


Abstract
In previous studies, gemfibrozil acyl-β-glucuronide, but not gemfibrozil, was found to be a mechanism-based inhibitor of cytochrome P450 2C8. To better understand whether this inhibition is specific for gemfibrozil acyl-β-glucuronide or whether other glucuronide conjugates are potential substrates for inhibition of this enzyme, we evaluated several pharmaceutical compounds (as their acyl glucuronides) as direct-acting and metabolism-dependent inhibitors of CYP2C8 in human liver microsomes. Of 11 compounds that were evaluated as their acyl glucuronide conjugates, only gemfibrozil acyl-β-glucuronide exhibited mechanism-based inhibition, indicating that CYP2C8 mechanism-based inhibition is very specific to certain glucuronide conjugates. Structural analogs of gemfibrozil were synthesized, and their glucuronide conjugates were prepared to further examine the mechanism of inhibition. When the aromatic methyl groups on the gemfibrozil moiety were substituted with trifluoromethyls, the resulting glucuronide conjugate was a weaker inhibitor of CYP2C8 and mechanism-based inhibition was abolished. However, the glucuronide conjugates of monomethyl gemfibrozil analogs were mechanism-based inhibitors of CYP2C8, although not as potent as gemfibrozil acyl-β-glucuronide itself. The ortho-monomethyl analog was a more potent inhibitor than the meta-monomethyl analog, indicating that CYP2C8 favors the ortho position for oxidation and potential inhibition. Molecular modeling of gemfibrozil acyl-β-glucuronide in the CYP2C8 active site is consistent with the ortho-methyl position being the favored site of covalent attachment to the heme. Moreover, hydrogen bonding to four residues (Ser100, Ser103, Gln214, and Asn217) is implicated.
Introduction
There have been several reports of clinical interactions between 5-(2,5-dimethylphenoxy)-2,2-dimethyl-pentanoic acid (gemfibrozil, Lopid; Parke-Davis, Ann Arbor, MI) and CYP2C8 substrates such as cerivastatin, repaglinide, rosiglitazone, and pioglitazone (Backman et al., 2002; Niemi et al., 2003a,b; Jaakkola et al., 2005). Although in vitro studies indicate that gemfibrozil is a more potent inhibitor of CYP2C9 than CYP2C8 (Wen et al., 2001), the results of clinical studies have shown that gemfibrozil is a more significant inhibitor of CYP2C8. This discrepancy was explained by demonstrating that the major metabolite of gemfibrozil, gemfibrozil acyl-β-glucuronide, is a potent inhibitor of CYP2C8 based on in vitro studies. These same authors demonstrated that gemfibrozil acyl-β-glucuronide inhibits the CYP2C8-mediated metabolism of cerivastatin in vitro, as well as the organic anion-transporting peptide mediated uptake of cerivastatin (Shitara et al., 2004). In subsequent studies, Ogilvie et al. (2006) found that gemfibrozil acyl-β-glucuronide was a direct-acting and metabolism-dependent inhibitor of CYP2C8 with a KI of 20 to 52 μM and a kinact of 0.21 min−1. When gemfibrozil was coadministered with repaglinide (a substrate for CYP3A4 and CYP2C8) in a clinical study, an increase in the area under the curve was observed for repaglinide that persisted for at least 12 h after the dose, indicating that this mechanism-based inhibition occurs in vivo (Tornio et al., 2008). The effect is rapid and is evident 1 h after gemfibrozil administration (Honkalammi et al., 2011). In a recent study, Baer et al. (2009) showed that gemfibrozil acyl-β-glucuronide was covalently bound to the heme of CYP2C8 and proposed P450-catalyzed oxidation of either of the benzylic methyl groups to form a benzyl radical intermediate (Fig. 1A).

A, reaction pathway for time-dependent inhibition of CYP2C8 by gemfibrozil acyl-β-glucuronide proposed by Baer et al. (2009). B, reaction pathway for time-dependent inhibition as proposed in this article, indicating that o-Me is the predominant configuration for oxidation to a reactive metabolite.
Other recent reports of glucuronide metabolites interacting directly with CYP2C8 have been published, including the CYP2C8-mediated hydroxylation of the acyl glucuronide of diclofenac and the 17β-O-glucuronide of estradiol (Kumar et al., 2002; Delaforge et al., 2005). Acyl glucuronides have long been known to be reactive, capable of undergoing reactions such as hydrolysis, rearrangement, and covalent binding to proteins, which possibly result in pharmacological or toxicological effects (Bailey and Dickinson, 2003).
The purpose of this study was to determine whether the acyl-β-glucuronide of gemfibrozil is a unique mechanism-based inhibitor of CYP2C8 or whether other glucuronide conjugates are capable of inhibiting this enzyme. Several compounds were evaluated as their acyl glucuronide conjugates in this regard. As follow-up to the work of Baer et al. (2009), gemfibrozil analogs and their glucuronide conjugates were synthesized and evaluated as inhibitors of CYP2C8 activity in HLM (Fig. 2). Molecular modeling was also conducted to better understand how the gemfibrozil acyl-β-glucuronide and its analogs interact with the CYP2C8 active site.
Materials and Methods
Chemicals and Reagents.
Amodiaquine, clotrimazole, diclofenac, gemfibrozil, ibuprofen, indomethacin, ketoconazole, ketoprofen, mefenamic acid, phenelzine, saccharic acid lactone, and simvastatin were purchased from Sigma-Aldrich (St. Louis, MO). Atorvastatin, dihydroketoprofen, montelukast, and all glucuronide conjugates (except those prepared below) were purchased from Toronto Research Chemicals (North York, ON, Canada). (R)- and (S)-Naproxen were purchased from VWR (Bridgeport, NJ). HLM and P450 microsomes (Supersomes) derived from baculovirus-infected insect cells were obtained from BD Biosciences (Woburn, MA). Liver 9000g supernatant fraction of Aroclor 1254-treated rats (ArRS9) was purchased from Moltox Inc. (Boone, NC). Alamethicin, bovine serum albumin, NADPH, and UDPGA were obtained from Sigma-Aldrich. MgCl2 was purchased from Ambion (Austin, Texas). NH4OAc was purchased from EM Science (Cherry Hill, NJ). HPLC-grade acetonitrile and water were purchased from Mallinckrodt Baker (Phillipsburg, NJ). All other reagents were obtained from commercial sources.
