


Abstract
P-glycoprotein (P-gp) is an ATP-dependent efflux transporter highly expressed in gastrointestinal tract and multidrug resistance tumor cells. Inhibition or induction of P-gp can cause drug-drug interactions and thus influence the effects of P-gp substrate drugs. Previous studies indicated that 20(S)-ginsenoside Rh2 [20(S)-Rh2] could synergistically enhance the anticancer effects of conventional chemotherapeutic agents at a nontoxic dose. The aim of present study was to investigate in vitro and in vivo whether 20(S)-Rh2 was a P-gp inhibitor and analyze the possible inhibitory mechanisms and potential herb-drug interactions. Results showed that in vitro, 20(S)-Rh2 significantly enhanced rhodamine 123 retention in cells and decreased the efflux ratio of digoxin, fexofenadine, and etoposide, which were comparable to the effects of the established P-gp inhibitor verapamil. However, the transport of 20(S)-Rh2 suggested that 20(S)-Rh2 was not a P-gp substrate. Furthermore, the inhibitory effect persisted for at least 3 h after removal of 20(S)-Rh2. Unlike P-gp substrates, 20(S)-Rh2 inhibited both basal and verapamil-stimulated P-gp ATPase activities. It also significantly decreased UIC2 binding fluorescence, a marker for conformational change of P-gp. In situ and in vivo experiments showed that 20(S)-Rh2 increased the area under the plasma concentration-time curve and maximum plasma concentration of digoxin, fexofenadine, and etoposide significantly without affecting terminal elimination half-time. Long-term treatment with 20(S)-Rh2 failed to affect intestinal P-gp expression in vitro and in vivo. In conclusion, 20(S)-Rh2 is a potent noncompetitive P-gp inhibitor, which indicates a potential herb-drug interaction when 20(S)-Rh2 is coadministered with P-gp substrate drugs. It could increase the absorption of P-gp substrate drugs without long-term induction of P-gp expression in rats.
Introduction
Traditional Chinese medicine has been an essential part of the health care system in several Asian countries for thousands of years (Qiu, 2007) and is also considered a complementary or alternative medical system in most Western countries. There is increasing evidence that traditional Chinese medicine has been widely used in combination with chemical drugs. Hence, herb-drug interactions are now emerging, and it has become necessary to investigate potential changes in drug efficacy and safety when herbs are coadministered (Chan et al., 2010). Pharmacokinetic herb-drug interactions related to metabolic enzymes have already attracted considerable attention (Rengelshausen et al., 2005). However, research on herb-drug interactions related to transporters is relatively limited, despite its important role in mediating drug disposition (Marchetti et al., 2007).
Ginsenosides are among the active ingredients of ginseng, the root of which has been used in traditional herbal remedies in Eastern Asia for more than 2000 years and have recently attracted attention worldwide. 20(S)-Ginsenoside Rh2 [20(S)-Rh2] (Fig. 1), a trace constituent in ginseng, was first isolated from red ginseng by Kitagawa et al. in 1983. It has a 20(S)-protopanaxadiol dammarane skeleton aglycone and exhibited anticancer effects in various cancer cells in vitro (Fei et al., 2002; Kim et al., 2004). However, the anticancer activity of 20(S)-Rh2 was rather weak compared with that of conventional chemotherapeutic agents. In addition, there are a few reports on the anticancer effect of Rh2 itself in vivo. Although it is not ideal to use Rh2 singly as a chemotherapeutic agent, some synergetic effects between Rh2 and chemotherapeutic agents have been reported from both in vitro and in vivo studies. Rh2 can reverse the resistance of daunomycin or vinblastine in P388/Adr cells (Hasegawa et al., 1995), synergistically enhance the activities of paclitaxel and mitoxantrone in prostate cancer cells (Jia et al., 2004; Xie et al., 2006), and increase the antitumor activity dose dependently while decreasing the genotoxic effects of cyclophosphamide (Wang et al., 2006). Most important, Rh2 enhanced the inhibitory effect of cisplatin on human ovarian cancer cells transplanted in nude mice at a noneffect dosage without any adverse effect (Kikuchi et al., 1991). A similar chemosensitizing effect toward paclitaxel was observed in nude mice bearing a human prostate cancer LNCaP xenograft (Xie et al., 2006). However, despite these compelling findings, the synergistic mechanisms of 20(S)-Rh2 remain unclear, and most studies have focused on pharmacodynamic responses in vitro, such as cell cycle arrest and apoptosis. Of note though, Hasegawa et al. (1995) suspected that these synergistic effects might be related to a change in the activity of the efflux transporter P-glycoprotein.
Structure of 20(S)-Rh2.
Our laboratory has focused on the pharmacokinetics of ginsenosides for a decade. In our previous studies, we found that the oral bioavailability of Rh2 was poor as a result of its low membrane permeability and the potential efflux mediated by ATP-binding cassette transporters; it is mainly distributed in the intestine (Gu et al., 2006, 2009, 2010) where P-gp is highly expressed. In consideration of the hypothesis of Hasegawa et al. (1995) that Rh2 might interact with P-gp, high levels of 20(S)-Rh2 in the intestine may result in some interactions with some oral P-gp substrate drugs, such as etoposide (Nishimura et al., 2008) and paclitaxel (Bardelmeijer et al., 2000). The present study was designed to investigate whether 20(S)-Rh2 is a P-gp inhibitor and, if so, to elucidate its underlying inhibitory mechanism. We also sought to further analyze the potential pharmacokinetic-based herb-drug interactions related to drug transporters. In the process, this work helped to elucidate the synergetic effects of Rh2 with some chemotherapeutic agents as part of a long-term effort to identify efficient and safe P-gp modulators from natural products. This is an attractive contemporary strategy to promote the efficacy of anticancer drugs with adjunctive therapy or alternative medication, following successful examples of schisandrin B from Schisandra chinensis (Turcz.) Baill (Sun et al., 2007) and tetrandrine from Stephania tetrandra S. Moore (Fu et al., 2002).
Materials and Methods
Animals.
Male healthy Sprague-Dawley rats (180–250 g) were obtained from the Experimental Animal Breeding Center, Nanjing General Hospital of Nanjing Military Command (Nanjing, China) and were kept at room temperature (22 ± 1°C), 50 to 60% relative humidity, and automatic day-night rhythm (12-h cycle). The animals were kept in these facilities for 1 week before the experiment. Before each experiment, the rats were fasted overnight (12 h) with free access to water. Animals were randomly assigned to different experimental groups. Animal experiments were carried out in accordance with the Guidelines for Animal Experimentation of China Pharmaceutical University (Nanjing, China), and the protocol was approved by the Animal Ethics Committee of this institution.
Materials.
20(S)-Ginsenoside Rh2 (purity >98%) was purchased from Jilin University (Changchun, China). Rhodamine 123, digoxin, fexofenadine, etoposide, and verapamil were purchased from Sigma-Aldrich (St. Louis, MO). Digitoxin and loratadine were purchased from the Chinese National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). [14C]Mannitol was purchased from GE Healthcare (Little Chalfont, Buckinghamshire, UK). HPLC-grade acetonitrile and methanol were purchased from Sigma-Aldrich. Deionized water was prepared by the Milli-Q system (Millipore Corporation, Billerica, MA) and was used throughout. Ethyl acetate and all other reagents and solvents were commercially available and of analytical grade.
Cell Culture.
Caco-2 cells (HTB-37) were obtained from American Type Culture Collection (Manassas, VA). Cells were routinely cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 1% nonessential amino acids, 1 mM sodium pyruvate, and 100 U/ml penicillin and streptomycin (Invitrogen, Carlsbad, CA). P-gp-overexpressing MCF-7/Adr cells derived from parental MCF-7 cells by Adriamycin selection were obtained from Institute of Hematology and Blood Diseases Hospital (Tianjin, China) and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 100 U/ml penicillin and streptomycin.
The cells were grown in an atmosphere of 5% CO2 at 37°C, and cell medium was changed every other day. Cells were passaged upon reaching ∼80% confluence. All cells used in this study were between passages 30 and 38 and were negative for mycoplasma infection.
Cellular Retention Studies of Rhodamine 123 by Caco-2 Cells.
The retention studies were performed with confluent epithelial monolayers of Caco-2 cells grown on 24-well plates. 20(S)-Rh2 and verapamil were prepared by dissolving in ethanol and dilution with HBSS. The final concentration of ethanol was 1% (v/v). In brief, cultured cells were washed and then preincubated in HBSS (37°C, pH 7.4) containing 20(S)-Rh2 (1, 5, or 10 μM) or 1% ethanol (control) for 1.5 h, followed by the addition of 5 μM rhodamine 123. Verapamil (10 μM) was used as a positive control inhibitor. After incubation for another 2 h, the retention was stopped by rinsing the cells three times with ice-cold HBSS. Cells were lysed by three freeze-thaw cycles, and protein concentrations were measured by the method of Bradford (1976). Rhodamine 123 concentration was determined by HPLC-fluorescence detection. All experiments were conducted in triplicate.
Inhibition of P-gp Substrate Transport by 20(S)-Rh2 in Caco-2 Cell Monolayers.
