


Abstract
More than 60 human immunodeficiency virus protease inhibitors were examined for the structure-activity relationship between metabolic stability, CYP3A4 inhibitory potency, and substrate-induced binding spectra with a ferric form of P450 in human liver microsomes. A positive relationship was found between CYP3A4 inhibitory potency and metabolic stability; namely, compounds that were more potent for the CYP3A4 inhibition generally were more metabolically stable. In addition, the compounds formed two clusters defined by the distinct type of substrate-induced P450 binding spectra: the compounds with type II binding spectra were more stable metabolically and more potent for the CYP3A4 inhibition than those with type I binding spectra. The structure-activity relationship suggested that the presence and position of heterocyclic nitrogen on the pyridine moiety play an important role in determining the manner of interaction with P450 and the magnitude of CYP3A4 inhibition/metabolic stability in the series of structurally related human immunodeficiency virus protease inhibitors under development.
In vitro metabolic stability has been routinely examined by using human liver microsomes for every candidate entering into the pharmacokinetics studies to identify (metabolically) stable HIV1 protease inhibitor candidates. CYP3A4 is known to play a major role in the metabolism of indinavir (Chiba et al., 1996) and other HIV protease inhibitors, including nelfinavir (Lillibridge et al., 1998), ritonavir (Kumar et al., 1996), and saquinavir (Eagling et al., 1997). In addition to the extensive metabolism by CYP3A4, HIV protease inhibitors (ritonavir, indinavir, and saquinavir) are known to be very potent CYP3A4 inhibitors with IC50 values of 0.02 to 3 μM (Eagling et al., 1997). Minor structural modification has been demonstrated to dramatically affect inhibitory potency of ritonavir and its analogs for CYP3A4 (Kempf et al., 1997). Therefore, in the discovery stage of drug development for HIV protease inhibitors, in vitro CYP3A4 inhibition study is also routinely conducted to evaluate the inhibitory potency of a drug candidate. Clinically, it is important to balance the potentially harmful adverse effects by a CYP3A4-mediated drug-drug interaction and the beneficial enhancement of pharmacological effects by a combination therapy for the AIDS patients (Barry et al., 1997). Despite CYP3A4 being one of the most prevalent isoforms in the human liver and its importance in the metabolism of pharmaceuticals, little is yet known about the active site structure (Smith et al., 1997). In the course of searching a backup candidate for indinavir, most of the HIV protease inhibitors tested in our laboratory were highly metabolically unstable due to the extensive metabolism by CYP3A4. It was therefore important to identify the factors that can provide rational drug design to improve metabolic stability. The purpose of the present study was to establish the structure-activity relationship between metabolic stability and CYP3A4 inhibitory potency of HIV protease inhibitor candidates in human liver microsomes.
Materials and Methods
Indinavir (Crixivan, MK-0639, L-735524) and all the other HIV protease inhibitors were synthesized either in the Department of Medicinal Chemistry at Merck Research Laboratories (West Point, PA) or in the Department of Molecular Design and Diversity at Merck Research Laboratories (Rahway, NJ). Testosterone and 6β-hydroxylated testosterone were obtained from Sigma Chemical Co. (St. Louis, MO). Pooled human liver microsomes were obtained from Keystone Skin Bank (Exton, PA). Monoclonal anti-CYP3A4 antibody was prepared at Merck Research Laboratories (West Point, PA). All other reagents were of analytical grade.
