


Abstract
Several in vitro criteria were used to assess whether three methylenedioxyphenyl (MDP) compounds, the isoquinoline alkaloids bulbocapnine, canadine, and protopine, are mechanism-based inactivators of CYP2C19. The recently reported fluorometric CYP2C19 progress curve analysis approach was applied first to determine whether these alkaloids demonstrate time-dependent inhibition. In this experiment, bulbocapnine, canadine, and protopine displayed time dependence and saturation in their inactivation kinetics with KI and kinact values of 72.4 ± 14.7 μM and 0.38 ± 0.036 min−1, 2.1 ± 0.63 μM and 0.18 ± 0.015 min−1, and 7.1 ± 2.3 μM and 0.24 ± 0.021 min−1, respectively. Additional studies were performed to determine whether other specific criteria for mechanism-based inactivation were fulfilled: NADPH dependence, irreversibility, and involvement of a catalytic step in the enzyme inactivation. CYP2C19 activity was not significantly restored by dialysis when it had been inactivated by the alkaloids in the presence of a NADPH-regenerating system, and a metabolic-intermediate complex-associated increase in absorbance at approximately 455 nm was observed. In conclusion, the CYP2C19 progress curve analysis method revealed time-dependent inhibition by these alkaloids, and additional experiments confirmed its quasi-irreversible nature. This study revealed that the CYP2C19 progress curve analysis method is useful for identifying novel mechanism-based inactivators and yields a wealth of information in one run. The alkaloids bulbocapnine, canadine, and protopine, present in herbal medicines, are new mechanism-based inactivators and the first MDP compounds exhibiting quasi-irreversible inactivation of CYP2C19.
Introduction
Inhibition of cytochrome P450 (P450) enzymes is a major mechanism for metabolism-based drug interactions. Today many clinically relevant drug interactions are known to be due to impairment of metabolic clearance via mechanism-based inactivation (MBI) of various P450 forms. Mechanism-based inactivators are generally defined as compounds that are biotransformed by the catalytic mechanism of the enzyme into a species that can form heme or protein adducts or a metabolic inhibitory complex (MIC). MBI is generally of greater concern than reversible inhibition because it can result in a more profound and prolonged effect than might be anticipated from the therapeutic dose or exposure. There are some dramatic examples of this type of drug interaction, for example, the potent inhibition of CYP3A4 by the calcium channel blocker mibefradil (Wienkers and Heath, 2005; Ghanbari et al., 2006; Venkatakrishnan and Obach, 2007).
CYP2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 together metabolize more than 90% of known drugs (Wienkers and Heath, 2005; Guengerich, 2008). CYP2C19 plays an important role in the metabolism of many drugs; most of the proton pump inhibitors and some antidepressant, antipsychotic, antiepileptic, and antiplatelet drugs are metabolized by CYP2C19 (Rendic, 2002; Pelkonen et al., 2008; Wang et al., 2009). In comparison with the other major P450 forms, relatively little has been published concerning CYP2C19 inhibitors, although screening during the early phases of drug metabolism studies with new chemical entities generally relies on the availability of highly potent and selective “diagnostic” inhibitors of individual P450 forms (Pelkonen et al., 2008; Khojasteh et al., 2011). CYP2C19 is also a rare exception among the human principal drug-metabolizing cytochrome P450 enzymes in that its crystal structure is not yet available.
In vitro kinetic assessment and prediction of drug interactions attributable to reversible inhibition (e.g., competitive) and MBI rely on operationally and mechanistically distinct approaches. Time-dependent inhibition (TDI) is one of the major features that distinguish MBI from reversible inhibition. The current guidelines on investigation of drug interactions by the U.S. Food and Drug Administration (FDA) (Guidance for Industry: Drug Interaction Studies—Study Design, Data Analysis, and Implications for Dosing and Labeling, 2006; http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072101.pdf) and European Medicines Agency (Guideline on the Investigation of Drug Interactions, 2010; http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/05/WC500090112.pdf) recommend testing of drug candidates also for mechanism-based inactivator properties. A multitiered approach was recently recommended, i.e., abbreviated assays to determine at first whether new chemical entities demonstrate TDI, followed by more thorough inactivation studies for positive compounds (Grimm et al., 2009). However, development of effective screening methods for analyzing MBI is still at an early stage. The ability to use in vitro human P450 TDI data for in vivo predictions should be viewed as a prerequisite for generating the data. There are two important terms in such activity predictions, i.e., the maximal inactivation rate constant (kinact) and the inactivator concentration required for half-maximal inactivation (KI). However, first-line screening assays typically involve characterization of an IC50 value or a time-dependent shift in IC50 (Venkatakrishnan and Obach, 2007; Grime et al., 2009).