In brief, Caco-2 cells with a density of 1.2 × 105 cells/insert were seeded on a permeable polycarbonate insert (Millicell cell culture inserts, 0.4-μm pore size, 12 mm diameter; Millipore Corporation) in 24-well tissue culture plates and were used for the experiment 18 to 21 days after seeding. [14C]Mannitol permeability and transepithelial electrical resistance measurements (Millicell-ERS epithelial volt-ohm meter; Millipore Corporation) were used to evaluate the integrity of Caco-2 cell monolayers. The monolayers used in transport studies had transepithelial electrical resistance values exceeding 600 Ω · cm2, and the leakage of mannitol was less than 0.3% of the dose/h.
On the day of an experiment, before initiation of transport studies, the cell monolayers were first washed with warm HBSS (pH 7.4) twice. HBSS containing 20(S)-Rh2 (1, 5, and 10 μM) or 1% ethanol (control) was then loaded into both apical and basolateral chambers. After incubation at 37°C for 1.5 h, 5 μM digoxin or 10 μM fexofenadine or 10 μM etoposide was added to either the apical or basolateral side to evaluate the transport in absorptive and secretory directions, respectively, and the cell monolayers were incubated for another 2 h. Verapamil (10 μM) was used as a positive control. At the designated time point, samples were taken from the receiving chamber for analysis. Digoxin, fexofenadine, and etoposide levels were determined by LC-MS/MS. All experiments were conducted in triplicate.
Transport Studies of 20(S)-Rh2 across Caco-2 Cell Monolayers.
Caco-2 cells were cultured in the same manner as described earlier. HBSS containing 20(S)-Rh2 (1, 5, and 10 μM) or digoxin (positive P-gp substrate) was loaded into either the apical or basolateral chambers, in the absence or presence of 10 μM verapamil (positive P-gp inhibitor). After incubation at 37°C for 1.5 h, aliquots from the receiver compartment were collected for determination of 20(S)-Rh2 or digoxin. The concentration of 20(S)-Rh2 was quantitated by LC-mass spectrometry as described previously (Gu et al., 2006).
Duration of P-gp Inhibitory Effect of 20(S)-Rh2.
To evaluate the duration of action of 20(S)-Rh2 as a P-gp inhibitor, Caco-2 cells were incubated with 20(S)-Rh2 (10 μM) or verapamil (10 μM) for 1.5 h, then were washed to discard 20(S)-Rh2 or verapamil, and were incubated for the designated time (0, 30, 60, and 180 min) in the absence of 20(S)-Rh2 or verapamil, before the addition of 5 μM rhodamine 123. The accumulation of rhodamine 123 lasted for 2 h. The intracellular concentration of rhodamine 123 was quantitated as described earlier. All experiments were conducted in triplicate.
P-gp ATPase Activity Assay.
Human P-gp-overexpressing membranes obtained from baculovirus-infected insect cells (BD Biosciences, San Jose, CA) were analyzed for both vanadate-sensitive basal and drug-stimulated ATP consumption in the absence or presence of 20(S)-Rh2. The inorganic phosphate released from ATP was measured by colorimetric detection at 880 nm to evaluate the activity of P-gp ATPase. All steps in the procedure were based on the manufacturer's protocol (5 mg/ml human P-gp membranes; BD Gentest, Woburn, MA) with minor modification. In brief, membranes (15 μg/well) prepared in Tris/MES buffer, pH 6.8 (50 mM Tris/MES, pH 6.8, 50 mM KCl, 5 mM sodium azide, 2 mM EGTA, and 2 mM dl-dithiothreitol) were incubated at 37°C on a 96-well plate for 5 min with 20(S)-Rh2 (0.5, 1, 5, 10, and 20 μM) in the presence or absence of 100 μM sodium orthovanadate (in triplicate). Verapamil (20 μM) was used as a positive control. To initiate the reaction, 12 mM Mg-ATP (20 μl) was added. The 60-μl reaction mixture was incubated at 37°C for 60 min and then was terminated by addition of 10% SDS containing antifoam A (30 μl). The inorganic phosphate released was detected by incubation at 37°C for 20 min with 200 μl of detection reagent [1:4 (v/v) mixture of 35 mM ammonium molybdate in 15 mM zinc acetate (pH 5.0) and 10% ascorbic acid]. The drug-stimulated ATPase activity (nanomoles per minute per milligram of protein) was determined as the difference between the amounts of inorganic phosphate released from ATP in the absence and presence of vanadate. For the inhibition studies of 20(S)-Rh2, verapamil-stimulated activity was assayed in the presence of 20 μM verapamil plus or minus increasing concentrations of 20(S)-Rh2 (1, 5, and 10 μM), using the method mentioned above.
MDR1 Shift Assay.
The MDR1 shift assay was performed according to the manufacturer's protocol (MDR1 Shift Assay; Millipore Bioscience Research Reagents, Temecula, CA). Approximately 1 × 106 MCF-7/Adr cells suspended in UIC2 binding buffer (1× phosphate-buffered saline and 1% bovine serum albumin) were prewarmed in a water bath at 37°C for 10 min, followed by incubation in the absence or presence of 20(S)-Rh2 (1 and 10 μM), 10 μM vinblastine (as P-gp competitive inhibitor), or 1 mM sodium orthovanadate (as a P-gp noncompetitive inhibitor) at 37°C for another 15 min. Then 2.5 μg of monoclonal antibody UIC2 was added. After incubation at 37°C for another 15 min, 1 ml of ice-cold UIC2 binding buffer was added to stop the reaction. The cells were washed twice, and then 250 μl of ice-cold phycoerythrin-labeled anti-mouse IgG was added. After 15 min at 4°C in the dark, samples were washed and resuspended in 0.5 ml of ice-cold UIC2 binding buffer and further analyzed by flow cytometer (Cytomics FC 500; Beckman Coulter, Fullerton, CA).
In Situ Single-Pass Perfusion Experiment in Rats.
In brief, rats were anesthetized with an intraperitoneal injection of phenobarbital sodium and placed on a heating surface to maintain a body temperature at 37°C. The abdomen was opened by a midline incision of 3 to 4 cm. An ileal segment of approximately 10 cm was isolated and cannulated on two ends with flexible plastic tubing. The ileal segment was flushed with normal saline to clean out any residual debris. Blank Krebs-Ringer buffer (128.5 mM NaCl, 4.7 mM KCl, 2.3 mM MgCl2, 3.3 mM CaCl2, 1.87 mM NaH2PO4 · 2H2O, 16.3 mM NaHCO3, 7.8 mM glucose, and 20 μg/ml phenol red, adjusted to pH 6.8 with 1.0 M phosphoric acid, 37°C), was then preinfused for 15 min by an infusion pump at a flow rate of 1.0 ml/min. After that, Krebs-Ringer buffer containing 1 μM rhodamine 123 in the absence or presence of 20(S)-Rh2 (1, 5, and 10 μM) was perfused at a flow rate of 0.2 ml/min through the intestinal segment.
Perfusate samples obtained from the outlet of ileum were collected every 20 min for 2 h. The radius and the length of the ileal segment were measured at the end of the experiment. During the whole course, care was taken to avoid disturbance of the circulatory system, and the exposed segment was kept moist with 37°C normal saline solution. Phenol red, the nonabsorbable marker for measuring water flux and correcting for water changes across the incised ileal segment, was determined using a spectrophotometer.
In Vivo Pharmacokinetic Studies.
To evaluate the effect of a single dose of 20(S)-Rh2 on the pharmacokinetics of each P-gp substrate, rats were divided into two groups with five animals each. One group received a single dose of 20(S)-Rh2 intragastrically at 25 mg/kg suspended in 0.5% sodium CMC, whereas the other received the vehicle (0.5% sodium CMC), serving as the control. Two hours later, a P-gp substrate, digoxin (0.25 mg/kg), fexofenadine (10 mg/kg), or etoposide (10 mg/kg), was given to the rats by intragastric administration.
To evaluate the effect of long-term treatment with 20(S)-Rh2 on the pharmacokinetics of etoposide, rats were divided into two groups with five animals each. One group received a dose of 20(S)-Rh2 intragastrically at 25 mg/kg suspended in 0.5% sodium CMC, once a day, for 10 successive days, whereas the other group received the vehicle (0.5% sodium CMC), serving as the control. On the 11th day, etoposide was administered intragastrically to rats at a single dose of 10 mg/kg.
In both studies, blood samples were collected before the P-gp substrate dosing and at 5, 10, 15, 30, 45, 60, 120, 240, 360, and 480 min after digoxin dosing or 5, 10, 20, 30, 45, 60, 120, 240, 420, and 600 min after fexofenadine dosing or 5, 10, 15, 30, 45, 60, 90, 120, 240, 360, and 480 min after etoposide dosing into heparinized tubes. Plasma was obtained by centrifugation at 5000g for 10 min and stored at −20°C before analysis.
Effects of 20(S)-Rh2 on the Basal Intestinal P-gp Expression In Vitro and In Vivo.
Caco-2 cells cultured routinely in 75-cm2 flasks were exposed to 1 μM 20(S)-Rh2, 10 μM 20(S)-Rh2, or ethanol (1%, v/v) as vehicle control after reaching 70% confluence. After exposure for 24, 48, and 72 h, the cells were harvested and stored at −80°C for Western blotting analysis.
