the long-lived metabolite, BIA 3-270, with slower elimination that peaks 12 h after administration.
The results obtained herein indicate that the major metabolic pathway of nebicapone is glucuronidation in meta position to the nitro group of the catechol. The extensive glucuronidation of nebicapone was evaluated in vitro using human microsomes and recombinant UGT. It is clear that correlations of the activities determined with in vitro systems and the whole organism are highly limited, not only because of the differential expression of UGT in the various tissues but also because it is not possible to quantify the levels of the individual enzymes in the microsomal preparations. The use of recombinant systems further adds limitations because of the variability of recombinant enzyme expression levels. In addition, increasing evidence suggests that UGT can form dimers in vivo (Fisher et al., 2001; Miners et al., 2004), possibly heterodimers whose role is not evaluated with the recombinant enzymes.
The kinetic analysis of nebicapone glucuronidation in liver and intestine microsomes revealed an affinity for the conjugation within the same order of magnitude for the two organs, with liver having a 2-fold higher Km than intestine (87 ± 4 versus 42 ± 3 μM). The intrinsic clearance of intestine, on the other hand, was about half the one of the liver. These results suggest that first-pass glucuronidation of nebicapone may represent a significant contribution to BIA 3-476 in plasma, which is further corroborated with the rapid increase of BIA 3-476 in circulation after p.o. administration of nebicapone to humans. The efficient glucuronidation of some catechols by human stomach and intestine microsomes, similar to the one obtained in liver microsomes, has already been described (Antonio et al., 2003). There is no information available concerning glucuronidation of tolcapone or entacapone in intestine microsomes; however, both compounds are glucuronidated at high rates by human liver and kidney microsomes, with entacapone having an affinity for HLM glucuronidation (Km = 47 ± 5 μM) within the same order of magnitude of the one of nebicapone. Tolcapone was reported to have a lower affinity (Km = 201 ± 102 μM); however, the error is too large to draw any conclusion (Lautala et al., 2000).
The kinetics of nebicapone glucuronidation in HLM and HIM suggest the involvement of more than one UGT in glucuronidation. All the recombinant human UGT tested, namely, UGT1A1, UGT1A3, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B7, and UGT2B15, were capable of conjugating nebicapone, albeit at different levels. Nebicapone was shown to be a particularly good substrate for UGT1A9, suggesting a role for this enzyme in in vivo metabolism. UGT1A9 is suggested to be the major enzyme responsible for tolcapone and entacapone glucuronidation in vivo (Lautala et al., 2000). Although different recombinant enzyme sources were used for the determination of glucuronidation affinities (Lautala et al., 2000), the UGT1A9 Km for tolcapone (66 ± 12.0 μM) is similar to the Km of nebicapone obtained in this study, which is in accordance with structure similarity of these two compounds.
The comparison of the apparent Km values obtained for the recombinant enzymes evaluated with those for human microsomes did not clarify which enzymes were involved in microsomal glucuronidation. Among the hepatic-expressed UGT, UGT1A9 had the highest formation rate of BIA 3-476; however, its affinity was superior to the one of liver microsomes (50 ± 2 versus 87 ± 4 μM, respectively), clearly indicating the involvement of one or more of the lower affinity enzymes in this sample. Regarding the intestinal-expressed UGT, UGT1A9 is the enzyme with the same binding affinity in the glucuronidation of nebicapone as intestine microsomes (50 ± 2 versus 42 ± 3 μM, respectively), suggesting that it could be the major enzyme responsible for nebicapone conjugation in the intestinal microsomal sample. With the available data it is not possible to discriminate the enzymes involved in in vivo glucuronidation of nebicapone. Besides, there are other UGT that were not evaluated in the present study and may be involved. Furthermore, the enzymes involved will depend not only on the kinetics of the reaction but also on the amount of compound that reaches the respective tissue and most significantly on the enzyme levels present in the tissues.
In conclusion, glucuronidation is an important feature of nebicapone pharmacokinetics, contributing undoubtedly to nebicapone elimination, and several UGT are involved in this reaction.
Acknowledgments
We thank Nuno Palma for helpful discussions on enzyme kinetics.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.010447.
-
ABBREVIATIONS:l-DOPA, l-3,4-dihydroxyphenylalanine; PD, Parkinson's disease; AADC, aromatic l-amino acid decarboxylase; COMT, catechol-O-methyltransferase; UGT, UDP-glucuronosyltransferase(s); HPLC/MS, high-performance liquid chromatography/mass spectrometry; nebicapone, BIA 3-202, 1-[3,4-dihydroxy-5-nitrophenyl]-2-phenyl-ethanone; BIA 3-467, 1-(5-acetamido-3,4-dihydroxyphenyl)-2-phenyl-ethanone; BIA 3-476, 1-(3-O-β-D-glucopyranuronosido-4-hydroxy-5-nitrophenyl)-2-phenyl-ethanone; BIA 3-465, 1-(4-hydroxy-5-nitro-3-O-sulfatophenyl)-2-phenyl-ethanone; BIA 3-270, 1-(4-hydroxy-3-methoxy-5-nitrophenyl)-2-phenyl-ethanone; HLM, human liver microsome(s); HIM, human intestinal microsome(s); AUC, area under the curve.
- Received April 5, 2006.
- Accepted June 19, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}