Compounds containing a methylenedioxyphenyl (MDP) or 1,3-benzodioxole moiety can inactivate P450 enzymes via MIC formation with the enzyme (Casida, 1970; Franklin, 1971). The MDP moiety is found in several drugs, pesticides, and naturally occurring compounds (Fukuto et al., 1991; Murray, 2000; Fontana et al., 2005; Kalgutkar et al., 2007). The phosphodiesterase-5 inhibitor tadalafil and the selective serotonin reuptake inhibitor paroxetine are examples of drugs that contain MDP. These two drugs are known to form MIC with human CYP3A4 and CYP2D6, respectively, resulting in mechanism-based inactivation of these enzymes (Bertelsen et al., 2003; Ring et al., 2005; Kalgutkar et al., 2007).
We reported recently a novel fluorometric progress curve analysis approach for rapid identification of TDI of CYP2C19 (Salminen et al., 2011a). The isoquinoline alkaloids bulbocapnine, canadine, and protopine contain the MDP moiety and inhibit CYP2C19 activity (Salminen et al., 2011b). The purpose of this study was to investigate the inhibition mechanism of CYP2C19 by these herbal medicine alkaloids and to apply the progress curve analysis method for evaluating the potential MBI mode of inhibition. This method was shown to yield a wealth of information in one run, and additional experiments demonstrated the quasi-irreversible nature of inhibition by these alkaloids.
Materials and Methods
Materials.
The origin of the isoquinoline alkaloids tested was described previously (Salminen et al., 2011b): protopine was isolated from Bocconia cordata Willd., bulbocapnine was isolated from Corydalis cava L., and canadine is a semisynthetic compound from berberine. Isoniazid and tranylcypromine were purchased from Sigma-Aldrich (St. Louis, MO) and were of the highest purity available. Dibenzylfluorescein (DBF) (purity >99%) and cDNA-expressed human wild-type CYP2C19 (Supersomes) (with cytochrome b5) were purchased from BD Biosciences Discovery Labware (Bedford, MA).
Progress Curve Analysis.
The progress curve analyses were performed as described recently (Salminen et al., 2011a). This analysis uses an “all-in” approach in which the enzyme (1.5 pmol of CYP2C19) is exposed simultaneously to the probe substrate dibenzylfluorescein (1 μM DBF), inhibitor or the vehicle, and the NADPH-regenerating system in a 150-μl total volume in microplate wells. The enzyme activity is monitored by measuring the fluorescence intensity in a continuous mode at 1-min intervals for 45 min at excitation and emission wavelengths of 485 and 535 nm, respectively. DBF at 1 μM and seven different concentrations of each alkaloid were used: 0.274 to 200 μM for protopine, 6.25 to 400 μM for bulbocapnine, and 0.082 to 60 μM for canadine. These inhibitor concentrations were selected to ensure a wide range of inactivation across the 45-min time course. Controls were treated similarly but without addition of inhibitor. The fluorescence data obtained were analyzed to determine the key kinetic parameters (kobs, kinact, and KI) and mechanistic information on the inactivation process. All results represent the mean of duplicate determinations.
Irreversibility of Inactivation.
