


Abstract
Estimation of unbound fraction of substrate in microsomal incubation media is important in accurately predicting hepatic intrinsic clearance and drug-drug interactions. In this study, the unbound fraction of 1223 drug-like molecules in human liver microsomal incubation media has been determined using equilibrium dialysis. These compounds, which include 27 marketed drug molecules, cover a much broader range of physiochemical properties such as hydrophobicity, molecular weight, ionization state, and degree of binding than those examined in previous work. In developing the in silico model, we have used two-dimensional molecular descriptors including cLogP, Kier connectivity, shape, and E-state indices, a subset of MOE descriptors, and a set of absorption, disposition, metabolism, and excretion structural keys used for our in-house absorption, disposition, metabolism, excretion, and toxicity modeling. Hydrophobicity is the most important molecular property contributing to the nonspecific binding of substrate to microsomes. The prediction accuracy of the model is validated using a subset of 100 compounds, and 92% of the variance is accounted for by the model with a root mean square error (RMSE) of 0.10. For the training set of compounds, 99% of variance is accounted for by the model with a RMSE of 0.02. The performance of the developed model has been further tested using the 27 marketed drug molecules with a RMSE of 0.10 between the observed and the predicted unbound fraction values.
In drug discovery, the ultimate goal of a medicinal chemist is to rationally design compounds that are safe and efficacious at a marketable dose regimen. Such chemical design efforts frequently rely upon intrinsic clearance (CLint) and P450 inhibitory potency (IC50) estimates derived from human liver microsomal incubations. With relatively few assumptions, these estimates can be expected to have proportional implications for oral dose requirements and therapeutic windows against pharmacokinetic drug-drug interactions. For example, the classic well stirred model of hepatic clearance can be used to illustrate the directly proportional relationship between CLint and oral dose by eq. 1 (Wagner et al., 1965). Other oral dose determinants include the unbound steady-state average concentration necessary for the desired pharmacologic response (Css, u), dose interval (τ), and fraction of dose absorbed (fa):  In addition, by assuming that inhibition of P450 occurs via a competitive mechanism, eq. 2 is a valid and commonly used approach to calculate the therapeutic window against pharmacokinetic drug-drug interactions:
In addition, by assuming that inhibition of P450 occurs via a competitive mechanism, eq. 2 is a valid and commonly used approach to calculate the therapeutic window against pharmacokinetic drug-drug interactions: 
It has now been well established in the scientific literature that nonspecific binding of compounds into microsomal phospholipids will confound the experimental estimation of CLint and IC50 by decreasing the fraction of unbound drug (fumic) to metabolizing enzymes within the incubation (Kalvass et al., 2001; Margolis and Obach, 2003; Obach, 1997, 1999; Tran et al., 2002). In such cases, estimates of CLint and IC50 derived from microsomes are best deemed “apparent” (i.e., CLint, app and IC50, app) because they represent a mixture of kinetic determinants with both relevant (CLint and IC50) and irrelevant (fumic) implications for drug design by eqs. 3 and 4 (Obach, 1996): 

From these relationships, it follows that design efforts based simply on CLint, app and IC50, app values will be misguided in the presence of significant nonspecific binding (i.e., fumic « 1). This can be particularly damaging, considering the general influence of lipophilicity over CLint (increases), IC50 (decreases), and fumic (decreases). Thus, it is important to note that microsomal binding may mask worsening values of CLint and IC50 and entice design efforts into increasingly lipophilic space. It is also important to note that extensive nonspecific microsomal binding is common among both marketed drugs and chemical libraries of the pharmaceutical industry, particularly among lipophilic chemical entities. Thus, it is essential for these kinetic parameters to be corrected for fumic to ensure effective design in drug discovery (Obach, 1996; Tucker et al., 2001; Bjornsson et al., 2003; Grime and Riley, 2006). Unfortunately, experimental methods for the determination of fumic (e.g., equilibrium dialysis and ultracentrifugation) are far less amenable to high throughput screening than are microsomal stability incubations, thereby limiting the flow of information available to support design efforts. The need for this information has led to the development of several in silico methods for predicting fumic from physicochemical properties (most notably, LogD and LogP) (Austin et al., 2002, 2005; Hallifax and Houston, 2006; Sykes et al., 2006). Although the number of compounds used to develop these published models is limited (e.g., 56) and the data are gathered from a variety of sources (e.g., different laboratories, conditions, and species), the results indicate that fumic is highly amenable to prediction via physiochemical properties. This finding is perhaps not surprising, given that fumic probably represents a passive partitioning into microsomal phospholipids. In this work, we developed a novel in silico model using Cubist with human fumic data generated in our laboratory for 1223 compounds including 27 commercial available chemicals via a uniform 96-well equilibrium dialysis methodology. In addition to a relatively larger number of compounds, this data set encompasses a much broader range of physiochemical properties such as lipophilicity (LogP and LogD), molecular weight, ionization state (pKa), and degree of binding (log[fumic/(1 – fumic)]) than were examined in previous work.