The rats receiving etoposide after long-term treatment with 20(S)-Rh2 were euthanized after blood collection. Ileal segments of the intestines were immediately excised, washed with normal saline, and stored at −80°C until Western blotting analysis.
Western Blotting Analysis.
Cells or intestinal homogenates were lysed in ice-cold radioimmunoprecipitation assay lysis buffer with 0.02 mM phenylmethanesulfonyl fluoride for 30 min and then ultrasonicated for 60-s intervals in an ice bath. Samples were then centrifuged at 500g for 10 min at 4°C. The supernatant was transferred to a new tube and centrifuged at 15,000g for 60 min at 4°C. The supernatant (cytosol protein, such as β-actin) and precipitant (membrane protein, such as P-gp) were both collected and stored at −80°C until use. Protein concentrations were measured using a BCA protein assay kit (Pierce Chemical, Rockford, IL) according to the manufacturer's instructions. Samples reconstituted in SDS-polyacrylamide gel electrophoresis sample loading buffer were boiled for 5 min for protein denaturation. Protein samples were separated on an 8% SDS-polyacrylamide gel and transferred onto a polyvinylidene difluoride membrane (Millipore Corporation). After blotting, the membrane was blocked with 10% bovine serum albumin in Tris-buffered saline-Tween 20 (TBS-T) for 1 h at 37°C. Immunoblots were incubated with the primary monoclonal antibody to P-gp (1:800; clone3C3.2; Millipore Corporation) or β-actin (1:800; Bioworld Technology, St. Louis Park, MN) for 24 h at 4°C. The membrane was washed (4 × 10 min), incubated with the secondary antibody horseradish peroxidase-conjugated goat anti-mouse IgG (1:800; Boster Biological Technology, Wuhan, China) for 1 h at 37°C and then washed three times with TBS-T. The signals were detected using an enhanced chemiluminescence kit (Pierce Chemical). The P-gp protein band intensity was normalized to that of β-actin.
Analytical Methodology.
Analysis of rhodamine 123 in cell lysates and buffer samples.
In brief, an aliquot of 100 μl of sample was protein-precipitated with 300 μl of acetonitrile. After centrifugation, 20 μl of the supernatant was injected into a Shimadzu LC-10Avp system (Shimadzu, Kyoto, Japan) coupled with a fluorescence detector (RF-10AXL) with the excitation wavelength set at 485 nm and emission wavelength at 546 nm. The chromatographic conditions were as follows: column, Kromasil ODS (KR100-5C18, 250 × 4.6 mm, 5 μm; Eka Chemicals AB, Bohus, Sweden); mobile phase, 0.1% acetic acid-acetonitrile (65:35, v/v); column temperature, 40°C; and flow rate, 1 ml/min. Quantitation was based on the external standard method.
Analysis of digoxin, fexofenadine, and etoposide in cell lysates, buffer samples, and rat plasma.
An aliquot of 100 μl of sample with 10 μl of internal standard spiked was extracted by 1 ml of ethyl acetate. After centrifugation, 900 μl of the extracted organic layer was evaporated to dryness with a Thermo Savant SPD 2010 SpeedVac system (Thermo Fisher Scientific, Waltham, MA). The residue was reconstituted in 100 μl of acetonitrile, followed by another centrifugation. Then 10 μl of the supernatant was injected for LC-MS/MS analysis. The system consisted of a Finnigan Surveyor HPLC system (Thermo Fisher Scientific) and Finnigan TSQ Quantum Discovery Max system (Thermo Fisher Scientific), equipped with an electrospray ionization source. Data acquisition was performed with Xcalibur 2.0 software (Thermo Fisher Scientific). The analytical columns were a Gemini C18 column (150 × 2.0 mm, 3 μm; Phenomenex, Torrance, CA) for analysis of digoxin and a Cosmosil ODS column (5C18-MS-II, 150 × 2.0 mm, 5 μm; Nacalai Tesque, Japan) for analysis of fexofenadine and etoposide. The column and autosampler tray temperatures were 40 and 4°C, respectively. Detailed LC conditions and mass parameters are shown in Table 1.
Analytical conditions in LC-MS/MS analysis
Data Analysis.
For the transport assay, the apparent permeability coefficient (Papp) is presented in centimeters per second and calculated as in eq. 1:
 where ΔQ/Δt is the permeability rate (nanomoles per second), A is the surface area of the membrane (centimeters squared); and C0 is the initial concentration in the donor chamber (nanomolar). The efflux ratio (ER) was calculated as in eq. 2:
where ΔQ/Δt is the permeability rate (nanomoles per second), A is the surface area of the membrane (centimeters squared); and C0 is the initial concentration in the donor chamber (nanomolar). The efflux ratio (ER) was calculated as in eq. 2:
 In the single-pass perfusion assay, the absorption rate constant ka and Cout (corr) were calculated from eqs. 3 and 4:
In the single-pass perfusion assay, the absorption rate constant ka and Cout (corr) were calculated from eqs. 3 and 4:

 Drug permeability across rat ileum (Peff) was calculated from eq. 5:
Drug permeability across rat ileum (Peff) was calculated from eq. 5:
 C(PR)in and C(PR)out are the inlet and outlet phenol red (PR) concentrations, respectively, Cout (corr) is the water flux corrected outlet concentration of rhodamine 123 at specific time interval, Cin denotes the inlet rhodamine 123 concentration, Q is the flow rate through the ileal segment, and V and A are the volume and surface area of perfused segment, respectively.
C(PR)in and C(PR)out are the inlet and outlet phenol red (PR) concentrations, respectively, Cout (corr) is the water flux corrected outlet concentration of rhodamine 123 at specific time interval, Cin denotes the inlet rhodamine 123 concentration, Q is the flow rate through the ileal segment, and V and A are the volume and surface area of perfused segment, respectively.
Pharmacokinetic parameters of P-gp substrates were obtained by noncompartmental analysis using DAS (Drug and Statistics, version 2.1; Chinese Pharmacological Society, Beijing, China). The area under the plasma concentration-time curve (AUC) was calculated using the trapezoidal method. The terminal half-life (t1/2) was calculated as ln2/k, where k, the elimination rate constant, was determined from the slope of the terminal regression line.
Data are expressed as means ± S.E. Between-group comparisons were performed using Student's t test. For multiple comparisons, one-way analysis of variance followed by a post hoc test was used. The difference was considered to be statistically significant if p < 0.05.
Results
20(S)-Rh2 Increased the Cellular Retention of Rhodamine 123 by Caco-2 Cells.
Intracellular accumulation of rhodamine 123, a classic P-gp fluorescence substrate for rapid screening, was tested to explore the inhibitory potential of 20(S)-Rh2 on P-gp. 20(S)-Rh2 concentration dependently increased cellular retention of rhodamine 123 in Caco-2 cells by 1.26-, 1.92-, and 2.53-fold in the presence of 1, 5, and 10 μM 20(S)-Rh2, respectively (p < 0.01) (Fig. 2). The established P-gp inhibitor verapamil (10 μM) exhibited a potent inhibitory effect on P-gp, resulting in a significant 1.92-fold increase of intracellular accumulation of rhodamine 123 (p < 0.01).

Effects of 20(S)-Rh2 on the accumulation of rhodamine 123 in Caco-2 cells. Cells were preincubated with 20(S)-Rh2 for 1.5 h followed by a further 2-h incubation in the presence of 5 μM rhodamine. Data are the mean ± S.E. of three independent experiments. **, p < 0.01 versus control.