Synthesis of Gemfibrozil Analogs and Isolation of Glucuronide Conjugates.
See supplemental data and Fig. 2.

Chemical preparation of gemfibrozil analogs. DMF, dimethylformamide.
Time-Dependent Inhibition of CYP2C8 in HLM.
Test compounds and positive controls (60 nl, in dimethyl sulfoxide) at 10 concentrations were incubated with 15 μl of HLM (0.1 mg/ml HLM, 1 mM NADPH, 100 mM potassium phosphate buffer, pH 7.4, and 2 mM MgCl2) for 30 min (or as specified) at 37°C. This mixture was then diluted with an equal volume (15 μl) of substrate mixture (4 μM amodiaquine, 1 mM NADPH, 100 mM potassium phosphate buffer, pH 7.4, and 2 mM MgCl2), and the reaction continued for 10 min at 37°C. To determine the IC50 value of the test compounds at the 0-min time point, test compounds were first mixed with 15 μl of the substrate mixture and then were immediately diluted with an equal volume (15 μl) of HLM mixture and incubated for 10 min at 37°C. At the end of 10-min incubation, these reactions were terminated by the addition of 30 μl of quench buffer (94% water-5% acetonitrile-1% formic acid) containing internal standard (0.15 μM [2H5]-N-desethylamodiaquine).
In one experiment, the test compounds were first preincubated (30 min at 37°C) with alamethicin-treated (25 μg/ml) HLM (0.1 mg/ml) with and without 2 mM UDPGA, followed by addition of NADPH (1 mM final) and further incubation for 0, 15, and 30 min at 37°C. At the end of the incubation time with NADPH, the reactions were continued by addition of the probe substrate mixture, containing 4 μM amodiaquine, 1 mM NADPH, 100 mM phosphate buffer (pH 7.4), and 2 mM MgCl2. After 10 min of incubation at 37°C, the reaction was terminated by addition of the quench buffer containing internal standard (described above).
Immediately before sample analysis, the denatured protein was precipitated by centrifugation for 10 min at 2500g, and the supernatant was used to perform sample analysis. The amount of N-desethylamodiaquine produced in each reaction was determined by RapidFire mass spectrometry (BIOCIUS Life Sciences, Wakefield, MA) using on-line solid-phase extraction (SPE) with tandem mass spectrometry (MS/MS). Four compounds, known CYP2C8 inhibitors, were tested as positive controls in each experiment. They included three reversible CYP2C8 inhibitors (montelukast, clotrimazole, and ketoconazole) and one time-dependent inhibitor (phenelzine).
Metabolic Profiling.
Gemfibrozil, gemfibrozil glucuronide, 5-(2,5-bis(trifluoromethyl)phenoxy)-2,2-dimethylpentanoic acid (BTFM gemfibrozil), and BTFM gemfibrozil glucuronide (50 μM) were individually incubated at 37°C with pooled HLM (1 mg/ml) or recombinant CYP2C8 (∼50 pmol/ml) in phosphate buffer (100 mM, pH 7.4). Reactions were started by addition of NADPH (3 mM) in phosphate buffer. Sample aliquots were taken at various times: 0, 30, and 60 min. Reactions were quenched by addition of equal volumes of acetonitrile containing 4% formic acid, and the precipitated proteins were removed by centrifugation (1000g for 5 min). The supernatant was analyzed by direct injection onto the LC-UV-MSn system for metabolic profiling.
Analytical Procedures.
Synthesis of gemfibrozil analogs.
Microwave reactions were performed using a CEM Discover microwave (ramp, 1 min; stirring, On; power Max, On). Column chromatography was performed using a flash chromatography system (Biotage, LLC, Charlotte, NC). HPLC analysis was performed using a ProStar diode array detector (model 335; Varian, Inc., Palo Alto, CA) with ProStar pumps (model 215; Varian) with 25-ml pump heads and a SunFire C18 column (3.5 μm, 4.6 × 150 mm; Waters, Milford, MA). High-resolution mass spectrometry analysis was performed with an Exactive Fourier transform Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA) in negative ionization electrospray mode using Xcalibur software (Xcalibur, Inc., Reston, VA). Proton NMR spectra were recorded on a 400-MHz DPX400A spectrometer (Bruker, Newark, DE).
Isolation of glucuronides.
In the course of isolation, chromatographic fractions were screened by LC/MS for glucuronide products with a Thermo Finnigan Deca LCQ LC/MS system and SunFire C18 column (5 μm, 4.6 × 150 mm). The mobile phase was 10 mM NH4OAc with CH3CN (95:5; solvent A) and 10 mM NH4OAc with CH3CN (5:95; solvent B). Conditions were as follows: start, 85:15 (A:B) to 0:100 (A:B) over 25 min, held at 100% solvent B for 5 min, and then back to the initial conditions over 2 min (32 min total); flow rate, 1.2 ml/min; wavelength, 254 nm; and ESI-MS, negative ion mode. NMR data were recorded in CD3OD (5-mm tube) using a Bruker Avance III 500 MHz spectrometer.
Metabolite identification.
HPLC analyses were performed using an 1100 series separation module (Agilent Technologies, Santa Clara, CA) and a Synergi Hydro-RP column (2.0 × 150 mm, 4 μm; Phenomenex, Torrance, CA) maintained at room temperature. The mobile phase consisted of a 98:2 mixture of 10 mM NH4OAc and CH3CN (solvent A) and a 70:30 mixture of CH3CN and CH3OH (solvent B). Separations of components present in the incubation mixture was achieved under the following gradient conditions: initially solvents were held isocratically at 0% solvent B for 1 min, followed by a linear gradient to 100% solvent B over 30 min. Solvent B was then held isocratic at 100% for 3 min. Thereafter, solvents were immediately brought back to the initial conditions, 0% solvent B in 0.1 min and reequilibrated column for 4.9 min with a total chromatographic run time of 38 min.
The eluent from the HPLC column was routed in-line to an 1100 series photodiode-array detector (scanning 200–600 nm at 5 Hz; Agilent) and then to a LCQ Deca Xp Plus ion trap mass spectrometer (Thermo Fisher Scientific). The electrospray ion source was operated in negative ionization mode with data-dependent product ion scanning. The product ion MSn spectra were obtained by collision-induced dissociation using normalized energy of 35% and an isolation width of 3 Da.