Materials and Methods
Materials. Human liver microsome mix (HL-Mix-101) was generated from a pool of 59 individual livers and was obtained from BD Gentest (Woburn, MA). The protein content of the microsome mix was 20.4 mg/ml, and the cytochrome P450 concentration was determined to be 0.33 nmol/mg. The same batch of microsomes was used for all of the studies and was stored at –80°C. Albendazole, amitriptyline, carbamazepine, chlorimipramine, chlorpromazine, clozapine, colchicine, desipramine, diazepam, erythromycin, imipramine, indomethacin, ketoprofen, ketoconazole, midazolam, pimozide, piroxicam, prednisone, promethazine, propranolol, quinidine, sulindac, tenidap, tolbutamide, trimeprazine, verapamil, and warfarin were obtained from the Pfizer compound collection (Table 1). All of the Pfizer proprietary compounds were obtained from the Pfizer internal sample bank (Groton, CT). A 96-well dialysis apparatus and cellulose dialysis membrane strips with a molecular weight cutoff at 12,000 to 14,000 were purchased from HTDialysis (Gales Ferry, CT). Breathe Easy gas-permeable sealing membranes were obtained from Diversified Biotech (Boston, MA).
Physicochemical properties and observed fumic versus predicted fumic for 27 commercial compounds
Experimental Methods.Microsomal binding incubations. Microsomal binding in human liver microsomes was performed in triplicate using a 96-well equilibrium dialysis as adapted from a previous described method (Banker et al., 2003). Each dialysis membrane strip was conditioned sequentially for 15 min in deionized water, 30% ethanol, and then 0.1 M potassium phosphate buffer, pH 7.4. A conditioned membrane strip was then loaded into a 96-well dialysis apparatus. Experimental compound was diluted in dimethyl sulfoxide to a concentration of 100 μM. Human liver microsomes were thawed and diluted with 0.1 M potassium phosphate buffer (pH 7.4) to a final protein concentration of 0.76 mg/ml. Experimental compound was then further diluted 100-fold to yield 1 μM final concentration with the diluted microsomal solution. The compound and microsomes were thoroughly mixed, and 150 μl of the resulting solution was added to one side of the dialysis well as a donor side and 20 μl of this solution was retained for analysis. An equivalent 150 μl of 0.1 M potassium phosphate buffer (pH 7.4) was added to the other side of the dialysis well as a receiving side. The 96-well HTDialysis apparatus was sealed with a gas-permeable membrane and incubated at 37°C with 10% CO2 for 5 h on an orbital shaker rotating at 200 rpm. After the 5-h incubation period, the final microsomal solution and buffer samples (20 and 80 μl, respectively) were removed from each side of dialysis unit. Then 20 μlofthe microsomal solution from the donor side was mixed with 80 μl of phosphate buffer and 80 μl of buffer sample from the receiving side was mixed with 20 μl of control diluted microsomes to generate an equivalent matrix before sample extraction. Samples were frozen at –20°C until LC-MS/MS analysis.