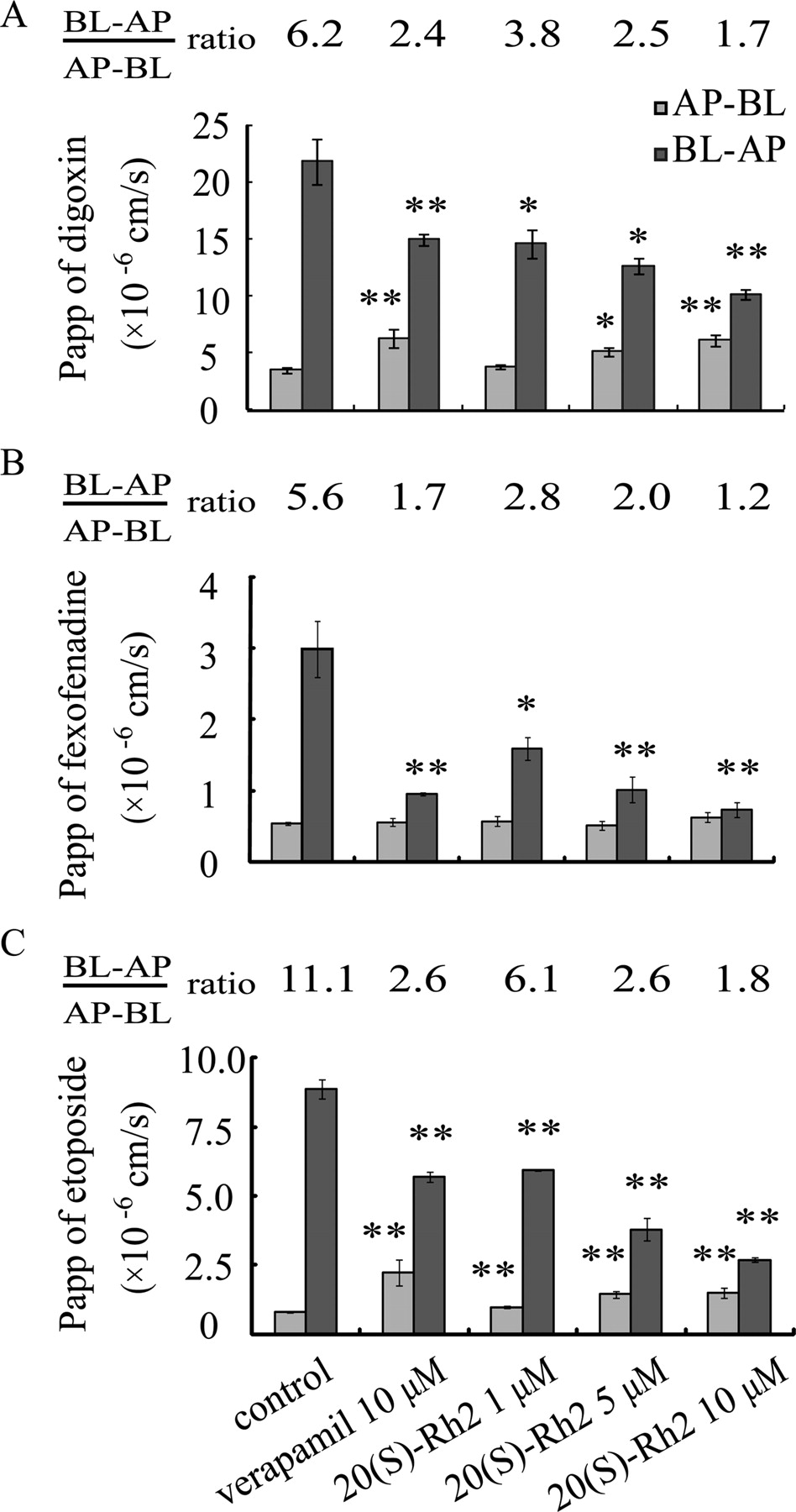
20(S)-Rh2 Decreased the Efflux Ratio of Digoxin, Fexofenadine, and Etoposide across Caco-2 Monolayers.
The flux of digoxin (5 μM), fexofenadine (10 μM), and etoposide (10 μM) across Caco-2 cell monolayers in the absorptive (AP-BL) and secretory (BL-AP) directions and the corresponding Papp values are shown in Fig. 3. Digoxin, fexofenadine, and etoposide exhibited highly polarized transport across Caco-2 cell monolayers with marked efflux ratios. The presence of 20(S)-Rh2 or verapamil (the positive control inhibitor) significantly and in a concentration-dependent manner decreased the transport of digoxin, fexofenadine, or etoposide across Caco-2 monolayers in the BL-AP direction but increased digoxin or etoposide transport in the AP-BL direction, which correspondingly led to the decrease in efflux ratios.

Effects of 20(S)-Rh2 on the transport of P-gp substrates across Caco-2 monolayers. Cells were preincubated for 1.5 h in the presence of 20(S)-Rh2 and followed by coincubation for 2 h in the presence of 5 μM digoxin (A) or 10 μM fexofenadine (B) or 10 μM etoposide (C). Data are the mean ± S.E. of three independent experiments. *, p < 0.05 versus control; **, p < 0.01 versus control.
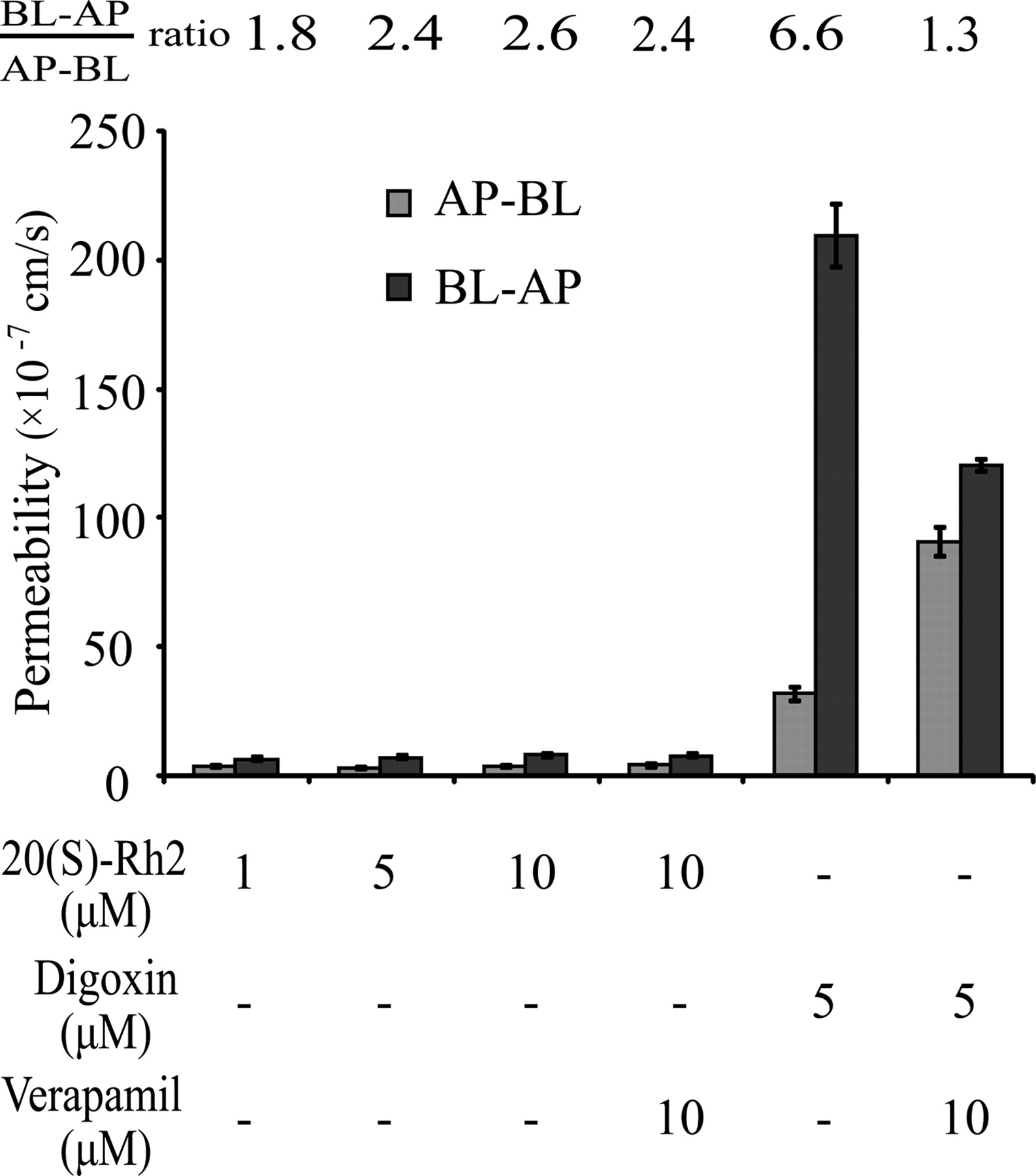
20(S)-Rh2 Exhibited No Polarized Transport Mediated by P-gp across Caco-2 Cell Monolayers.
The Papp values of 20(S)-Rh2 in both AP-BL and BL-AP directions at all concentrations were approximately 10−7 cm/s, which indicated poor transmembrane permeability of 20(S)-Rh2 (Fig. 4). In addition, the efflux ratio of 20(S)-Rh2 was approximately 2.0, which is considered a threshold for defining whether a drug is a substrate of P-gp or not. Furthermore, the efflux ratio of 20(S)-Rh2 was not changed in the presence of 10 μM verapamil. In contrast, the classic P-gp substrate digoxin possessed an efflux ratio of 6.6, which was far greater than 2.0, and this high efflux ratio of digoxin decreased significantly to 1.3 in the presence of 10 μM verapamil. Thus, 20(S)-Rh2 is not a substrate of P-gp.