Quantification of N-desethylamodiaquine (CYP2C8 inhibition).
The amount of substrate probe metabolite (N-desethylamodiaquine) was measured using SPE-MS/MS analysis on a RapidFire MS/MS system, which consisted of a RapidFire 200 HT System (BIOCIUS Life Sciences) and a 4000 QTRAP hybrid triple quadrupole linear ion trap mass spectrometer (AB Sciex, Foster City, CA) using a Turbo V source with an ESI probe. The samples (20 μl) were loaded onto the BIOCIUS extraction SPE C4 column with mobile phase A (water with 0.01% trifluoroacetic acid-0.09% formic acid) at 1.5 ml/min for 3.2 s and were eluted with mobile phase B (CH3CN with 0.01% trifluoroacetic acid-0.09% formic acid) at 1.25 ml/min for 3 s, followed by a reequilibration of the SPE cartridge with mobile phase A at 1.5 ml/min for 0.5 s. The total cycle time was ∼10 s/injection. The SPE eluent was then introduced to a 4000 QTRAP mass spectrometer. Selected reaction monitoring was used for analysis of metabolite and internal standard. The selected reaction monitoring instrument parameters were as follows: ESI in positive ion mode; m/z 328.2 → 282.8 transition for N-desethylamodiaquine; m/z 331.2 → 282.8 transition for [2H5]-N-desethylamodiaquine; 55 V declustering potential; 26 eV collision energy; and 80 ms dwell time. The acquired data were processed with RapidFire peak integration software, and the results were exported as an Excel file for IC50 calculation.
Data Analysis.
The signal intensity of the N-desethylamodiaquine (from MS/MS analysis) was normalized to the signal of internal standard, [2H5]-N-desethylamodiaquine; thus, signal intensity in each reaction was expressed as signal ratio. The sample signal ratios were then normalized to the average signal ratio of the reactions performed in the absence of the test substance (solvent control, 0%inhibition) and in the absence of enzyme (background, 100% inhibition). Results were expressed as percentage inhibition of the enzyme activity with solvent control, calculated as
 where S is sample, T is average solvent control, and B is average background signals.
where S is sample, T is average solvent control, and B is average background signals.
The results were then imported into in-house curve fitting software (CurveMaster), which uses MathIQ (ID Business Solutions, Ltd., Guildford, Surrey, UK) to determine the IC50 value for each test compound. The IC50 is defined as the concentration corresponding to 50% inhibition of the enzyme activity observed with solvent control, derived from the fitted 10-point curve as determined using a four-parameter logistic regression model:
 where A is minimum inhibition, B is maximum inhibition, X is inhibitor concentration, C = X, at which Y = A+ (B − A)/2 (i.e., concentration of inhibitor at which half of the maximal inhibition is observed), and D is the Hill coefficient (slope). IC50 values (where %inhibition = 50) for each compound at each time point were determined. IC50 values and percent inhibition at the highest concentration are reported for each tested time point.
where A is minimum inhibition, B is maximum inhibition, X is inhibitor concentration, C = X, at which Y = A+ (B − A)/2 (i.e., concentration of inhibitor at which half of the maximal inhibition is observed), and D is the Hill coefficient (slope). IC50 values (where %inhibition = 50) for each compound at each time point were determined. IC50 values and percent inhibition at the highest concentration are reported for each tested time point.
Molecular Modeling.
All compounds were sketched using ChemDraw and converted to three-dimensional structures using Omega (version 2.2; OpenEye Scientific Software, LLC, Santa Fe, NM). The structures were minimized using Batchmin (Schrödinger, Inc., New York, NY) using the OPLS2005 forcefield with implicit solvation. The CYP2C8 X-ray crystal structure was retrieved from the Research Collaboratory for Structural Bioinformatics (Protein Data Bank code 2NNI). The protein was prepared with the Protein Prep Wizard within Maestro (version 8.5; Schrödinger, Inc.) using the standard defaults for metal treatment and amino acid deletions, among others. Docking was performed with Glide (Schrödinger, Inc.) using flexible conformations for the ligand. Grids were prepared using the Glide Grid generation tool by selecting montelukast to specify the binding site. No constraints were applied for docking. Poses were evaluated using the Glide score and by visual inspection. Poses that included an interaction with the heme group were preferred.
Results
Effect of Various Glucuronide Conjugates on CYP2C8 Activity.
The acyl glucuronide conjugates of 11 compounds were evaluated as direct-acting and time/mechanism-dependent inhibitors of CYP2C8 activity in HLM. Activities were assessed with a marker substrate (amodiaquine) at 2 μM, a concentration close to its reported Km of 1.86 μM (Walsky and Obach, 2004). The results are summarized in Table 1. Of the compounds tested, only gemfibrozil acyl-β-glucuronide elicited a time/mechanism-dependent effect, with a 16-fold shift in IC50 at the 0-min time point [IC50(0) = 21 μM] to the 30-min time point [IC50(30) = 1.4 μM]. These results are similar to those reported by Ogilvie et al. (2006): IC50(0) = 24 ± 5 μM and IC50(30) = 1.8 ± 0.5 μM. No time/mechanism-dependent effect was observed when gemfibrozil itself was tested in this assay [IC50(0)/IC50(30) ratio ∼1.0], also similar to results reported by Ogilvie et al. Therefore, gemfibrozil itself is not a metabolism-dependent inhibitor of CYP2C8. Controls for the CYP2C8 inhibition experiments included three reversible inhibitors, montelukast (IC50 = 0.03 μM), clotrimazole (IC50 = 0.7 μM), and ketoconazole (IC50 = 2.8 μM), and one time-dependent inhibitor, phenelzine [IC50(0) = >200 μM, IC50(30) = 60 μM]. Control values were in agreement with published results (Polasek et al., 2004; Walsky et al., 2005) and were included for all CYP2C8 inhibition experiments.