LC-MS/MS analysis. LC-MS/MS analysis was performed by using a Shimadzu LC-20 AD gradient high-performance liquid chromatograph pump with a Shimadzu DGU-20A5 degasser (Shimadzu, Columbia, MD), a CTC PAL autoinjector (Leap Technologies, Carrboro, NC), and a PE Sciex API 3000 triple quadrupole mass spectrometer (Sciex, Thornhill, Ontario, Canada) with a turbo ionspray interface. Separation of the analyte was achieved by using an ACE C18 2.1 × 30 mm column with 5-μm particle size operated under room temperature (ACE, Chadds Ford, PA). Mobile phase system A consisted of 95% 10 mM ammonium formate (adjusted to pH 3.2 with formic acid) with 5% of acetonitrile. Mobile phase system B consisted of 95% acetonitrile with 5% 10 mM ammonium formate (adjusted to pH 3.2 with formic acid). The proportion of mobile phase B was held at 0% for 0.45 min and then linearly increased to 95% over next 0.75 min. Then mobile phase B was held at 95% for 0.2 min and returned to 0% over 0.1 min. After each injection the column was allowed to re-equilibrate for 0.2 min at 0% of mobile phase B. The system was operated with a flow rate of 0.75 ml/min.
The frozen samples were thawed at room temperature and quenched by 200 μl of acetonitrile containing an appropriate internal standard compound. The plate of quenched samples was then centrifuged at 3000 rpm at 4°C for 20 min to precipitate proteins, and 10 μl of supernatant was injected into the liquid chromatograph-mass spectrometer. Approximately 10% of the eluent was introduced into the mass spectrometer source. The source temperature of the mass spectrometer was maintained at 450°C. The most abundant product ion of each molecular ion (M + H+) was chosen in multiple reaction monitoring mode on the basis of collision-induced dissociations of precursor occurring at a collision energy of 25, 35, or 45 eV, respectively, for accommodating the needs of a large quantity of compounds and the detection sensitivity of the mass spectrometer. Quantitation of each compound was achieved by comparison of the analyte/internal standard peak area ratios of the donor side (microsomal solution) and the receiving side (buffer). The unbound fraction of each compound in human liver microsomes (fumic) was calculated according to eq. 5:  where Cb and Cm denote peak area ratio of a compound in the dialysis apparatus from buffer and microsomal solution chambers, respectively.