To assess whether removal of unbound inhibitor from the enzyme solution would reverse the CYP2C19 inactivation, samples were dialyzed after inactivation. A single concentration of each inhibitor (causing >90% inactivation on the basis of the progress curve analysis) was incubated with CYP2C19 enzyme in the presence and absence of the NADPH-regenerating system at 37°C for 30 min before dialysis. Controls were treated similarly but with no inhibitors included. The incubation mixtures contained 0.1 M potassium phosphate buffer (pH 7.4), the inhibitor (200 μM bulbocapnine, 100 μM canadine, 150 μM protopine, 2 mM isoniazid, or 150 μM tranylcypromine), 0.1 μM CYP2C19, and the NADPH-regenerating system or the vehicle. After the 30-min incubation, the samples were dialyzed in Slide-A-Lyzer mini-dialysis units (molecular weight cutoff 3500; Thermo Fisher Scientific, Waltham, MA) against 2.0 liters of 0.1 M potassium phosphate buffer, pH 7.4 (four 500-ml treatments for 2 h each) for 10 to 12 h at 4°C. Equivalents before the dialysis and from the dialyzed samples were collected into 96-well plates for enzyme activity measurements. The reactions were initiated by addition of the NADPH-regenerating system and the substrate (1 μM), and the reactions were measured at 1-min intervals for 60 min. The known mechanism-based inactivator, isoniazid, was used as a positive control, and the known reversible inhibitor, tranylcypromine, was used as a negative control.
Spectroscopic Determination of MIC Formation.
CYP2C19 Supersomes were used to characterize MIC formation associated with the metabolism of bulbocapnine (166.7 μM), canadine (100 μM), protopine (100 μM), and tranylcypromine (100 μM). Tranylcypromine was used as a negative control because it is a known reversible inhibitor. The incubation mixtures contained 0.1 M potassium phosphate buffer (pH 7.4), 0.13 μM CYP2C19 enzyme, the inactivator, and the NADPH-regenerating system, whereas the reference sample contained the vehicle used to dissolve the inhibitor. All MIC formation experiments were initiated by the addition of the NADPH-regenerating system and maintained at 37°C for 15 min. MIC formation was observed with an EnVision 2104 multilabel plate reader (PerkinElmer Life and Analytical Sciences, Waltham, MA) by scanning from 400 to 500 nm to monitor changes in the absorbance spectra. A similar assay procedure but in the absence of the NADPH-regenerating system was also performed. All experiments were performed in duplicate. Data were analyzed using Prism 5.0 software (GraphPad Software Inc., San Diego, CA).
Determination of IC50.
IC50 values for bulbocapnine, canadine, and protopine were determined essentially as described previously (Salminen et al., 2011a,b) using the kinetic assay procedure with seven different concentrations of each inhibitor. The mixture containing the inhibitor, enzyme, and NADPH-regenerating system was first preincubated for 30 min at 37°C, after which the reactions were initiated by addition of the substrate, and enzyme activity was monitored at 1-min intervals for 30 min. All IC50 values were determined as means from duplicate determinations. Linear regions of the progress curves were used for the reaction velocity calculations. Percentages of remaining enzyme activity were plotted as a function of the logarithm of the molar concentration of inhibitor, and the curves were fitted to a sigmoid dose-response equation with Prism 4.0 software (GraphPad Software Inc.).
Results
Progress Curve Analysis.
The reaction progress curves for each of the alkaloids are shown in Fig. 1. All progress curves without the inactivator displayed linear kinetics and confirmed that the measurements were being made during the linear steady-state phase of the reaction. Significant time- and concentration-dependent inactivation of CYP2C19 was observed for all three alkaloids as deduced from the curvilinear function of the curves (Fig. 1). Thus, data analysis was performed as described previously (Salminen et al., 2011a) to obtain estimates of kobs, vi, and vs values (Fig. 1) and of KI and kinact values (Fig. 2). The plots in Fig. 2 reveal a two-step binding and inactivation mechanism as seen by the saturation at higher concentrations. The KI and kinact values determined are presented in Table 1.

Representative progress curves of CYP2C19 inactivation by bulbocapnine (A), canadine (B), and protopine (C), indicating time-dependent inhibition.