IC50 values for inhibition of CYP2C8-catalyzed amodiaquine N-deethylation by various glucuronide conjugates after incubation with NADPH-fortified HLM for 0 and 30 min
Although no other compounds were time-dependent inhibitors of CYP2C8 as their acyl glucuronides, three could be considered “strong” inhibitors (simvastatin glucuronide > mefenamic glucuronide > diclofenac glucuronide) with IC50 values less than 20 μM and greater than 90% inhibition. Three compounds were “weak” inhibitors (ketoprofen glucuronide > indomethacin glucuronide > atorvastatin glucuronide) with IC50 values between 25 and 50 μM and greater than 65% inhibition. Four compounds did not exhibit CYP2C8 inhibition (ibuprofen glucuronide, (R)- and (S)-naproxen glucuronide, and dihydroketoprofen glucuronide) (Table 1).
The aglycone (parent) compounds were also tested as inhibitors of CYP2C8 (Table 2). Simvastatin, mefenamic acid, and atorvastatin were moderate to weak inhibitors of CYP2C8, with IC50 values similar to those of their glucuronide conjugates. Ibuprofen, (R)- and (S)-naproxen, and dihydroketoprofen did not show CYP2C8 inhibition, also similar to their glucuronide conjugates. However, the glucuronide conjugates of diclofenac and indomethacin were approximately 2-fold more potent as CYP2C8 inhibitors compared with the corresponding aglycones. The glucuronide conjugate of ketoprofen showed the greatest inhibition (versus its aglycone), with a greater than 5-fold lower IC50. No time-dependent CYP2C8 inhibition was observed for the parent aglycones.
IC50 values for inhibition of CYP2C8-catalyzed amodiaquine N-deethylation by various aglycones after incubation with NADPH-fortified HLM for 0 and 30 min
Metabolism-Dependent Inhibition of CYP2C8 by Gemfibrozil and BTFM Analog.
Gemfibrozil, BTFM gemfibrozil, and gemfibrozil acyl-β-glucuronide were preincubated with HLM in the presence and absence of UDPGA and NADPH (0, 15, and 30 min) before addition of amodiaquine to monitor CYP2C8 activity (data not shown). BTFM gemfibrozil was a CYP2C8 inhibitor (IC50 15–26 μM), but the inhibition was not dependent on UDPGA or NADPH, indicative of direct (reversible) inhibition of CYP2C8 by this analog. In the presence of UDPGA and NADPH, gemfibrozil was a time/mechanism-dependent inhibitor of CYP2C8 after 30 min of incubation [IC50(30) = 25 μM], indicating that glucuronidation of gemfibrozil is necessary for inhibition of CYP2C8. In the absence of NADPH at 0 min, gemfibrozil acyl-β-glucuronide did not demonstrate CYP2C8 inhibition, but showed increasing inhibition after a longer preincubation time in the presence of NADPH [IC50(30) = 4.0 μM], indicating the