where Cb and Cm denote peak area ratio of a compound in the dialysis apparatus from buffer and microsomal solution chambers, respectively.

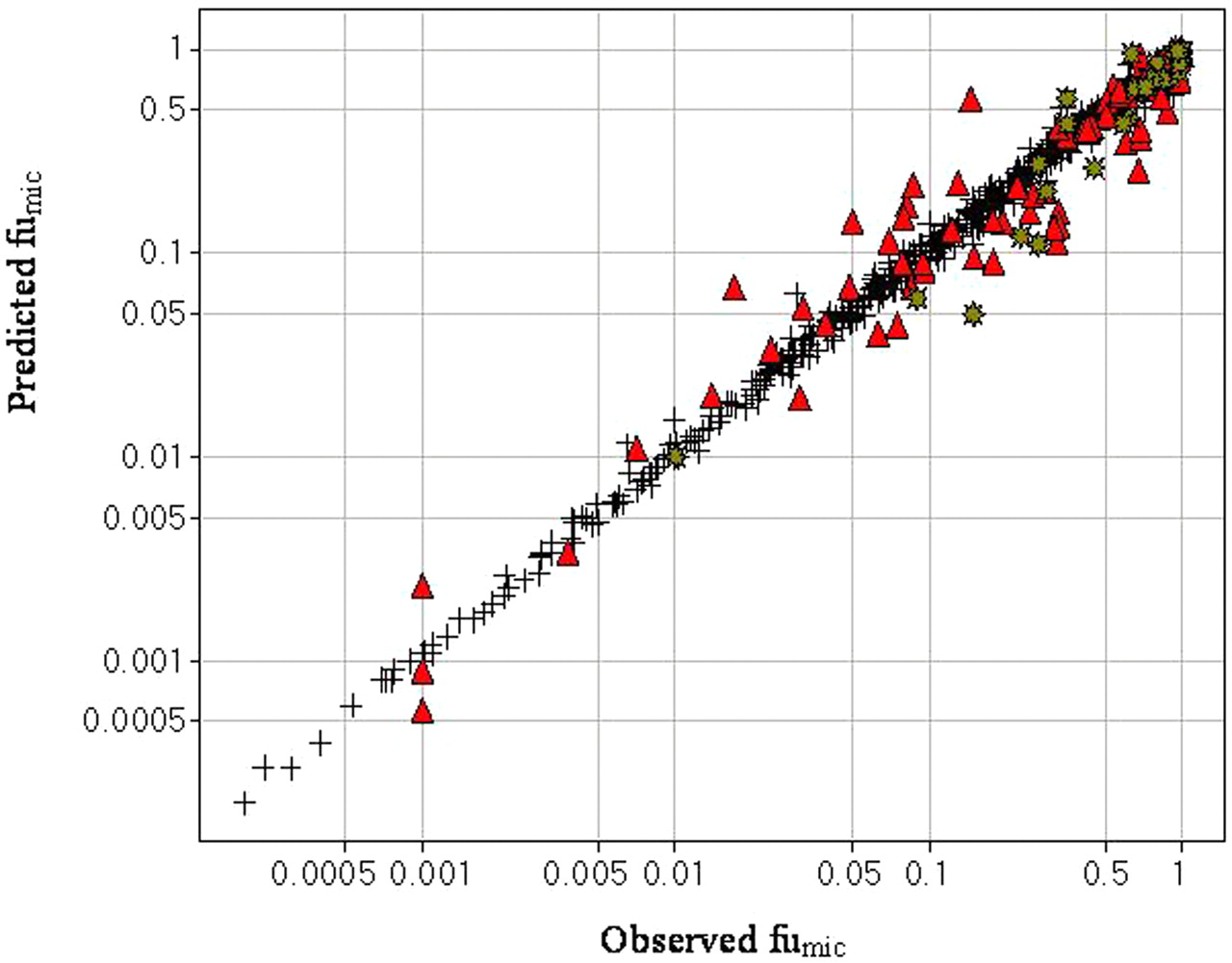
The observed versus predicted microsomal unbound fraction (fumic)(+, training set; red ▴, test set; green *, marketed drugs).
Model Development. For compounds with measurements done at different microsomal protein concentrations (internal historical data), the unbound fraction was normalized to a value at a microsomal protein concentration of 0.76 mg/ml using a modified version of an equation described previously (Kalvass and Maurer, 2002) (eq. 6):  where fumic2 is the unbound fraction at a microsomal concentration of 0.76 mg/ml and fumic1 is the unbound fraction at a microsomal concentration of C1. A logit transformation of all the data was performed according to eq. 7, which will enable the predicted fumic within a boundary of 0 to 1:
where fumic2 is the unbound fraction at a microsomal concentration of 0.76 mg/ml and fumic1 is the unbound fraction at a microsomal concentration of C1. A logit transformation of all the data was performed according to eq. 7, which will enable the predicted fumic within a boundary of 0 to 1: 
The whole set of 1223 compounds was randomly divided into two subsets. A subset of 1123 compounds was selected as a training set to develop a model, and a subset of 100 compounds was used as a validation set to validate the developed model. In addition, a set of 27 drug molecules (Table 1) was used as an external validation set to test the model performance.
In this study, we have used two-dimensional molecular descriptors including cLogP (BioByte Corp., Claremont, CA), Kier connectivity, shape, and E-state indices (Kier, 1987, 1989, 1999), a subset of MOE descriptors (Chemical Computing Group Inc., 2004, MOE 2004.03, http://www.chemcomp.com), and a set of ADME keys that are structural features used for our in-house ADMET modeling (Tu and Li, 2004; Lee et al., 2007). Some of the descriptors such as Kier shape indices contain implicit three-dimensional information. Explicit three-dimensional molecular descriptors were not used to avoid bias of the analysis due to predicted conformational effects and speed of calculation for fast prediction as stated in a previous publication (Gao et al., 1999). cLogD and pKa calculations in our analysis were performed using ACD software (version 9.03; ACD/Labs, Toronto, ON, Canada).