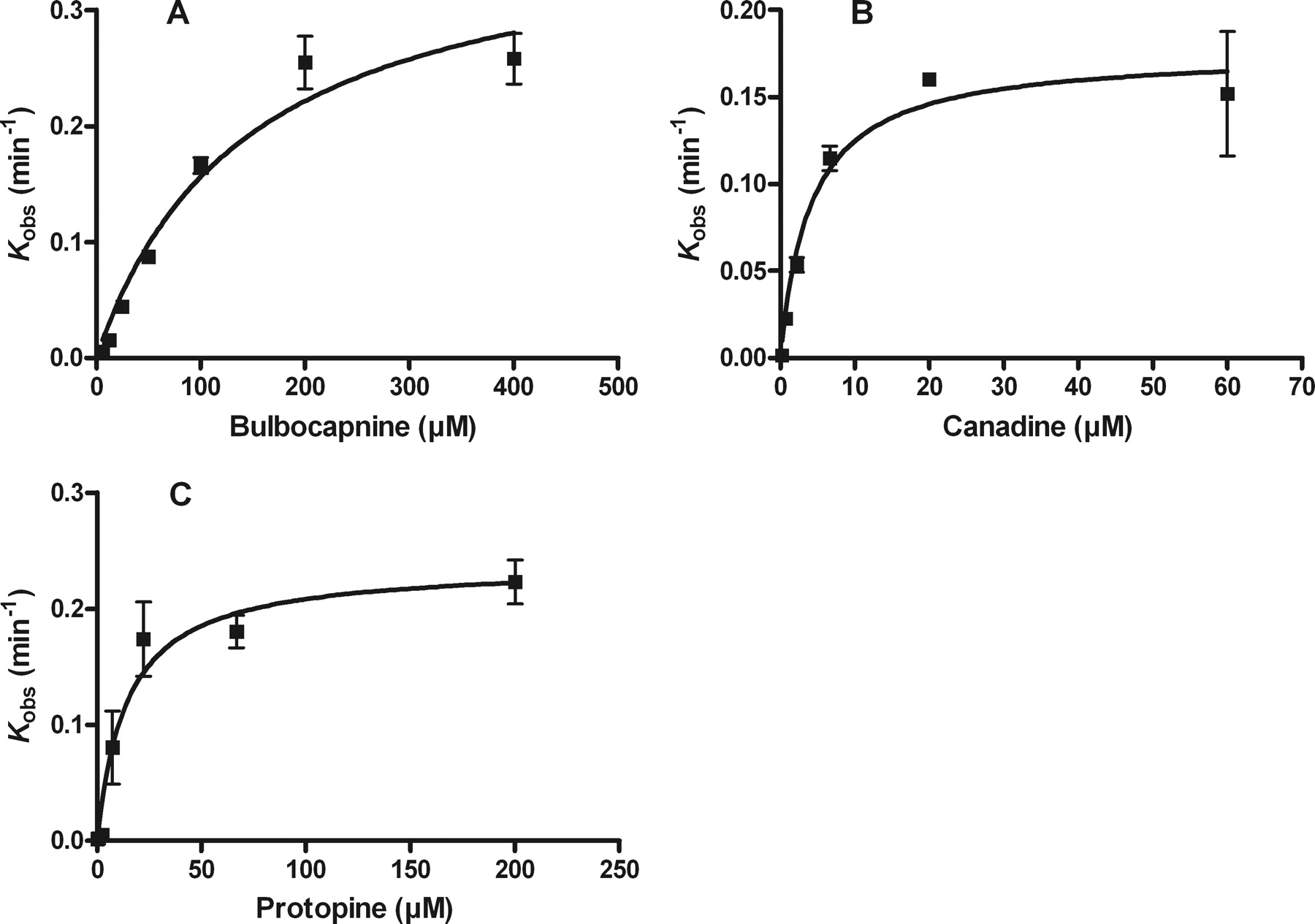
Plots of kobs against inactivator concentrations for bulbocapnine (A), canadine (B), and protopine (C), indicating a two-step binding and inactivation mechanism due to a saturable step before inactivation.
Inactivation kinetic constants for the alkaloids
Irreversibility of Inactivation.
The alkaloids investigated displayed characteristics in the dialysis experiment similar to those of isoniazid. The activity of CYP2C19, which had been inactivated by bulbocapnine, canadine, protopine, or isoniazid was not restored by dialysis or recovered only slightly when the NADPH-regenerating system was included in the inactivation mixture. In contrast, a dramatic recovery of enzyme activity could be achieved in the absence of the NADPH-regenerating system in the inactivation mixture. The activity of CYP2C19 inactivated by tranylcypromine was restored by dialysis in the presence and absence of the NADPH-regenerating system in the inactivation mixture. The results are summarized in Table 2.
CYP2C19 activity before and after dialysis
Spectroscopic Determination of MIC Formation.
The MIC- associated increase in absorbance at approximately 455 nm was observed in the case of canadine and protopine and at approximately 450 nm in the case of bulbocapnine, but no MIC-associated increase in absorbance was observed for tranylcypromine. None of the compounds produced the characteristic peak at ∼455 nm in the absence of the NADPH-regenerating system (Fig. 3).