dependence on metabolism for inhibition. Although the metabolism of gemfibrozil to gemfibrozil acyl-β-glucuronide was not monitored in this experiment, it appears that incomplete formation of the glucuronide conjugate occurred, as indicated by the discrepancy in IC50 in the two experiments. These results indicate that the glucuronide conjugate has to be prepared before its incubation with HLM and that insufficient glucuronide would be formed in a reaction mixture containing the aglycone and cofactor-fortified HLM. For this reason, glucuronides of gemfibrozil analogs were biosynthesized from their respective aglycones, either by incubation with HLM (BTFM gemfibrozil) or rat liver ArRS9 (o-Me gemfibrozil and m-Me gemfibrozil) in the presence of UDPGA.
Effect of Gemfibrozil and Analog Glucuronide Conjugates on CYP2C8 Activity.
The IC50 values of the gemfibrozil analogs as well as their glucuronide conjugates were evaluated for time/mechanism-dependent inhibition as shown in Table 3. As observed previously, gemfibrozil acyl-β-glucuronide behaved as a time- and metabolism-dependent inhibitor of CYP2C8. In comparison, BTFM gemfibrozil was an inhibitor of CYP2C8 (IC50 of 13–17 μM), but its acyl-β-glucuronide was not a time-dependent inhibitor after a 30-min preincubation with NADPH. The o-Me and m-Me gemfibrozil analogs exhibited almost no CYP2C8 inhibition and their corresponding acyl-β-glucuronides demonstrated weak inhibition without preincubation with NADPH (Fig. 3). However, after a 30-min preincubation, the o-Me and m-Me gemfibrozil acyl-β-glucuronides trended toward time-dependent inhibition, with the o-Me gemfibrozil glucuronide more potent an inhibitor than the m-Me gemfibrozil glucuronide (Table 3).
IC50 values for inhibition of CYP2C8-catalyzed amodiaquine N-deethylation by gemfibrozil analogs and their glucuronide conjugates after incubation with NADPH-fortified HLM for 0 and 30 min
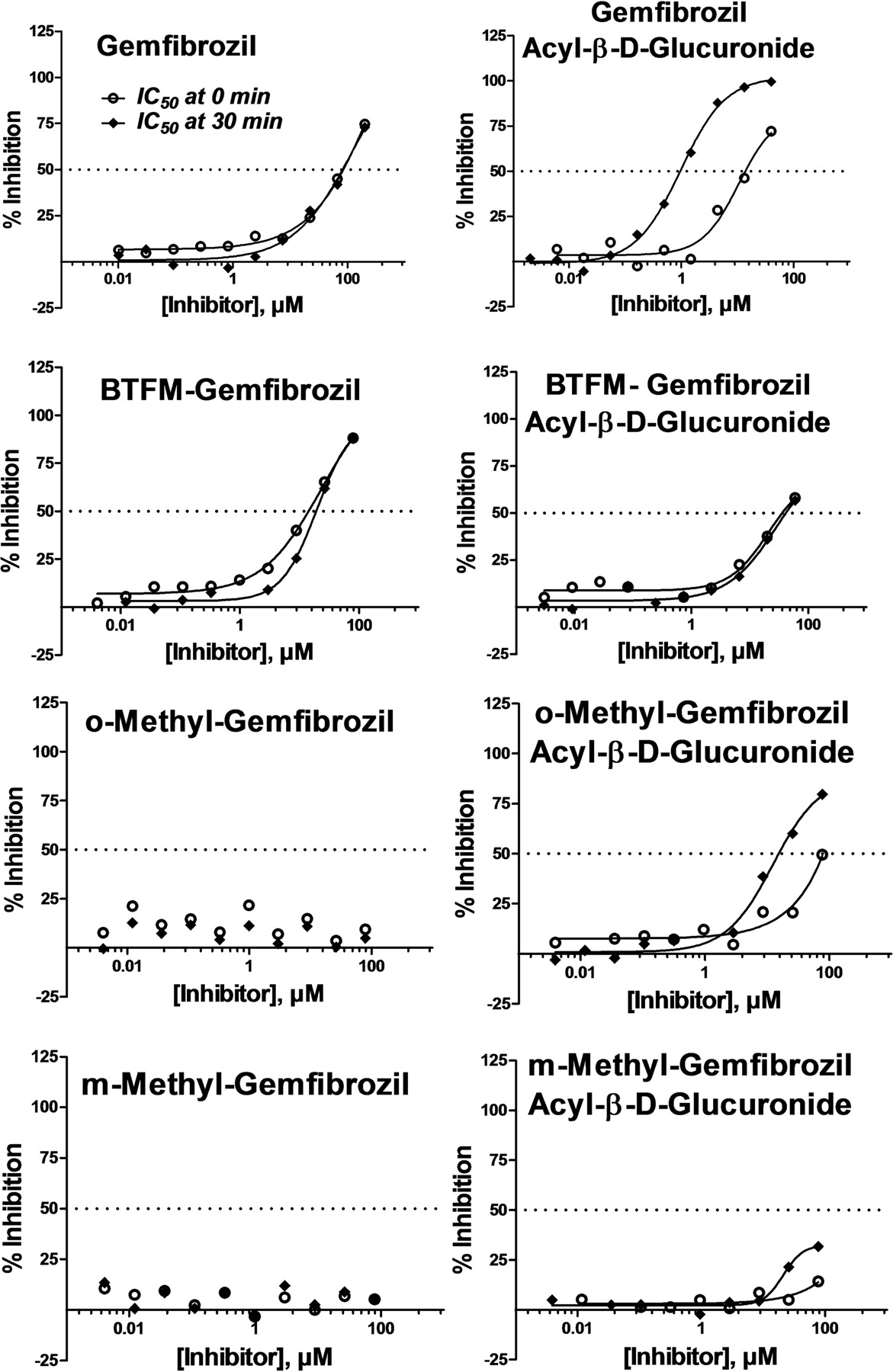
IC50 curves for inhibition of CYP2C8-mediated amodiaquine N-deethylation by gemfibrozil and analog compounds and their respective acyl-β-d-glucuronides after incubation with NADPH-fortified HLM for 0 min (○) and 30 min (♦).
Metabolism of Gemfibrozil Acyl-β-Glucuronide and BTFM Gemfibrozil Glucuronide in the Presence of HLM and Human Recombinant CYP2C8.