The statistical model was derived using Cubist committee plus composite models (Rulequest Research, 2006, http://www.rulequest.com). Cubist is a tool for generating rule-based predictive models and resembles a piecewise linear regression model, except that the rules can overlap. It does this by building a model containing one or more rules, where each rule is a conjunction of conditions associated with a linear regression. If a new case satisfies all the conditions, then the associated linear regression model is used to predict the target value. If two or more rules apply to a case, then the values are averaged to arrive at a final prediction. The predictive accuracy of a rule-based model can be improved by combining it with an instance-based or nearest-neighbor model. The latter predicts the target value of a new case by finding the n most similar cases in the training data and averaging their target values. Then Cubist combines the rule-based prediction with instance-based prediction to give a final predicted value. Prediction reliability was estimated using similarity to the training set of compounds. The similarity matrix used in the study is atom pair similarity (Carhart et al., 1985; Sheridan et al., 2004).

Distribution of measured microsomal unbound fraction (fumic) (gray, neutral compounds; blue, basic compounds; red, acidic compounds).

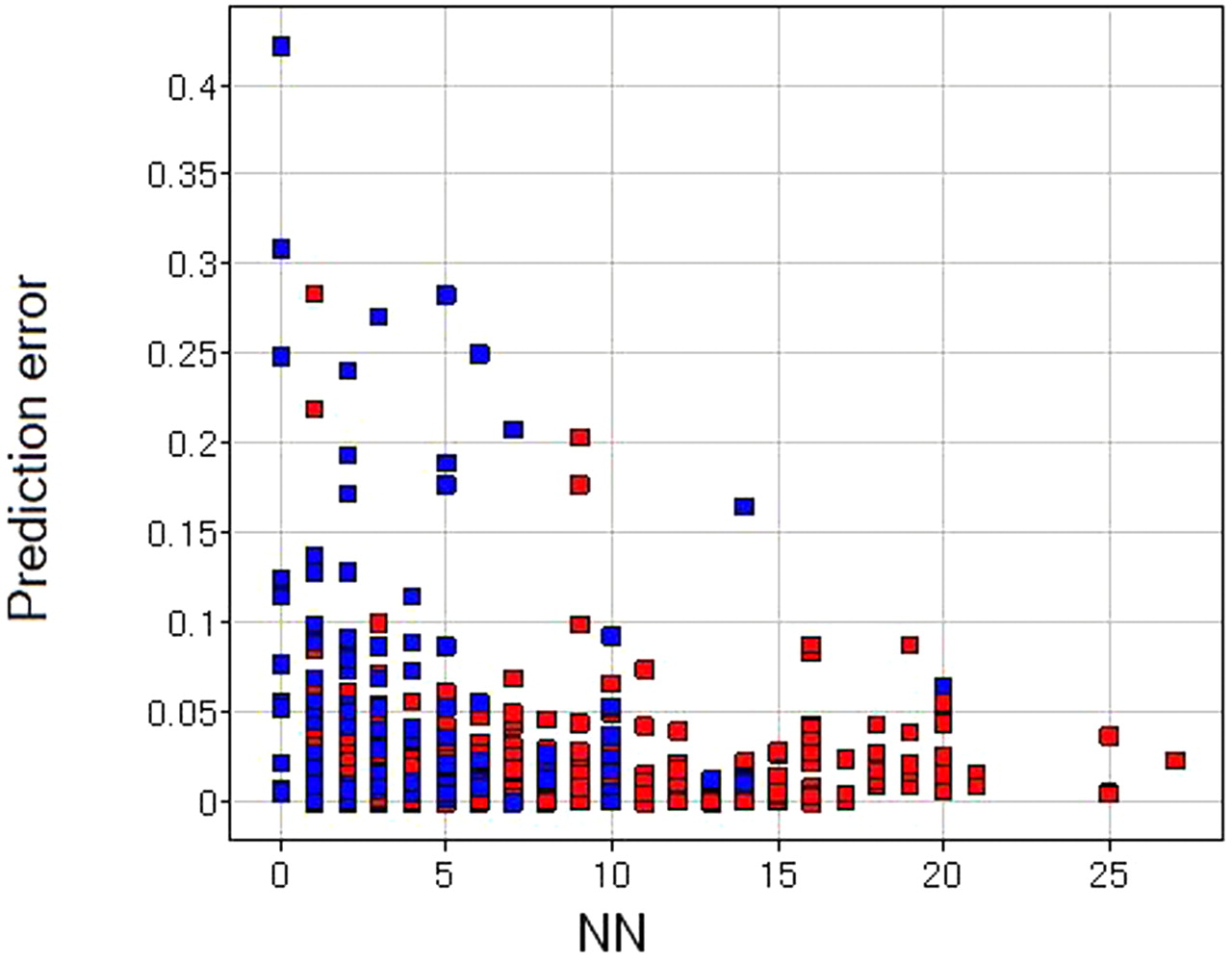
Plot of absolute prediction error versus number of NNs to the training set (red ▪, training set; blue ▪, test set).
Results
A set of 1223 Pfizer proprietary compounds was selected to cover a range of lipophilicity (cLogP from –1.45 to 9.20 and cLogD from –4.6 to 9.0), molecular weight (from 200 to 650), ionization class (from an acidic pKa of 2.0 to a basic pKa of 11.2), and degree of binding (log[fumic/(1 – fumic)] from 6 to –8). The 27 commercial compounds selected cover a range of lipophilicity (cLogP from 1.2 to 6.4 and cLogD from –1.2 to 4.8) and molecular weight (from 236 to 734), ionization class (from an acidic pKa of 3.96 to a basic