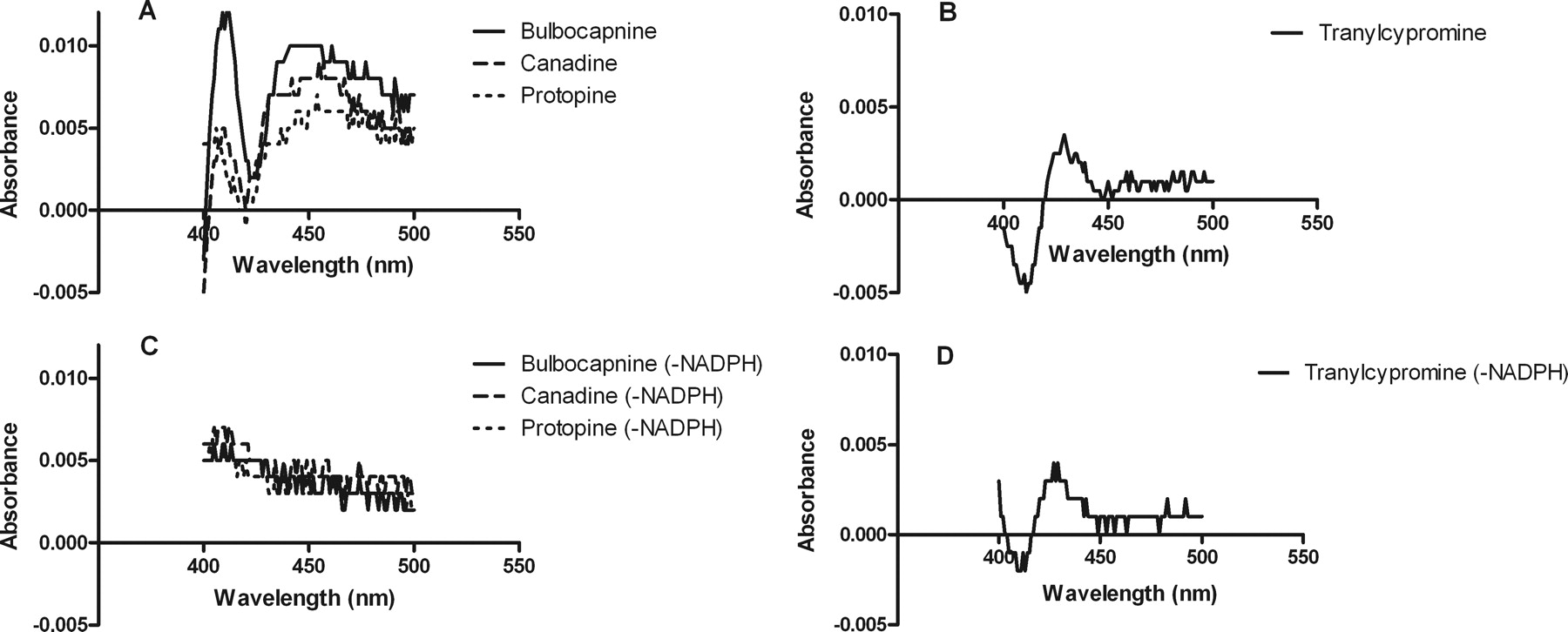
Representative difference spectra for incubations of human CYP2C19 with isoquinoline alkaloids and the negative control, tranylcypromine, respectively, in the presence (A and B) and absence (C and D) of the NADPH-regenerating system.
Determination of IC50.
The IC50 values of bulbocapnine, canadine, and protopine were assessed using the standard 30-min preincubation as recommended by the FDA (Guidance for Industry: Drug Interaction Studies—Study Design, Data Analysis, and Implications for Dosing and Labeling,, 2006, http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072101.pdf) concerning TDI, and were compared with the inhibition potency category screened earlier without actual preincubation (Salminen et al., 2011b). The inhibition potency categories of bulbocapnine and canadine changed from weak (17.3 μM) to moderately potent (8.4 μM) and from moderately potent (2.2 μM) to potent (0.29 μM), respectively (Table 3). The IC50 value determination with the 30-min preincubation did not change the inhibition potency category of protopine (2.6 versus 1.8 μM). In addition, the IC50 values of bulbocapnine, canadine, and protopine were also found to increase according to increasing substrate (DBF) concentration, indicating substrate protection against inhibition (data not shown).