Pooled human liver microsomes (final concentration = 0.5 mg/ml) were incubated with 1 μM HIV protease inhibitor candidate in a reaction mixture consisting of 0.15 M Tris-HCl buffer (pH 7.4), 1 mM EDTA, and NADPH-generating system (10 mM G6P, 2 IU/ml G6P dehydrogenase, 10 mM MgCl2) at 37°C. Samples were taken at the designated time points, and the unmetabolized substrate concentration was measured by an LC-MS (described below). The in vitro metabolic clearance (CLmet, ml/min/kg) was calculated by D (amount of substrate, nmol/mg of protein), AUC (area under the curve of unmetabolized substrate extrapolated to the infinity, nmol·min/ml), MC (microsomal content, mg of protein/g of liver), and LW (liver weight, g of liver/kg of body weight) with the following equation: CLmet = D · MC · LW/AUC, where MC = 50 (mg of protein/g of liver) and LW = 20 (g of liver/kg of body weight) were assumed for humans (Lin et al., 1996). For inhibition studies, HIV protease inhibitor candidate was added at various concentrations (0, 0.1, 0.2, 0.5, 1, 2, 5, 10, 20, 50, and 100 μM) to a microsomal reaction mixture (final volume = 250 μl in 0.15 M Tris-HCl buffer, pH 7.4) containing an NADPH-generating system (10 mM G6P, 2 IU/ml G6P dehydrogenase, 10 mM MgCl2), 0.5 mg/ml protein, 1 mM EDTA, and 20 μM testosterone. Incubation was carried out at 37°C for 20 min, followed by an HPLC analysis. The HPLC method for testosterone metabolism is described elsewhere (Chiba et al., 1997). The inhibition constant (IC50) was calculated from the relationship between the inhibitor concentration (I) and the percentage of control activity of testosterone 6β-hydroxylase activity atI with the aid of a nonlinear regression program in SCIENTIST (MicroMath, Salt Lake City, UT).
Spectral titrations were conducted using a double-beam spectrophotometer (Lambda 20, Perkin-Elmer, Norwalk, CT). Microliter volumes of ethanol solutions of HIV protease inhibitors were added to the experimental cuvette with an equal volume of ethanol added to the reference cuvette. Each cuvette contained a 0.5-ml incubation mixture consisting of 0.15 M Tris-HCl buffer (pH 7.4), 1 mM EDTA, and 1 mg/ml pooled human liver microsomes. After each dilution, the difference spectrum was scanned at 20°C from 350 to 500 nm. The type of substrate-induced binding spectra with a ferric form of P450 heme was determined by the position of wavelengths for peak (λmax) and minimum (λmin) on the spectrum (Jefcoate, 1978).
For antibody study, an aliquot of a different volume of monoclonal anti-CYP3A4 antibody (0–20 μl) was added to the human liver microsomes (0.05 mg), and the mixture was preincubated for 15 min on ice. Metabolism was initiated by the addition of HIV protease inhibitor (10 or 50 μM) in a reaction mixture also containing 1 mM NADPH, 1 mM EDTA plus generating system (the same content described above). Final protein concentration was 0.5 mg/ml. Incubations were conducted for 30 to 90 min at 37°C. The remaining substrate was measured by the LC-MS method described below.
HIV protease inhibitor candidates were assayed by an HPLC system (SYS-S200 HPLC-MS system, PE SCIEX, Norwalk, CT) with mass detector (API-150EX, PE SCIEX). The HPLC was performed on the column (Betasil C18, 50 × 3 mm, 5 μm; Keystone, Bellefonte, PA) at the flow rate of 0.6 ml/min. The mobile phase consisted of 5 mM ammonium acetate, pH 4.5 adjusted with glacial acetic acid (A) and acetonitrile (B). The ratio (A/B) of the mobile phase was adjusted according to the retention time of each HIV protease inhibitor candidate.
Results
We have examined substrate-induced P450 binding spectra with a ferric form of P450, as well as CLmet and CYP3A4 inhibition potency (IC50), for 70 HIV protease inhibitor candidates in human liver microsomes (Fig.1). A greater than 100-fold difference was found for the values of CLmet among candidates. Similarly, CYP3A4 inhibition data demonstrated a 2000-fold difference in IC50 values between the most and least potent inhibitors. There was a positive relationship between CLmet and IC50; namely, the more metabolically stable HIV protease inhibitor had a greater potency to inhibit CYP3A4. The drug candidates formed two clusters defined by the distinct type of P450 binding spectra: the compounds with type II binding spectra generally were more stable (average CLmet = 72 ml/min/kg) and potent for the CYP3A4 inhibition (average IC50 = 1.63 μM) than those with type I binding spectra (average CLmet = 402 ml/min/kg; average IC50 = 16.4 μM). Consistent with the difference in the inhibitory potency between type I and type II compounds, spectral kinetics studies revealed that theKs values for the compounds with type II binding spectra are markedly smaller (range = 0.06–0.13 μM; average = 0.10 μM) than those with type I binding spectra (range = 0.998–7.84 μM; average = 3.32 μM). Immunoinhibition studies with monoclonal anti-CYP3A4 antibody indicated that the metabolism of typical candidates was >90% inhibited by the antibody, suggesting that CYP3A4 plays a major role in the metabolism of HIV protease inhibitors tested.