Gemfibrozil acyl-β-glucuronide was stable to metabolism in the presence of both NADPH-fortified HLM and recombinant CYP2C8 (≥98% remaining after 60 min of incubation). Only three minor (<1%) metabolites, M1, M2, and M3, were detected by HPLC-UV-MS in both incubations; representative chromatograms (HLM) are shown (Fig. 4C). The structures of the metabolites were assigned on the basis of their MSn product ion mass spectra compared with that of gemfibrozil acyl-β-glucuronide (Fig. 4). Gemfibrozil acyl-β-glucuronide gave a protonated molecular ion at m/z 425. Subsequent MS2 fragmentation gave a daughter ion at m/z 249 indicating loss of glucuronide moiety (176 Da). Further fragmentation (MS3) of the ion at m/z 249 gave rise to an ion at m/z 121, which corresponded to the dimethyl-phenoxy part of the molecule. The three metabolites, M1, M2, and M3, produced similar full-scan mass spectra with a deprotonated molecular ion at m/z 441, a mass 16 Da higher than gemfibrozil acyl-β-glucuronide, denoting that a single oxidation had occurred. The MS2 spectrum of M1, M2, and M3 produced an abundant ion at m/z 265 (loss of glucuronide moiety). MS3 fragmentation of m/z 265 produced an ion at m/z 137, implying that oxidation occurred on the dimethyl-phenoxy moiety for all three metabolites. An attempt was made to further characterize these metabolites, but efforts were unsuccessful because of the very low amounts in the incubates. Under conditions in which gemfibrozil acyl-β-glucuronide metabolites M1 to M3 were detected, no metabolites of BTFM gemfibrozil glucuronide were detectable (data not shown).
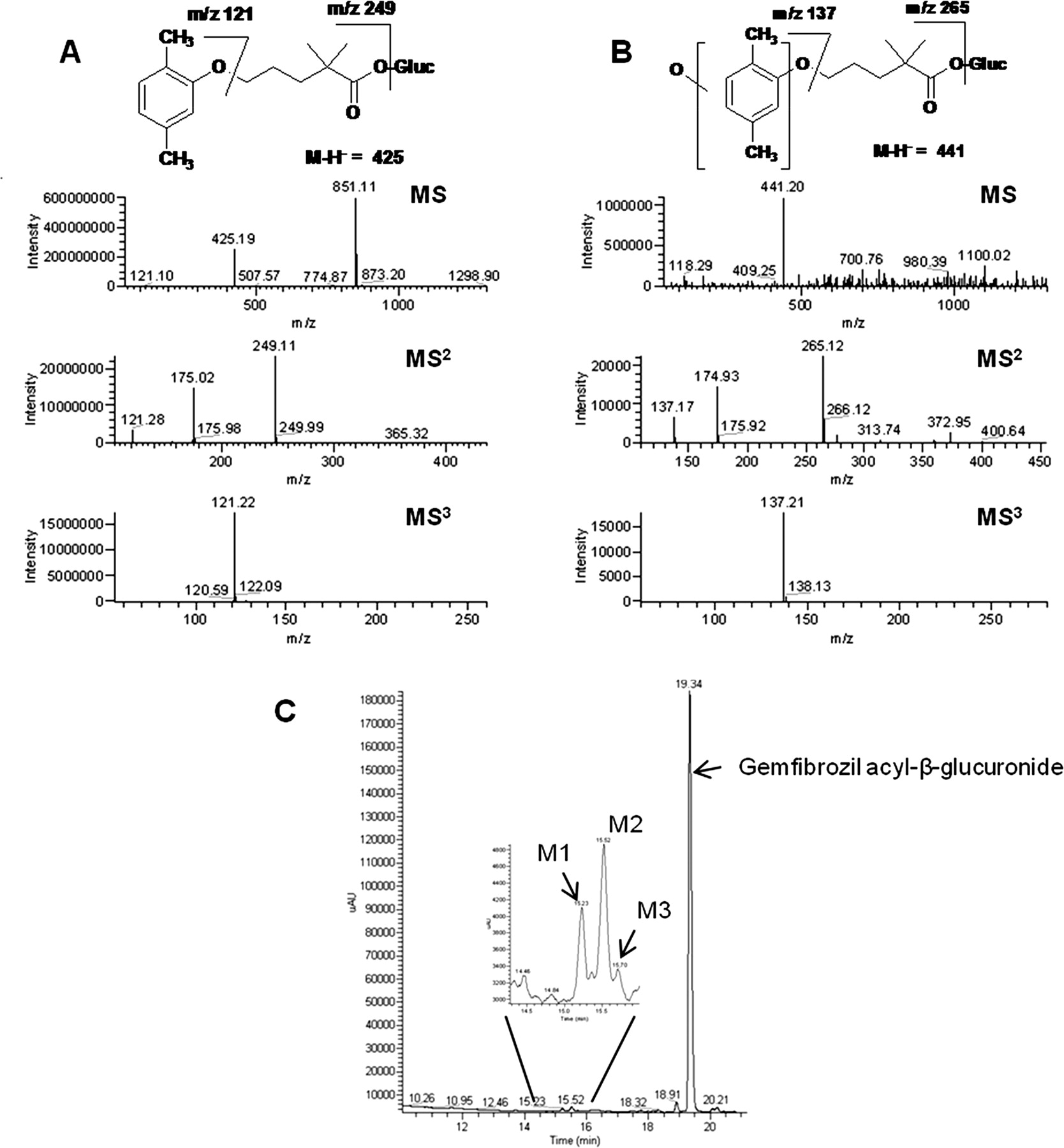
Negative LC-MS/MS analysis of (A) gemfibrozil acyl-β-glucuronide (B) oxidative metabolite after incubation with NADPH-fortified HLM for 60 min (Gluc, glucuronide) and (C) UV chromatogram (272 nm) of gemfibrozil acyl-β-glucuronide oxidative metabolites after incubation with HLM and NADPH for 60 min. Metabolites were also observed using recombinant CYP2C8 (results not shown).
Computational Docking Studies.
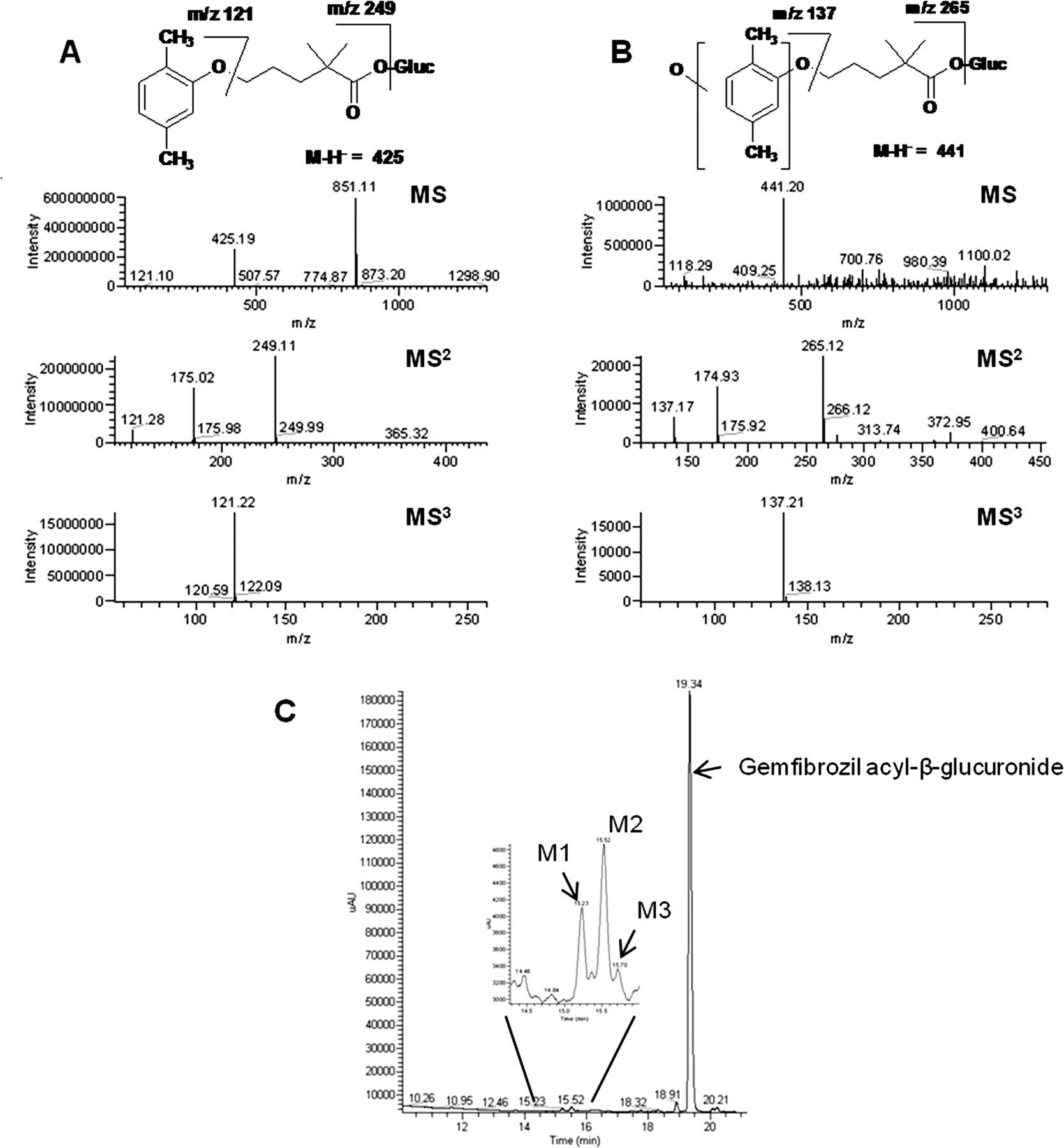
Computational docking studies were performed to investigate the orientation of the gemfibrozil acyl-β-glucuronide in the active site of CYP2C8 (Fig. 6). Docking poses with the o-Me position oriented toward the γ-meso position of the heme were superior to those with the m-Me oriented toward the heme. Both orientations include numerous hydrogen bonds between the glucuronide and the CYP2C8 active site. The carboxylate of the glucuronide forms hydrogen bonds to side chains of Asn217 and Ser100 and to the backbone NH of Ser103. Additional hydrogen bonds are formed to Gln214 and Ser100. The pose orienting the o-Me toward the heme (Fig. 5A) forms numerous hydrophobic contacts to the site, including Thr301, Ile113, Val366, Ser114, and Val477. In addition, the 5-position methyl fits into small hydrophobic recess between Val477 and Phe205, contributing to the orientation of the o-Me position toward the heme.