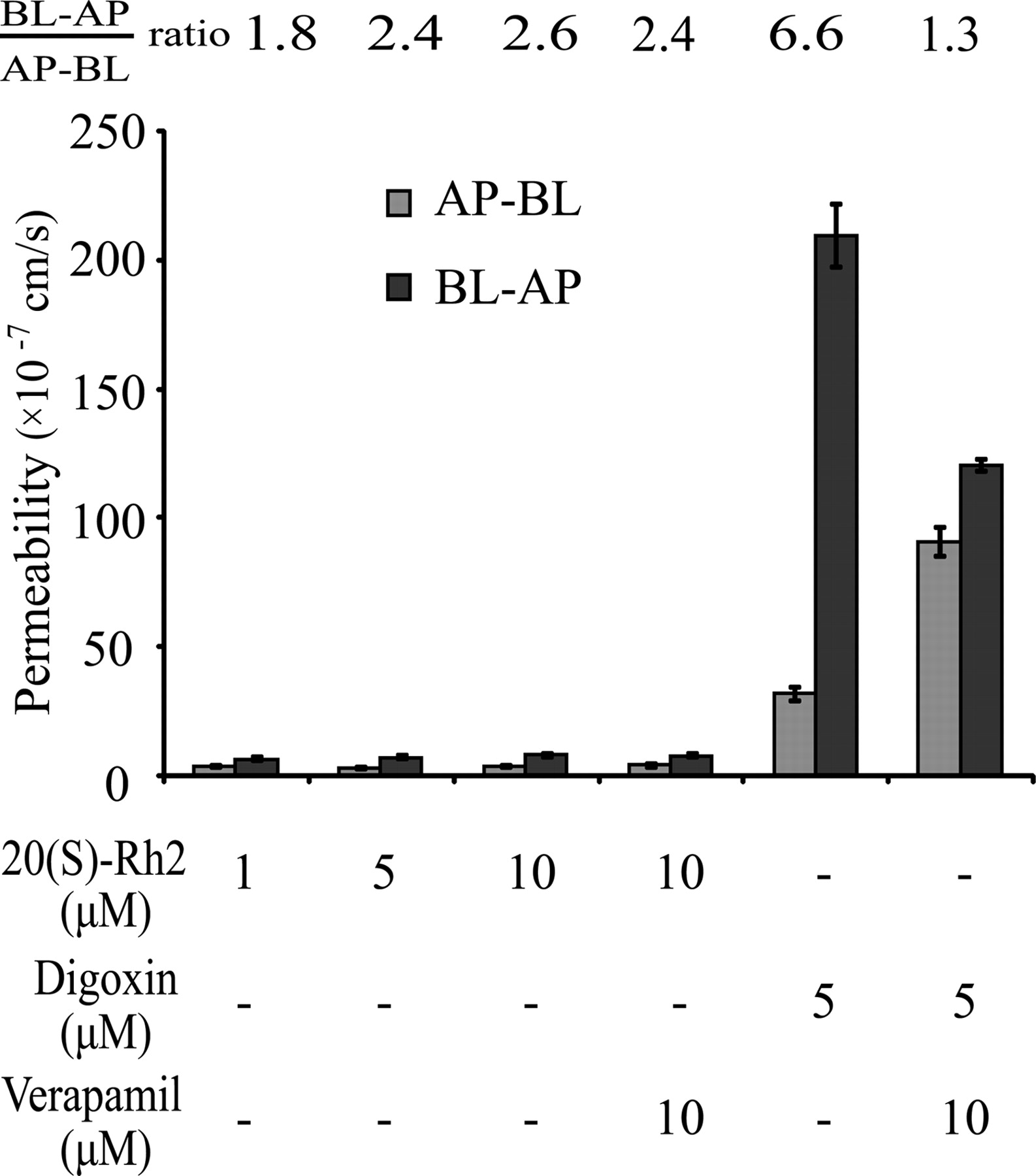
Transport characteristics of 20(S)-Rh2 across Caco-2 cell monolayers. 20(S)-Rh2 (1, 5, 10 μM) was loaded on either the AP or BL side and incubated for 1.5 h. Digoxin was used as a positive P-gp substrate control. Verapamil was used as a positive P-gp inhibitor control. Data are the mean ± S.E. of three independent experiments.
20(S)-Rh2 Inhibited P-gp Function for at Least 3 h after Being Removed.
After incubation with Caco-2 cells for 1.5 h, 20(S)-Rh2 was removed from the cell culture. Throughout the following 0 to 180 min, the P-gp inhibitory effect was still present, because the intracellular accumulations of rhodamine 123 at various time points were nearly the same as that at T0 (Fig. 5). In contrast, the competitive P-gp inhibitor verapamil lost its effect rapidly as soon as it was removed, and the intracellular accumulation of rhodamine 123 was not more than half of that at T0.

Duration of the P-gp inhibitory effect of 20(S)-Rh2. Caco-2 cells were exposed to 10 μM 20(S)-Rh2 for 1.5 h, washed, and incubated in 20(S)-Rh2-free medium for the indicated periods before the addition of 5 μM rhodamine 123 and further incubation for 2 h. T0 represents the end of the 20(S)-Rh2 incubation phase. Verapamil was used as a positive control. Data are the mean ± S.E. of three independent experiments. **, p < 0.01 versus T0.
20(S)-Rh2 Inhibited the Basal and Verapamil-Stimulated P-gp ATPase Activity.
The efflux activity of P-gp is ATP-dependent, and expression of P-gp results in the appearance of vanadate-sensitive drug-stimulated ATPase activity. Figure 6A shows the basal and verapamil-stimulated P-gp ATPase activity and also the effects of various concentrations of 20(S)-Rh2 on P-gp ATPase. Verapamil, a known substrate of P-gp, significantly stimulated ATPase activity at 20 μM. In contrast, 20(S)-Rh2 did not show stimulation at 0.5, 1, and 5 μM but exhibited an inhibition at 10 and 20 μM (p < 0.05). This result suggested that 20(S)-Rh2 may be an inhibitor but not a substrate of P-gp (Chang et al., 2006).
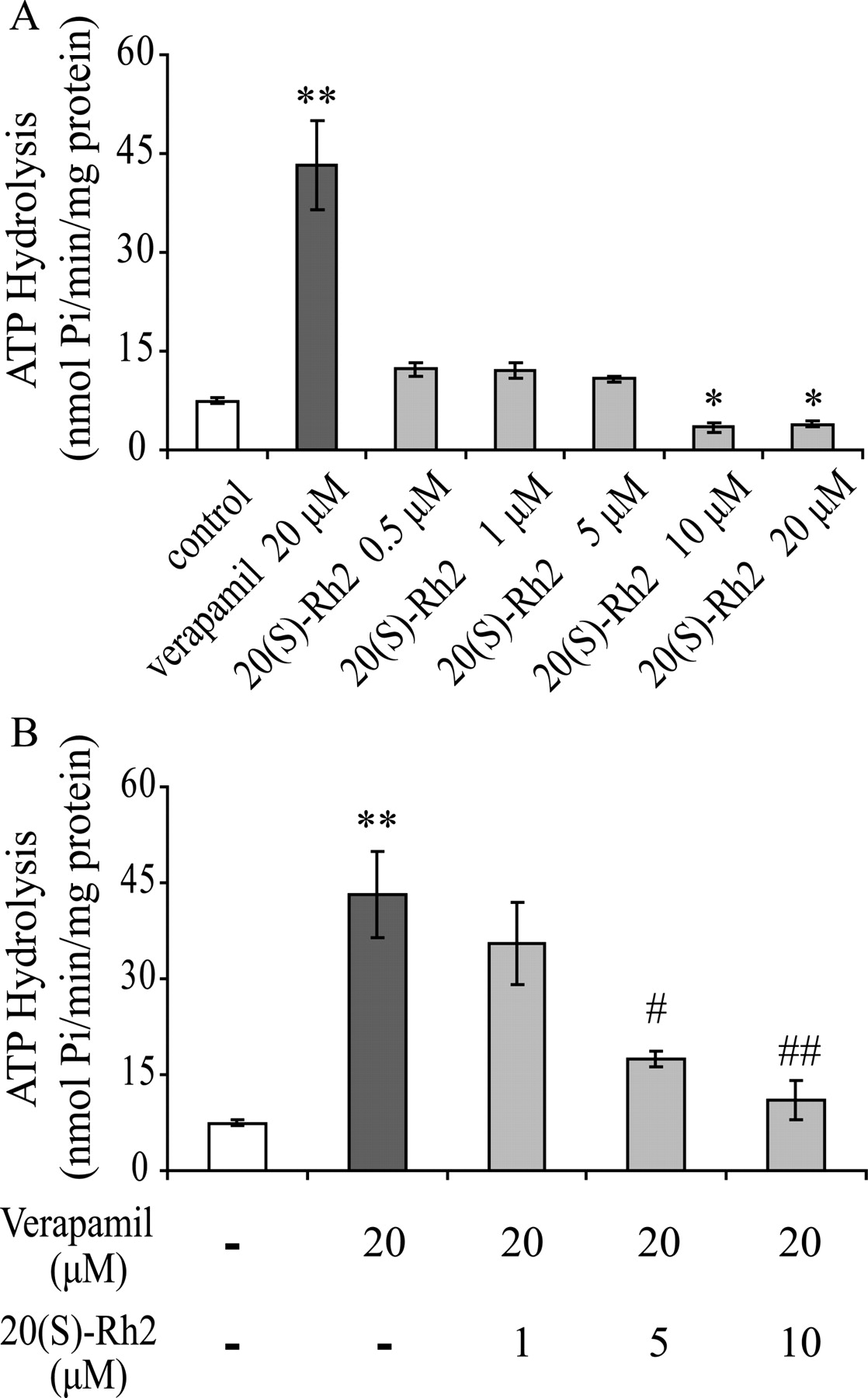
Effects of 20(S)-Rh2 on basal and verapamil-stimulated ATPase activity of P-gp. Human P-gp-overexpressing membranes obtained from baculovirus-infected insect cells were incubated with the indicated concentrations of 20(S)-Rh2 in the absence (A) or presence (B) of 20 μM verapamil. Data are the mean ± S.E. of three independent experiments. *, p < 0.05 versus control; **, p < 0.01 versus control; #, p < 0.05 versus 20 μM verapamil; ##, p < 0.01 versus 20 μM verapamil.