IC50 values for the alkaloids
Discussion
The present work demonstrated that the recently reported (Salminen et al., 2011a) progress curve analysis is a useful screening approach for identifying novel mechanism-based inactivators of CYP2C19. Three new mechanism-based inactivators of CYP2C19 were introduced. The alkaloids bulbocapnine, canadine, and protopine displayed significant time- and concentration-dependent inactivation of CYP2C19 in the progress curve analysis, allowing for the determination of the two principal kinetic constants kinact and KI. The analysis also offered mechanistic insights into the inactivation process by revealing saturation of inactivation kinetics, indicative of a two-step inactivation mechanism.
In general, mechanism-based inactivators of various enzymes including P450s have very specific features, which make them recognizable in in vitro tests. Several methods for investigating MBI properties have appeared during the last few decades (Abeles and Maycock, 1976; Silverman, 1995; Kent et al., 2001; Fontana et al., 2005; Polasek and Miners, 2007). However, effective and sophisticated methods for the screening of MBI in P450s are still at an early stage and in vitro-in vivo correlations are limited or impossible without reasonable estimates of kinact and KI (Ghanbari et al., 2006; Grime et al., 2009; Grimm et al., 2009).
Additional experiments were performed to confirm the MBI nature of inhibition by these alkaloids. The dialysis experiment detected an NADPH-dependent difference in recovery of the enzyme activity after inactivation with bulbocapnine, canadine, and protopine, displaying characteristics similar to those of the positive control isoniazid. It is known that P450 enzymes can oxidize 1,1-disubstituted hydrazines and acyl hydrazines, such as isoniazid, to products that coordinate tightly to the heme iron atom, causing quasi-irreversible inactivation (Hines and Prough, 1980; Muakkassah et al., 1981; Correia and Ortiz de Montellano, 2005), and clinically relevant drug interactions with CYP2C19 substrate drugs have been reported (Nishimura et al., 2003; Kalgutkar et al., 2007). In contrast to irreversible inactivation, quasi-irreversible inactivation in vitro via MIC formation can be reversible after dialysis, because MIC formation does not actually destroy the enzyme. Catalytically active enzymes can also be regenerated under appropriate experimental conditions, i.e., the activity of alkylamine MICs can be disrupted in vitro by oxidation of P450 back to the ferric state using potassium ferricyanide. Under physiological conditions, however, the MIC complexes are known to be so stable that the P450 enzyme(s) involved in the complex formation would be unavailable for drug metabolism, and, hence, the pharmacokinetic impact is indistinguishable from irreversible (suicide) inhibition (Kalgutkar et al., 2007; Polasek and Miners, 2007; Riley et al., 2007; Grimm et al., 2009). For tranylcypromine, the dialysis experiment did not detect any NADPH-dependent difference either in inhibition or recovery of the enzyme activity.
Incubation of bulbocapnine, canadine, and protopine caused a NADPH-dependent increase of absorbance at approximately 455 nm (450 nm in the case of bulbocapnine), which is characteristic of the resulting ferrous complex between the heme iron atom and the inhibitory product after the catalytic action of the P450 enzyme, i.e., MIC formation (Kalgutkar et al., 2007). This is presumably due to coordination of a carbene intermediate of methylenedioxy group to heme as is the case with the selective serotonin reuptake inhibitor, paroxetine (Bertelsen et al., 2003; Correia and Ortiz de Montellano, 2005). The absorption spectrum observed with tranylcypromine is characterized by a “type II” binding spectrum with a Soret maximum at 425 to 435 nm and a trough at 390 to 405 nm, and this was independent of the NADPH-regenerating system (Fig. 3). This finding is consistent with previous reports of tranylcypromine interactions being able to elicit a type II spectral response with P450 enzymes (Taavitsainen et al., 2001; Wada et al., 2004). These results indicate that the mechanism of CYP2C19 inhibition by bulbocapnine, canadine, and protopine is quasi-irreversible and mediated via MIC formation.