Gemfibrozil acyl-β-glucuronide orientation in CYP2C8 orientation of o-Me toward heme (A) and orientation of m-Me toward heme (B). Key contact residues are highlighted in the figure. C, pose of gemfibrozil (slate blue carbons) in CYP2C8. The pose of gemfibrozil acyl-β-glucuronide from A is shown for spatial reference (cyan carbons). Note that the only contact residue in common is a hydrogen bond to Ser103.
In addition to the glucuronide, gemfibrozil itself was also docked into CYP2C8. The docking pose (Fig. 5C) for gemfibrozil (shown with slate blue carbons) is substantially different compared with that for the glucuronide metabolite. The pose of gemfibrozil acyl-β-glucuronide is shown for reference with cyan carbons. The only contact residue in common for the poses is Ser103. The dimethyl-phenyl ring of gemfibrozil is placed in a largely hydrophobic pocket formed by Ile106, Phe201, Val237, and Ala292. Because gemfibrozil is not a particularly potent inhibitor of CYP2C8 (Tables 2 and 3), it is likely that multiple weak binding poses are probable.
Inhibition of CYP2C8 (IC50 ∼10 μM) was observed with mefenamic acyl-β-glucuronide and diclofenac acyl-β-glucuronide, so the docking studies described herein were expanded to include these two glucuronides. In both cases, poses were generated forming hydrogen bonds to the glucuronide similar to those for the gemfibrozil acyl-β-glucuronide. In addition, good hydrophobic contacts are formed with hydrophobic residues proximal to the heme. However, in neither case were any functional groups oriented toward the heme, which would imply probable mechanism-based inhibition through covalent attachment. The methyl substituents of mefenamic acyl-β-glucuronide were oriented away from the heme in the favored binding pose (Fig. 6). A similar result was obtained with diclofenac acyl-β-glucuronide (data not shown).

Mefenamic acyl-β-glucuronide docked into the CYP2C8 active site. The molecule has binding between the protein and the glucuronide similar to that for gemfibrozil acyl-β-glucuronide. The methyl substituents are oriented away from the heme, making covalent attachment unlikely. No poses were identified that would orient the methyl groups toward the heme.
Discussion
Although it is known that gemfibrozil acyl-β-glucuronide is a mechanism-based inhibitor of CYP2C8 (Ogilvie et al., 2006), it is not known whether gemfibrozil acyl-β-glucuronide is unique in its ability to inhibit CYP2C8 or whether other acyl glucuronides are able to inhibit this enzyme, either reversibly or mechanistically. Of the acyl glucuronide conjugates tested in this study, 7 of 11 showed some degree of reversible CYP2C8 inhibition. Although some of these compounds show significant inhibition, the impact on CYP2C8 in vivo is not known. For example, Hatorp et al. (2003) have assessed the impact of simvastatin on the pharmacokinetics of repaglinide. Unfortunately, given the role of CYP2C8 and CYP3A4 in the clearance of repaglinide, the results are difficult to interpret. Additional clinical data are needed to place the results described herein in context. It is worth noting that when the aglycone (parent) compounds were tested for CYP2C8 inhibition, the IC50 values were similar to those of the corresponding glucuronide conjugates, indicating that glucuronide conjugates may fit into the CYP2C8 pocket just as well as the parent molecule. Also noteworthy is the fact that ketoprofen acyl glucuronide was even more potent than ketoprofen itself. Ketoprofen acyl glucuronide is present in human plasma and excreted in the urine (Upton et al., 1980), although CYP2C8 inhibition in a clinical setting has not been reported. Only gemfibrozil acyl-β-glucuronide was a time-dependent inhibitor of CYP2C8, indicating that there is a particular metabolic liability for this glucuronide conjugate; gemfibrozil itself was not a time-dependent inhibitor of CYP2C8 (Table 1).
This study confirms the previous finding that gemfibrozil causes time-dependent inhibition of CYP2C8 when incubated in the presence of UDPGA and NADPH. However, generation of the glucuronide in the incubation resulted in less potent inhibition of CYP2C8 compared with inhibition observed upon incubation with the gemfibrozil acyl-β-glucuronide (IC50 after 30 min of incubation is 25 versus 4.0 μM). It appears that although generation of the glucuronide conjugate in the incubation mixture indicates the presence of time-dependent inhibition, incubation with the glucuronide conjugate itself is necessary to accurately determine the time-dependent shift in IC50. Therefore, further studies with gemfibrozil analogs were also conducted with their respective glucuronide conjugates.
NADPH was also required to observe time-dependent inhibition of CYP2C8 in the presence of gemfibrozil acyl-β-glucuronide, indicating that the gemfibrozil glucuronide is probably oxidized by P450. Such a hypothesis is supported by the fact that incubation of gemfibrozil acyl-β-glucuronide with NADPH-fortified HLM produced three minor hydroxylation products (Fig. 4). It is unfortunate that turnover was low and the oxidative metabolites could not be further characterized (metabolism on the benzylic moiety of gemfibrozil is implicated). CYP2C8-catalyzed oxidation of glucuronide is not a novel finding, because it has been known for some time that the enzyme is capable of hydroxylating estradiol-17β-glucuronide (Delaforge et al., 2005). After a thorough docking of the molecule within the CYP2C8 crystal structure, the same authors concluded that the active site is large enough to accommodate a glucuronide conjugate. Likewise, Kumar et al. (2002) have described CYP2C8-mediated glucuronide metabolism: oxidation of diclofenac acyl glucuronide to 4′-hydroxy diclofenac acyl glucuronide. On the basis of the results presented herein, such metabolism does not render pronounced time-dependent inhibition of CYP2C8 in HLM (Table 1), in contrast to what is observed for gemfibrozil acyl-β-glucuronide.