The effect of 20(S)-Rh2 on verapamil-stimulated ATPase activity was also investigated. The production of inorganic phosphate stimulated by 20 μM verapamil in the absence or presence of 20(S)-Rh2 (1, 5, and 10 μM) was measured. The data showed that 20(S)-Rh2 inhibited verapamil-stimulated P-gp ATPase significantly and in a concentration-dependent manner (Fig. 6B). Moreover, 10 μM 20(S)-Rh2 reduced the P-gp ATPase activity close to the basal level.
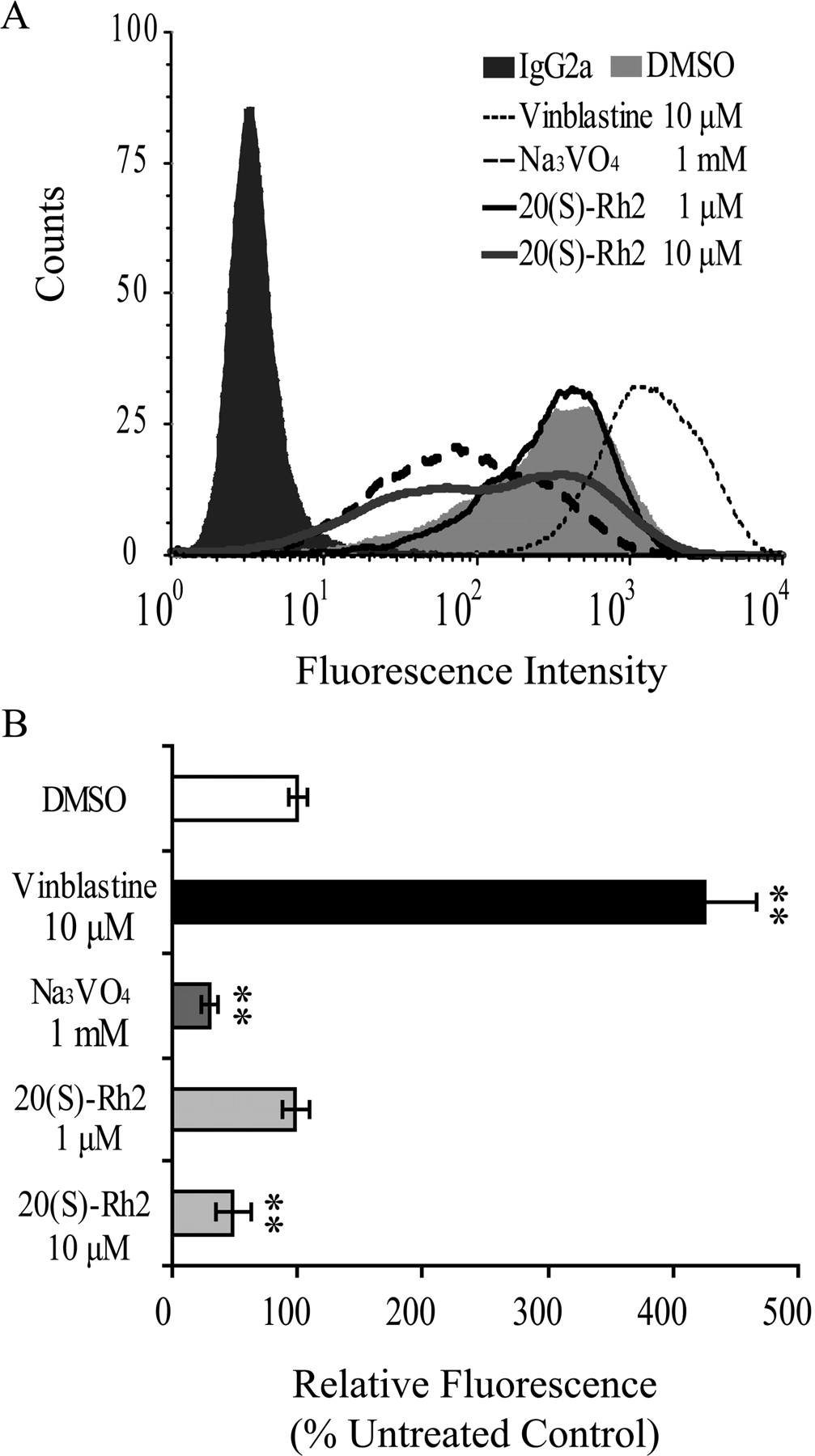
20(S)-Rh2 Changed the Binding of Conformation-Sensitive Antibody to P-gp.
UIC2, which reacts with an extracellular domain of P-gp, is a conformation-sensitive mouse monoclonal antibody against human MDR1. There is evidence that the presence of P-gp substrates and/or competitive inhibitors increases the reactivity of UIC2 drastically, whereas allosteric modulators that interfere with substrate binding indirectly reduced UIC2 reactivity (Mechetner et al., 1997; Maki et al., 2003). The results showed that the P-gp substrate and competitive inhibitor vinblastine at 10 μM increased UIC2 binding remarkably, whereas the allosteric inhibitor sodium orthovanadate decreased UIC2 binding significantly at 1 mM. As shown in Fig. 7, 20(S)-Rh2 did not influence UIC2 binding at 1 μM but caused a decrease at 10 μM, similar to sodium orthovanadate, which inhibits P-gp function by trapping it as a P-gp-ADP-vanadate complex.
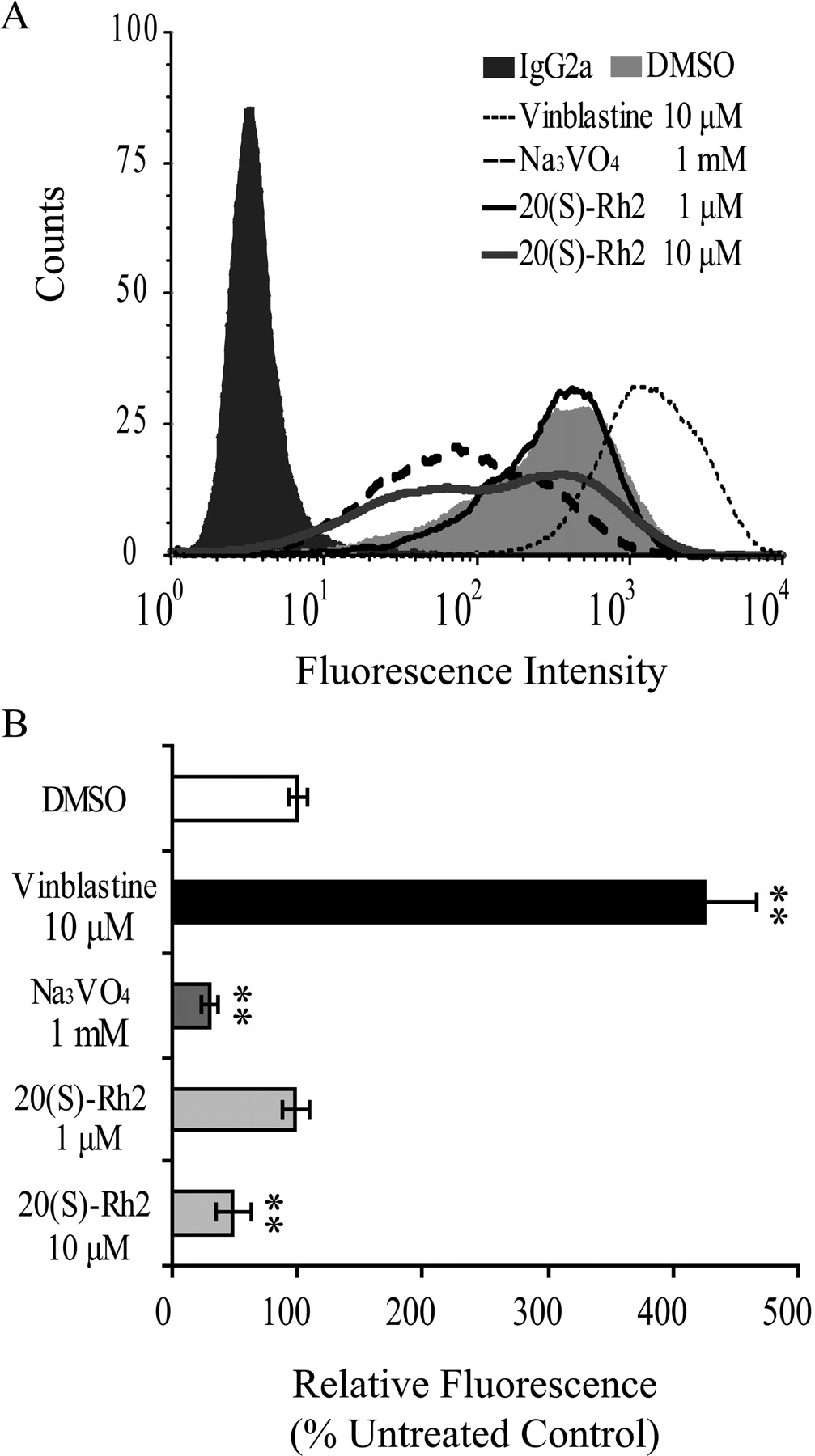
Effects of 20(S)-Rh2 on the binding of conformation-sensitive antibody UIC2 to P-gp. A, effects of 1 and 10 μM 20(S)-Rh2, 10 μM vinblastine (normal substrate control), and 1 mM Na3VO4 (ATPase inhibitor control) on UIC2 binding were studied. Cells incubated with normal IgG2a were included as a background control, and dimethyl sulfoxide (DMSO) treatment was used as a solvent control. B, results quantified from three independent experiments are presented as the mean ± S.E. **, p < 0.01 versus control.