The IC50 determinations were performed according to the guidelines provided by FDA (Guidance for Industry: Drug Interaction Studies—Study Design, Data Analysis, and Implications for Dosing and Labeling, 2006; http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072101.pdf) concerning TDI and potential MBI, i.e., using a preincubation step before initiation of the reactions. Increases in potency of CYP2C19 inhibition were observed after this preincubation, causing a ∼2-fold reduction in the IC50 value of bulbocapnine and protopine and a ∼7-fold reduction in the IC50 value of canadine. An increase in potency of the P450 inhibitor in the in vitro incubation or dosing period in vivo is a typical feature of TDI (Riley et al., 2007). However, this experiment does not actually provide any additional value for this research, and in the case of reversible inhibition, IC50 values could have been determined from the progress curve analysis approach, because this allows investigation of both reversible and irreversible components of the reaction mechanisms (Salminen et al., 2011a).
Several MDP compounds, for example, two marketed drugs, tadalafil and paroxetine, inhibit P450 enzymes CYP3A4 and CYP2D6 via MBI. The thienopyridine antiplatelet drug, ticlopidine, is the first and best-known mechanism-based inactivator of CYP2C19; the inactivation, which occurs by covalent binding of a reactive metabolite of ticlopidine, leads to clinically relevant drug interactions with CYP2C19 substrate drugs (Donahue et al., 1997; Tateishi et al., 1999; Ha-Duong et al., 2001; Venkatakrishnan and Obach, 2007). Bulbocapnine, canadine, and protopine are widely distributed in plants of the family of Papaveraceae (or Fumariaceae, depending on botanical classification), with occurrences in a few species of other families. Hence, they are components present in variable amounts in numerous herbal medicine preparations (Vacek et al., 2010; Gotti, 2011). Protopine is present, e.g., in preparations from the medicinal plant Fumaria officinalis, which is included in the European Pharmacopoeia (Vrba et al., 2011). A wide spectrum of biological activities have been reported for all three alkaloids, and their core structures have been used to develop lead molecules for potential treatments against a variety of diseases (Zhang et al., 2007; Jadhav et al., 2010). For example, the protoberberine skeleton has been used to develop antibacterial agents (Jadhav et al., 2010), and bulbocapnine possesses dopamine D2 receptor antagonist properties (Zhang et al., 2007). These three alkaloids are also present in several traditional Chinese medicine preparations (Salminen et al., 2011b). Thus, products containing these alkaloids have the capacity to inhibit CYP2C19 and interfere with the elimination of the drugs that are substrates of this enzyme.
In conclusion, specific in vitro criteria were used to assess whether three MDP compounds, bulbocapnine, canadine, and protopine, are mechanism-based inactivators of CYP2C19. The applied CYP2C19 progress curve analysis yielded a wealth of information in one run because the quasi-irreversible inactivation pattern by these compounds explained the TDI observed in this analysis, and it provided the principal kinetic constants and mechanistic information of the inactivation process. These herbal medicine alkaloids are novel mechanism-based inactivators for CYP2C19 and may thus interact with drugs that are CYP2C19 substrates. In addition, because a variety of different mechanism-based inactivators are known to be useful in identifying active site amino acid residues involved in substrate binding and catalysis, these alkaloids can serve as good tools for studying structural and functional elements of CYP2C19, especially in the absence of the crystal structure of CYP2C19.
Authorship Contributions
Participated in research design: Salminen and Raunio.
Conducted experiments: Salminen.
Contributed new reagents or analytic tools: Meyer and Imming.
Performed data analysis: Salminen.
Wrote or contributed to the writing of the manuscript: Salminen, Meyer, Imming, and Raunio.
Acknowledgments
We thank Hannele Jaatinen for technical help, Dr. Jarkko I. Venäläinen and Prof. Markku Pasanen for reviewing the manuscript, and Dr. Ewen MacDonald for linguistic advice and revision of the manuscript.
Footnotes
This work was supported by the Finnish Graduate School in Toxicology.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.111.041319.
-
ABBREVIATIONS:
- P450
- cytochrome P450
- MBI
- mechanism-based inactivation
- MIC
- metabolic-intermediate complex
- TDI
- time-dependent inhibition
- FDA
- U.S. Food and Drug Administration
- MDP
- methylenedioxyphenyl
- DBF
- dibenzylfluorescein.
- Received June 22, 2011.
- Accepted September 9, 2011.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}