pKa of 10.4), and degree of binding (log[fumic/(1 – fumic)] from –4.6 to 5.0) (Table 1). An equilibrium dialysis in a 96-well format was used to determine the extent of binding in triplicate for all of the compounds in human liver microsomes. The 27 commercial molecules were used to validate the accuracy and precision of the experimental design and were used as quality control during the 4-month study period (S.E. <10% from quadruplicates). Data from studies with recovery >80%, and coefficient of variation <20% from triplicates were included in the data set.

Relationship of microsomal unbound fraction with hydrophobicity (a, ACD LogD; b, clogP) (blue +, basic; red ▴, acidic; ○, neutral).
A predictive model was developed using Cubist. The observed and predicted microsomal unbound fractions using the developed model are plotted in Fig. 1. Because the model was built using committee plus composite models, a molecular descriptor can be used in multiple regression equations. It is not possible to describe the importance of a particular descriptor using correlation coefficients. The 9 most important molecular descriptors are cLogP, ADME Key 179 (smarts: [O,o]!:*.*), ADME Key 171 (smarts: [NH]C=[O,S,N]), kS_dO (Kier E-state for =O), vsa_don (VDW donor surface area), ADME Key 10 (smarts: *:,=[c,C;H0,H1]O[C;H1,H2,H3]), ADME Key 302 (smarts: N∼[!H]∼[N,O]), number of carboxylic groups, and 3χvc (Kier connectivity index). Hydrophobicity (cLogP) is the most important molecular property in this model. For the test set of 100 compounds, 92% of the variance is accounted for by this model with a root mean square error of 0.10, and for the training set of compounds, 99% of variance is accounted for by this model with a root mean square error of 0.02. To further test the performance of the developed model, we determined the fumic for 27 drug molecules and predicted with the model. The observed and predicted fumic values for these 27 drugs are listed in Table 1 and graphed in Fig. 1, and 52% of them are predicted within an error of 0.10.