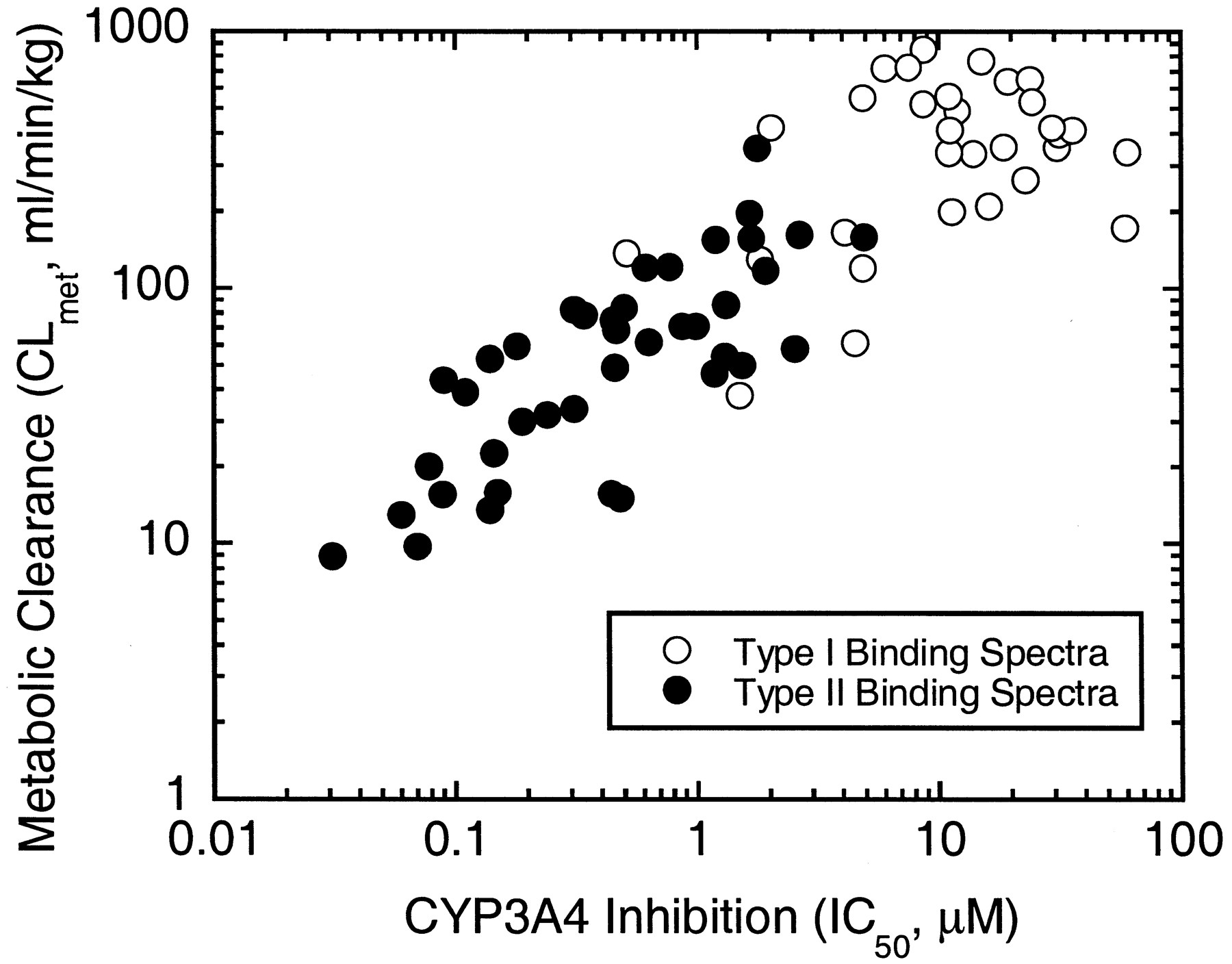
Correlation between metabolic stability (metabolic clearance) and CYP3A4 inhibitory potency (IC50) in HIV protease inhibitor candidates in pooled human liver microsomes.