In a recent study, Baer et al. (2009) have shown that after incubation with recombinant CYP2C8, the gemfibrozil acyl-β-glucuronide is covalently bound to the heme moiety, on the basis of mass spectrometry and deuterium isotope effects. Oxidation of the aromatic o-Me or p-Me group to form a benzyl radical was proposed as the reactive intermediate that binds to the γ-meso position of the heme. The authors concluded that because of the rotameric flexibility of the alkyl chain of gemfibrozil acyl-β-glucuronide, there was no indication that either the o-Me or the m-Me would have a favored orientation. To further probe this hypothesis, three gemfibrozil analogs and their corresponding glucuronides were evaluated in the CYP2C8 inhibition assay. These three analogs were BTFM gemfibrozil, o-Me gemfibrozil, and m-Me gemfibrozil.
When the gemfibrozil aromatic methyl groups were substituted with trifluoromethyls to give BTFM gemfibrozil, this compound was a competitive CYP2C8 inhibitor; however, time-dependent inhibition of CYP2C8 was not observed either for the analog or the corresponding glucuronide conjugate of this analog. This is to be expected, on the basis of the hypothesis that oxidation of the methyl groups is necessary for time-dependent inhibition of CYP2C8 and that such metabolism can be blocked by substitution with trifluoromethyls. In agreement, turnover of BTFM gemfibrozil and its acyl-β-glucuronide was not detectable after incubation with NADPH-fortified HLM (Fig. 5).
Time-dependent inhibition of CYP2C8 was not observed with the mono-methyl gemfibrozil analogs. However, the glucuronide conjugate of o-Me gemfibrozil was a time-dependent inhibitor of CYP2C8. The o-Me gemfibrozil glucuronide analog appears to be a more potent CYP2C8 inhibitor than the m-Me gemfibrozil glucuronide analog but less potent than gemfibrozil acyl-β-glucuronide itself (Table 3).
In contrast to what Baer et al. (2009) had observed in their computational docking studies, the orientation of the o-Me position of the gemfibrozil acyl-β-glucuronide appears to fit better in the CYP2C8-binding pocket compared with the m-Me position, based on our modeling studies (Fig. 6). The pose of orientation of the o-Me position with the methyl group oriented toward the heme appears to fit better in the CYP2C8-binding pocket compared with the m-Me being oriented toward the heme position. It appears that more hydrophobic interactions are present in the o-Me analog-oriented pose than in the m-Me-oriented pose. The loss of potency of the o-Me glucuronide analog compared with gemfibrozil acyl-β-glucuronide may be due to additional rotational freedom of the ortho-phenyl position in the active site. It appears that the additional methyl in the meta position allows the molecule to fit into a shallow pocket of the CYP2C8 molecule and appears to limit the rotation of the phenyl ring. Thus, the proposed reaction pathway for gemfibrozil acyl-β-glucuronide more likely involves oxidation of the o-Me position leading to covalent binding and inactivation of the CYP2C8 (Fig. 1B), rather than a lack of a favored oxidation position as indicated by Baer et al. (2009) (Fig. 1A). It is also possible that a dimethide analog could be formed as a reactive intermediate, which would help explain the loss of potency in the mono-methyl analogs, but there is no current evidence to indicate that this is occurring.
Although most glucuronide conjugates are eliminated without inhibiting major metabolic pathways, some have the potential to bring about drug-drug interactions (Faed 1984). Therefore, the 2008 U.S. Food and Drug Administration safety guidelines state that if a drug conjugate is reactive (e.g., acyl glucuronide), then additional safety assessment may be needed (U.S. Food and Drug Administration, 2008). Although P450 inhibition screening could be part of such an assessment, such studies are not conducted routinely (Bode 2010). It has been suggested that glucuronides may be generated by incubation of a test compound with alamethicin-treated (UDPGA-fortified) HLM to form the glucuronide, followed by incubation with NADPH and a probe substrate(s) to assess inhibition of one or more P450s (Ogilvie et al., 2006; Bode, 2010). However, a more accurate determination of time-dependent inhibition would need to be conducted with the isolated glucuronide once mechanism-based inhibition was suspected. In any case, it appears from this study that if the parent compound is an inhibitor of CYP2C8, the corresponding acyl glucuronide may also contribute to the inhibition. Although gemfibrozil acyl-β-glucuronide has a relatively unique configuration that renders mechanism-based inhibition and this mechanism is not applicable to all acyl glucuronides, P450 inhibition (including time-dependent inhibition) should be evaluated in the presence (and absence) of UDPGA for all compounds that produce acyl glucuronide conjugates. If inhibition is observed in the presence of UDPGA, the corresponding acyl glucuronide should be generated and tested for P450 inhibition. Although mechanism-based inhibition of additional P450s (beyond CYP2C8) by glucuronides has not been reported to date, in vitro testing of multiple human P450s is advisable.
Authorship Contributions
Participated in research design: Jenkins, Zvyaga, Burrell, Turley, and Rodrigues.
Conducted experiments: Zvyaga, Hurley, Turley, Wagner, Leet, and Philip.
Contributed new reagents or analytic tools: Burrell, Turley, and Leet.
Performed data analysis: Jenkins, Zvyaga, Johnson, Hurley, Wagner, and Philip.
Wrote or contributed to the writing of the manuscript: Jenkins, Zvyaga, Johnson, Turley, Leet, Philip, and Rodrigues.
Acknowledgments
We gratefully acknowledge helpful discussions and suggestions by Drs. Michael Sinz and Kenneth Santone (Bristol-Myers Squibb).
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.111.041947.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- HLM
- human liver microsomes
- BTFM gemfibrozil
- 5-(2,5-bis(trifluoromethyl)phenoxy)-2,2-dimethylpentanoic acid
- P450
- cytochrome P450
- UDPGA
- UDP-glucuronic acid
- HPLC
- high-performance liquid chromatography
- SPE
- solid-phase extraction
- MS/MS
- tandem mass spectrometry
- LC
- liquid chromatography
- MS
- mass spectrometry
- ESI
- electrospray ionization
- Me
- methyl.

- Received July 26, 2011.
- Accepted September 12, 2011.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}