Discussion
The distribution of measured fumic is shown in Fig. 2, and fumic from this study is evenly distributed across the whole range for neutral, basic, and acidic compounds. The applicability of quantitative structure-activity relationship models is limited by the chemistry space of data from which the models are derived. If a model is trained on a narrow chemistry space, we intuitively expect the model only to apply to a narrow range of chemical compounds. On another hand, if the training set is very diverse, covering broad chemistry spaces, we would expect the model to be applicable to a wider chemical scope and therefore more powerful in molecular design. Generally it is believed that if a new chemical structure is similar to the training set of compounds, it is more likely to be well predicted; otherwise there is a significant extrapolation and the estimated value contains high uncertainty. In this study, we have used atom-pair similarity as the similarity matrix to indicate whether a new molecule is similar to the training set of compounds. The numbers of nearest neighbors (NNs) were calculated using a similarity threshold of 0.7. This similarity index has been shown to perform well in many in-house ADMET models as well as in literature reports (Sheridan et al., 2004). For the test set of 100 compounds, 84% of them with 1 or 2 NNs are predicted within an error of ±0.1, whereas only 55% of the compounds with 0 NNs are predicted within an error of ±0.1. Similarly, 52% of the 27 commercial molecules are predicted within an error of ±0.10. The relationship of prediction error against number of NNs is plotted in Fig. 3, and it is apparent that the precision of the prediction increases with the increase of NNs.
The nonspecific binding of compounds to in vitro microsomal incubation matrix is believed to be mainly due to the partitioning of compounds to the hydrophobic phospholipids compartment. It is expected that the most important molecular property underlying the nonspecific binding is molecular hydrophobicity. The correlations of unbound fraction with calculated hydrophobicity parameters cLogD and cLogP are shown in Fig. 4. A similar observation has also been found for rat microsomal binding (Austin et al., 2002). Within a similar cLogD range, basic compounds tend to have lower unbound fractions than neutral and acidic compounds, and this is mainly due to the fact that positively charged molecules have higher affinity to phospholipids. It has also been noted that there