Open and closed circles represent the candidates showing type I and type II binding spectra, respectively.
Discussion
Compounds that can bind simultaneously to both the lipophilic region of the P450 protein and to the prosthetic heme iron are known to be inherently more potent inhibitors than those depending on only one of these binding interactions (Schenkman et al., 1981). The inhibitory potency of such compounds is governed not only by their hydrophobic character but also by the strength of the bond between their heteroatomic lone pair electrons and the prosthetic heme iron. The binding of inhibitors that are strong iron ligands gives rise to a type II difference spectrum. For the 70 HIV protease inhibitor candidates tested, the compounds with type II binding spectra all possess at least one nitrogen-containing heterocycle, such as a pyridine and furanopyridine. Strong ligands such as the heterocyclic nitrogen displace weak ligands (water) from the hexacoordinated heme of P450. This leads to the subsequent coordination to the pentacoordinated heme, resulting in the P450 shift from its high-spin to low-spin dominant form. This spin state change is accompanied by an increase in the redox potential of the P450, which makes P450 reduction (by NADPH P450 reductase) more difficult. Therefore, both the change in redox potential and the physical occupation of the sixth coordination site for oxygen by the strong iron ligand of HIV protease inhibitors may be responsible for more potent CYP3A4 inhibition and lower turnover rates observed in the compounds with type II binding spectra than in those with type I.
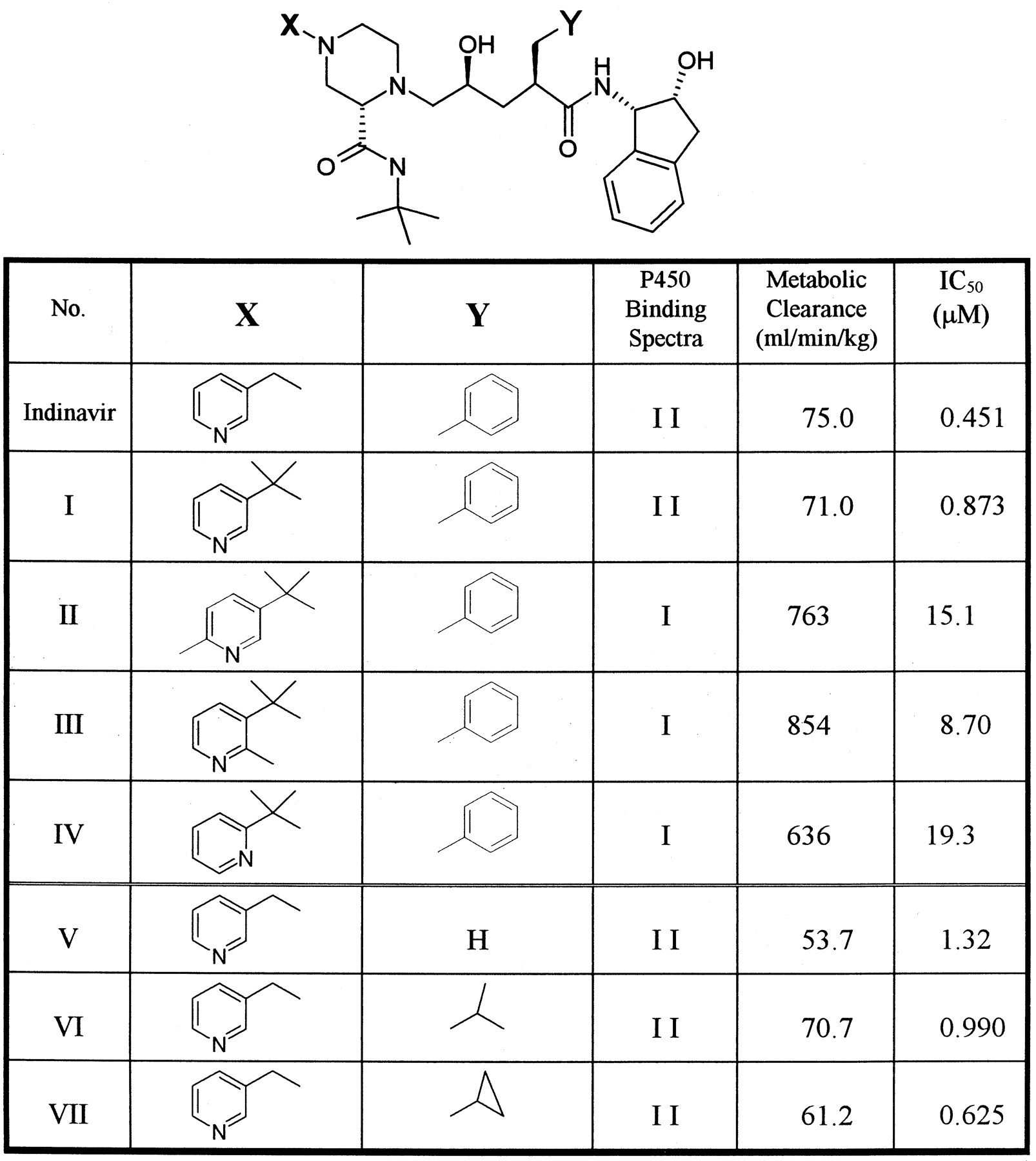
The comparison between compounds having the same structural template clearly demonstrated that minor structural modifications dramatically changed the CYP3A4 inhibitory potency as well as the in vitro metabolic stability. Examples are shown in Fig. 2. The gem-dimethyl modification to blockN-dealkylation (indinavir → compound I) did not change the type of P450 binding spectra (both type II) and had little effect on IC50 and metabolic stability, while one-methyl substitutions on the pyridine ring (compound I→ II or III) switched the binding spectra from type II to type I. This substitution dramatically decreased both CYP3A4 inhibitory potency and metabolic stability by a factor of >10 compared with indinavir. Also, the position of the nitrogen atom on the pyridine ring appears to be critical: compound