20(S)-Rh2 Increased Rhodamine 123 In Situ Ileal Permeability in the Single-Pass Intestinal Perfusion Model in Rats.
To further explore whether the inhibitory effect of 20(S)-Rh2 on P-gp could result in herb-drug interactions with P-gp substrates in the intestinal tract, in situ single-pass intestinal perfusion experiments with rat ileum were performed to calculate the absorption rate constant and intestinal permeability of rhodamine 123 in the presence or absence of various concentrations of 20(S)-Rh2. Steady-state fluxes of rhodamine 123 in the presence of 20(S)-Rh2 (1, 5, and 10 μM) are shown in Table 2. In the presence of 20(S)-Rh2, intestinal permeability and the absorption rate constant of rhodamine 123 significantly increased in a concentration-dependent manner. These results clearly showed that 20(S)-Rh2 promoted the intestinal absorption of the classic P-gp substrate rhodamine 123 by circumvention of P-gp-mediated efflux.
Effects of 20(S)-Rh2 on absorption rate constant ka and permeability of rhodamine 123 in the single-pass intestinal perfusion model in rats
Peff and ka values are presented as the mean ± S.E.; studies were conducted in triplicate in each experimental group.
Single-Dose 20(S)-Rh2 Increased Plasma Concentrations of P-gp Substrates in Rats.
To assess whether the in vitro P-gp inhibitory effect of 20(S)-Rh2 also occurred in vivo, the pharmacokinetics of three P-gp substrates after intragastric administration were determined in Sprague-Dawley male rats with or without 20(S)-Rh2. Plasma concentration versus time curves of digoxin, fexofenadine, or etoposide after intragastric administration in the absence and presence of 20(S)-Rh2 are depicted in Fig. 8. As shown in Fig. 8A, coadministration of 20(S)-Rh2 (25 mg/kg) significantly increased the AUC of digoxin 1.67-fold, from 929.6 ± 156.0 ng/ml · min in the vehicle-treated group to 1549.3 ± 158.6 ng/ml · min in the 20(S)-Rh2-treated group. The maximum plasma concentration (Cmax) increased 1.51-fold, from 9.9 ± 1.2 ng/ml in the vehicle-treated group to 14.9 ± 1.6 ng/ml in the 20(S)-Rh2-treated group. The terminal t1/2 was not significantly changed after 20(S)-Rh2 treatment.

Plasma concentration-time curves in semilog scale of digoxin (A), fexofenadine (B), or etoposide (C) in control and 20(S)-Rh2-pretreated male SD rats. The smaller figures represent the data in normal scale. The control groups were intragastrically administered a dose of 0.25 mg/kg digoxin, 10 mg/kg fexofenadine, or 10 mg/kg etoposide, respectively. The pretreated groups were intragastrically administered a dose of 25 mg/kg 20(S)-Rh2, 2 h before digoxin, fexofenadine, or etoposide. Data are expressed as the mean ± S.E.; n = 5/group.
Fexofenadine, the metabolite of terfenadine, had been reported to be a P-gp substrate, and its intestinal absorption is controlled in part by P-gp (Petri et al., 2004; Ujie et al., 2008). Administration of 20(S)-Rh2 (25 mg/kg) 2 h before fexofenadine in rats had significant effects on plasma pharmacokinetic parameters of fexofenadine (Fig. 8B). The AUC of fexofenadine in the 20(S)-Rh2-pretreated group was 39,181.6 ± 8923.7 ng/ml · min, significantly higher than that in control group (16,206.5 ± 1803.6 ng/ml · min) by 2.42-fold, and Cmax increased by 3.46-fold (405.2 ± 128.2 ng/ml in the 20(S)-Rh2-pretreated group versus 117.0 ± 32.1 ng/ml in control group). Other parameters were not significantly changed by 20(S)-Rh2.
Etoposide, a natural product used for the treatment of malignancies, has been widely used as a P-gp substrate in experiments (Takeuchi et al., 2006; Xu et al., 2006). The plasma etoposide concentrations after a single dose of 25 mg/kg 20(S)-Rh2 were increased compared with those of control group (Fig. 8C). The AUC of etoposide was increased by 4.52-fold after 20(S)-Rh2 treatment (111,337.1 ± 28,909.2 versus 503,646.8 ± 109,286.8 ng/ml · min). In addition, 20(S)-Rh2 increased the etoposide Cmax value by 2.54-fold compared with that of the control (1062.5 ± 202.9 versus 2693.9 ± 682.1 ng/ml). The terminal t1/2 showed no difference between 20(S)-Rh2-treated and control groups.
Long-Term Treatment with 20(S)-Rh2 Failed to Affect the Pharmacokinetics of Etoposide in Rats.
As shown in Fig. 9, no significant difference in the Cmax (750.7 ± 91.8 versus 738.9 ± 78.2 ng/ml), AUC (84,564.9 ± 8347.0 versus 80,965.5 ± 6263.2 ng/ml · min), and terminal t1/2 (107.8 ± 10.6 versus 103.4 ± 11.2 min) of etoposide was observed between the control group and 20(S)-Rh2–10 successive day-treated group.

Plasma concentration-time curves of etoposide in control and 20(S)-Rh2 long-term-treated male SD rats. The control group was given blank vehicle for 10 successive days, and the long-term-treated groups were intragastrically administered a dose of 25 mg/kg 20(S)-Rh2 for 10 successive days. On the 11th day, a dose of 10 mg/kg etoposide was given administered to both groups. Data are expressed as the mean ± S.E.; n = 5/group.
20(S)-Rh2 Did Not Influence the Basal P-Gp Expression In Vitro and In Vivo.
To investigate the effects of 20(S)-Rh2 on basal P-gp expression in vitro, after Caco-2 cells were treated with 20(S)-Rh2 for 24, 48, and 72 h, respectively, the lysates were recovered and subjected to Western blot analysis. As shown in Fig. 10A, compared with control cells that were not exposed to 20(S)-Rh2, the P-gp level in the 20(S)-Rh2-treated cells was not altered significantly. In an analysis of ileum homogenate samples collected from the rats treated with 20(S)-Rh2 for 10 successive days in Fig. 10B, there was no significant difference between the control group and the 20(S)-Rh2-treated groups, which also indicated that 20(S)-Rh2 had no effect on basal intestinal P-gp expression in vivo.

Effects of 20(S)-Rh2 on P-gp expression levels in Caco-2 cells and in rat ileum. A, Caco-2 cells were exposed to 1 and 10 μM 20(S)-Rh2 or vehicle for 24, 48, and 72 h. B, expression of P-gp protein in the ileum of rats intragastrically administered vehicle or 20(S)-Rh2 at a dose of 25 mg/kg for 10 successive days. Rats were sacrificed on day 11. Top, representative blots. Lower, bar graph that shows the quantification of band intensity. Data are expressed as the mean ± S.E.
Discussion
P-gp, an extruding transporter, is highly expressed in the gastrointestinal tract and can form a biological barrier against xenobiotic agents. Thus, it can be a major determinant of drug pharmacokinetic behavior. Inhibition or induction of P-gp has been reported to be one of the causes of drug-drug interactions (DDIs) in animals and humans. On the one hand, DDIs should be avoided because they might cause severe clinical side effects. On the other hand, a DDI sometimes might benefit the patients by producing an enhanced effect from increased systemic levels of the target drug. The synergistic effect of Rh2 on anticancer therapies has already been reported (Jia et al., 2004; Wang et al., 2006; Xie et al., 2006). In the present study, the pharmacokinetic basis for this synergism was elucidated.
Caco-2 cell monolayers, which originate from a human colonic adenocarcinoma cell line, not only have been used widely as a model for drug intestinal absorption studies but also are often used as a model to analyze the possible DDIs involving P-gp, because of its rich P-gp expression in the apical membrane. It was found that 20(S)-Rh2 markedly increased the intracellular fluorescence of the P-gp substrate rhodamine 123 and inhibited the transport of three typical P-gp substrates (digoxin, fexofenadine, and etoposide), confirming that 20(S)-Rh2 is a potent P-gp inhibitor, comparable to the established P-gp inhibitor, verapamil.
In a recent study, 20(S)-protopanaxadiol, the metabolite of 20(S)-Rh2, was shown to inhibit P-gp in multidrug-resistant cancer cells (Zhao et al., 2009). Although a possible contributor to the interaction, only minor biotransformation of 20(S)-Rh2 into 20(S)-protopanaxadiol in Caco-2 cells was seen in our previous study (Xie et al., 2005). Therefore, the inhibitory effects on P-gp function we observed should come mainly from 20(S)-Rh2 rather than from its metabolite, 20(S)-protopanaxadiol.