is no significant difference among the three classes of compounds when plotted against cLogP, which is different from the phenomena of plasma protein binding (fuplasma). In plasma protein, acidic compounds in general are more tightly bound than basic compounds. As Fig. 5 shows, fumic does not track well with fuplasma. This demonstrates that fumic and fuplasma are unlikely to cancel out each other in the prediction of human clearance using well stirred or parallel-tube models (Obach, 1996). Both the unbound fraction in microsomal incubation media and the unbound fraction in human plasma are important in estimating in vivo clearance from in vitro metabolism data.

Plot of unbound fraction in microsome (fumic) and in plasma (fuplasma).

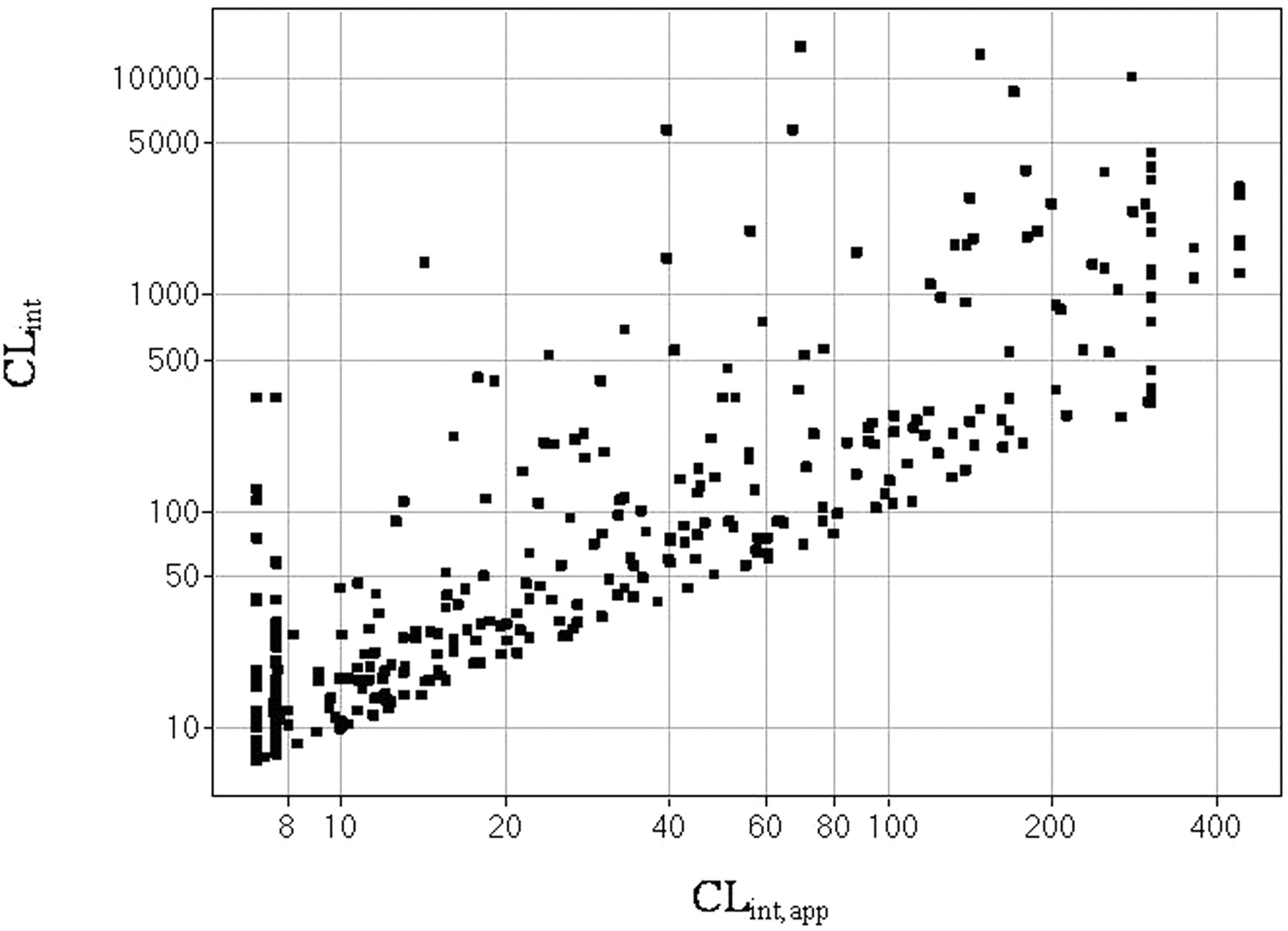
The observed in vitro apparent human microsomal clearance (CLint, app) versus calculated unbound clearance (CLint).
The free concentration of compound in microsomal incubation media significantly affects the observed in vitro CLint, app (eq. 3) and IC50, app (eq. 4). The observed CLint, app versus unbound CLint (corrected by fumic using eq. 3) is plotted in Fig. 6, and it has been noted that for compounds with low fumic («1), the difference can be more than 1000-fold, where the observed CLint, app was adapted from a method described previously (Obach, 1999). Nonspecific binding to incubation matrix renders fewer molecules available being metabolized by P450 enzymes, thus masking the true metabolic liability of a compound. A retrospective Pfizer internal analysis has shown that incorporation of human microsomal and plasma binding parameters could significantly increase the prediction accuracy of human pharmacokinetic parameters, including hepatic metabolic clearance and P450 inhibitory potency (data not shown). This finding provides a strong driving force to develop a predictive in silico model to estimate nonspecific binding to microsomal incubation media that will further assist molecule design with appropriate pharmacokinetic profiles to achieve a desirable efficacious concentration and safety window.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.020131.
-
ABBREVIATIONS: P450, cytochrome P450; LC, liquid chromatography; MS/MS, tandem mass spectroscopy; ADME, absorption, disposition, metabolism, and excretion; ADMET, absorption, disposition, metabolism, excretion, and toxicity; NN, nearest neighbor.
- Received December 13, 2007.
- Accepted July 3, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}