IV (nitrogen ortho to a linkage) did not produce type II binding spectrum, and the stability/inhibitory potency was much lower than compound I(nitrogen meta to a linkage). These results confirmed that the nitrogen atom on the pyridine moiety can coordinate with a ferric form of P450 heme iron. Substitutions on the nitrogen-containing heterocycles and the change of position of nitrogen clearly affect the manner/magnitude of coordination between the ligand and P450 heme, presumably due to the change in the steric environment for the interaction between substrate and P450 active site. Replacements on the benzene ring (compounds V–VII) had little effect on the type of spectra, inhibitory potency, and metabolic stability. This suggested that the functional group at the position of benzene may not be involved in the stabilization of the nitrogen (on the pyridine)-P450 heme coordination.

Effect of structure modifications on the type of P450 binding spectra, metabolic clearance, and CYP3A4 inhibitory potency (IC50).
In summary, the present data demonstrate that the manner of P450 (CYP3A4) interaction with HIV protease inhibitor candidates probed by the substrate-induced binding spectra plays an important role in simultaneously determining their metabolic stability and CYP3A4 inhibitory potency. Minor modification (or substitution) on the heterocycles dramatically affected their metabolic profiles, due likely to the change of steric environment involved in the interaction between substrate and P450 (CYP3A4) active site. The information on the structure-activity relationship successfully helped medicinal chemists to design more metabolically stable HIV protease inhibitor backup candidate.
Acknowledgments
We gratefully acknowledge the support and thoughtful discussion of Dr. Thomas A. Baillie during the course of this study. We also thank Joy A. Nishime for her technical support.
Footnotes
-
Send reprint requests to: Masato Chiba, Ph.D., WP75-200, Department of Drug Metabolism, Merck Research Laboratories, West Point, PA 19486. E-mail: chibam{at}banyu.co.jp
- Abbreviations used are::
- HIV
- human immunodeficiency virus
- P450
- human cytochrome P450
- CYP3A4
- human cytochrome P450 3A4
- G6P
- glucose 6-phosphate
- LC-MS
- liquid chromatography-mass spectroscopy
- CLmet
- in vitro metabolic clearance
- HPLC
- high-performance liquid chromatography.
- Received June 6, 2000.
- Accepted September 5, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}