Our earlier research showed that 100 μM verapamil and 10 μM cyclosporin A enhanced the accumulation of 20(S)-Rh2 in Caco-2 cells (Xie et al., 2005). We speculated at the time that 20(S)-Rh2 might be a substrate of P-gp. There was also an alternative possibility that 20(S)-Rh2 might be a substrate of other transporters in Caco-2 cells, which could be inhibited by verapamil and cyclosporin A. In fact, cyclosporin A and verapamil are reported to be inhibitors of breast cancer resistance protein (Ozvegy et al., 2001) and multidrug resistance proteins (Bai et al., 2004). Whether 20(S)-Rh2 is a substrate for these transporters or P-gp was unclear. Therefore, the transport of 20(S)-Rh2 in Caco-2 cells was investigated further. Unexpectedly, the efflux ratio of 20(S)-Rh2 was found to be approximately 2 and was not influenced by 10 μM verapamil, indicating that 20(S)-Rh2 is not a P-gp substrate. In agreement with this finding, Liu et al., (2009) reported an efflux ratio for 20(S)-Rh2 that was close to 1, which is similar to our results.
Furthermore, we examined the duration of action of 20(S)-Rh2. The third generation P-gp inhibitors N-[2-[[4-[2-(6,7-dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)ethyl]phenyl]carbamoyl]-4,5-dimethoxyphenyl]quinoline-3-carboxamide (XR9576, taraquidar) and N-(4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide (GF120918), which were not themselves substrates, exhibited slowly reversible inhibitory effects on P-gp function after removal of the modulator from the incubation medium. In contrast, cyclosporin A and verapamil, which were known to be both P-gp inhibitors and substrates, lost their inhibitory effects rapidly (Mistry et al., 2001; Pires et al., 2006). The action of 20(S)-Rh2 was different from the classic competitive inhibitor verapamil but was similar to that of XR9576 and GF120918, because the inhibitory effect of 20(S)-Rh2 persisted at least 3 h after removal from the media.
From a pharmacological perspective, noncompetitive inhibitors, such as XR9576, can bind with high affinity to P-gp but are not themselves substrates, which prevents ATP hydrolysis and transport of cytotoxic agent out of the cell, resulting in an increased intracellular concentration (Thomas and Coley, 2003).
In consideration of the protracted duration of the inhibitory effect, we tested whether 20(S)-Rh2 affected two key steps of P-gp function: ATPase hydrolysis and P-gp conformation change. Unlike P-gp substrates, 20(S)-Rh2 neither stimulated P-gp ATPase nor increased UIC2 binding. In fact, 20(S)-Rh2 inhibited P-gp ATPase and decreased UIC2 binding at high concentrations, unlike what is expected for a P-gp substrate. Many P-gp substrates, such as verapamil, vinblastine, and ritonavir, inhibit the function of P-gp by competing for the drug-binding site in P-gp with other P-gp substrates (Dantzig et al., 1996). They stimulate P-gp ATPase and increase UIC2 binding. With regard to other noncompetitive P-gp inhibitors, sodium orthovanadate, XR9576 and cis-(Z)-flupentixol, which are not P-gp substrates, exert their effects on P-gp function via allosteric modulation by interacting with other sites in P-gp (Maki et al., 2006). Their actions are completely different from those of the competitive inhibitors, as indicated by the inhibition of P-gp ATPase hydrolysis and decrease in UIC2 binding (Maki et al., 2003). In consideration of all of the above, 20(S)-Rh2 may be a noncompetitive inhibitor that interacts with P-gp allosterically by trapping P-gp in a transition-state conformation to inhibit P-gp ATPase without competing for the substrate site.
Because 20(S)-Rh2 was shown to be a potent noncompetitive P-gp inhibitor in vitro, we next examined its effects on P-gp function in vivo. In particular, we asked whether 20(S)-Rh2 could be used as an intestinal absorption enhancer for P-gp substrate drugs on the basis of an herb-drug interaction. To answer these questions, in situ and in vivo models were adopted. The ileum, with its high level of P-gp expression, was chosen for an in situ intestinal perfusion experiment and an evaluation of the effect of 20(S)-Rh2 on drug absorption. Rhodamine 123 was used as the P-gp substrate probe, given that it had much better water solubility than other P-gp substrates of interest. In the presence of 20(S)-Rh2, there was an obvious increase in the absorption rate constant and intestinal permeability by nearly 50%, compared with the control group, which indicated marked inhibition by 20(S)-Rh2 of intestinal P-gp.
To further investigate the interactions of 20(S)-Rh2 with P-gp in vivo, pharmacokinetic studies of P-gp substrates after intragastric administration were performed in Sprague-Dawley rats with or without coadministration of 20(S)-Rh2. Three P-gp substrates were chosen, among which digoxin and fexofenadine represented prototypical P-gp probes, whereas etoposide, as a clinical anticancer drug, provided a clinically relevant case example. The oral bioavailability of Rh2 is low (Gu et al., 2009), which is a common characteristic for most botanic origin drugs such as the flavonoid chrysin (Wang and Morris, 2007). Therefore, a relatively high dose of Rh2 (25 mg/kg; but far below the toxic dose in rats) was chosen for our in vivo experiment. Results revealed that intragastric administration of 25 mg/kg 20(S)-Rh2 significantly enhanced the AUC and Cmax of all three P-gp substrates without a notable influence on the terminal elimination half-life, which suggested that 20(S)-Rh2 increased intestinal absorption but not the systemic elimination of P-gp substrates. This finding was in accordance with our in situ results. Similar transporter-based absorption enhancement in vivo was also found between the flavonoid chrysin and nitrofurantoin (Wang and Morris, 2007).
Herb-drug interactions based on P-gp inhibition are of great clinical concern. Inhibition of P-gp not only can enhance the oral bioavailability of P-gp substrates, including many anticancer drugs, but it also reversed multidrug resistance induced by chemotherapeutic agents (Lancet et al., 2009; Kwak et al., 2010). In the present study, 20(S)-Rh2 enhanced the oral bioavailability of etoposide by 4.72-fold, suggesting that 20(S)-Rh2 may have promise as an adjuvant for chemotherapeutic agents on the basis of P-gp inhibition. However, further investigation is needed to determine whether the combination of 20(S)-Rh2 with P-gp substrate drugs can induce some unexpected systemic effect.
Function and protein expression are often tightly linked. Many P-gp inhibitors can strongly induce the expression of P-gp, in addition to the direct inhibitory effect they have on P-gp function (Hou et al., 2008). We tested whether 20(S)-Rh2 could exert a similar dual action effect at the intestinal barrier that protects the body from exposure to toxins. In our experiment, intragastric administration of 20(S)-Rh2 for 10 successive days failed to affect the pharmacokinetic parameters of etoposide 24 h after the last 20(S)-Rh2 dose, which indirectly indicated that long-term treatment of 20(S)-Rh2 did not alter the expression of P-gp in the intestine. Results from a Western blotting assay directly confirmed that 20(S)-Rh2 did not influence basal P-gp expression in the intestine. Therefore, the effects of 20(S)-Rh2 after long-time treatment should be restricted to P-gp inhibition.
In summary, our results suggest that 20(S)-Rh2 is a potent noncompetitive P-gp inhibitor. It can increase the absorption/bioavailability of P-gp substrate drugs including some chemotherapeutic agents when coadministered by the oral route. Moreover, long-term treatment with 20(S)-Rh2 does not affect the intestinal expression of P-gp in rats, indicating that P-gp inhibition should be sustained with multiple dose administration.
Acknowledgments.
We sincerely thank postgraduates Xiaozhou Wen, Ying Zhou, Fang Niu, Yuan Sun, and Xuefang Cheng (Key Laboratory of Drug Metabolism and Pharmacokinetics, China Pharmaceutical University, Nanjing, China) for their assistance.
Footnotes
This work was supported by the China National Nature Science Foundation [Grants 30973583, 30801411]; China “Creation of New Drugs” Key Technology Projects [Grants 2009ZX09304-001, 2009ZX09502-004]; and the Jiangsu Province Nature Science Foundation [Grant BK2008038].
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.034793.
-
ABBREVIATIONS:
- 20(S)-Rh2
- 20(S)-ginsenoside Rh2
- P-gp
- P-glycoprotein
- HPLC
- high-performance liquid chromatography
- HBSS
- Hank's balanced salt solution
- LC
- liquid chromatography
- MS/MS
- tandem mass spectrometry
- MES
- 4-morpholineethanesulfonic acid
- MDR
- multidrug resistance
- CMC
- carboxymethylcellulose
- AUC
- the area under the concentration-time curve
- AP
- apical
- BL
- basolateral
- DDI
- drug-drug interaction
- XR9576
- N-[2-[[4-[2-(6,7-dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)ethyl]phenyl]carbamoyl]-4,5-dimethoxyphenyl]quinoline-3-carboxamide
- GF120918
- N-(4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide.
- Received May 29, 2010.
- Accepted September 13, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}