


Abstract
In this study we have evaluated the application and reliability of using fluorescence (FLUO)-based high throughput screening assays with recombinant CYPs (rCYP). This was accomplished by screening 29 clinically important antiparasitic drugs for inhibition of the five major drug-metabolizing CYPs (-1A2, -2C9, -2C19, -2D6, and -3A4). Data from FLUO/rCYP assays were compared with that obtained by conventional HPLC assays using human liver microsomes (HLM) and rCYPs. TheKi values showed good correlations: FLUO/rCYP and HPLC/rCYP (r2 = 0.81), HPLC/rCYP and HPLC/HLM (r2 = 0.82), and FLUO/rCYP and HPLC/HLM (r2 = 0.72). Niclosamide had substrate-dependent contrasting effects on CYP2C9 activity with an apparent activation (400%) of 7-methoxy-4-trifluoromethylcoumarin demethylase activity and potent inhibition (Ki = 6.00 μM) of diclofenac 4-hydroxylase activity. Potent inhibitors of CYP1A2 were artemisinin, dihydroartemisinin, thiabendazole, primaquine, and niclosamide (Ki = 0.43, 3.67, 1.54, 0.22, and 2.70 μM, respectively). Proguanil, cycloguanil, amodiaquine, and desethylamodiaquine inhibited CYP2D6 (Ki = 6.76, 5.97, 2.1, and 4.13 μM, respectively). Considering the Cmax of these drugs, artemisinin, thiabendazole, primaquine, amodiaquine, and desethylamodiaquine may cause clinically important interactions because they are predicted to inhibit 67 to 99% of the activities of the CYPs they interact with. In addition, our results suggest CYP1A2 inhibition as the mechanism behind the observed thiabendazole/theophylline and primaquine/antipyrine interactions in vivo.
The increased availability of drugs, both prescription and over-the-counter, has resulted in people taking many drugs at any one time. Although multiple drug therapy has several advantages such as the simultaneous treatment of many ailments and achievement of better outcomes for difficult diseases such as acquired immunodeficiency syndrome, it brings with it an increased risk for drug-drug interactions (Stockley, 1996). Adverse drug reactions have recently been reported as the fourth to sixth most common cause of death in hospitalized patients in the United States (Lazarou et al., 1998). Most adverse effects are pharmacokinetically based and those involving metabolism are very important for many drugs (Kedderis, 1997). The cytochrome P450 (CYP1) superfamily of enzymes plays a crucial role in metabolism and, therefore, has a significant impact on the occurrence of drug-drug interactions (Gonzalez, 1997).
Members of the cytochrome P450 superfamily, CYP1A2, -2A6, -2C9, -2C19, -2D6, and -3A4, are responsible for the metabolism and disposition of more than 90% of therapeutics on the market (Bertz and Granneman, 1997). Drug-drug interactions involving CYPs are due to enzyme induction or inhibition, with those involving inhibition being more common (Lin and Lu, 1998). The withdrawal of terfenadine and mibefradil from the market due to inhibition-based interactions involving CYP3A4 and -2D6 exemplify the medical relevance of CYP inhibition (von Moltke et al., 1994; SoRelle, 1998). As a result, many pharmaceutical companies are now using in vitro systems to evaluate the potential of test compounds to cause clinically significant drug-drug interactions. These activities in the early drug discovery and development process are aimed at reducing attrition rates of new chemical entities to develop better medicines more quickly for the future. Similar studies on already marketed drugs that did not go through such characterization during their development are also necessary to understand any observed interactions and rationalize their clinical use.
The important in vitro enzyme kinetic parameters used to evaluate inhibition are the IC50 (inhibitor concentration causing a reduction of enzyme activity by half) andKi (inhibition constant). Until recently, these parameters were only generated using the laborious and expensive HPLC-based CYP marker reaction assays (Pearce et al., 1996;Masimirembwa et al., 1999). HTS inhibition assays that make use of recombinant CYPs (rCYP) and substrates that produce fluorescent metabolites have now been developed (Crespi et al., 1997). In this study, the fluorescence (FLUO) assays were evaluated by comparing data from FLUO/rCYP assays with that obtained by conventional HPLC-based assays using human liver microsomes (HLM) and rCYPs.
Multiple drug therapy is particularly common in tropical medicine. The relatively slow development of new antiparasitic drugs (Rosenblatt, 1999) has made it necessary to prolong the efficacy of currently available drugs (Bloland and Ettling, 1999), hence the use of combination therapy that helps reduce the loss of drugs to parasite-resistance (White, 1999). The high incidence of parasitic diseases together with the continued spread of human immunodeficiency virus and associated infections in these areas means increased exposure to multiple drugs, thus increasing the risk of drug-drug interactions. In this study the inhibitory effects of antiparasitic drugs on human CYPs were, therefore, also investigated to enable the prediction of potential interactions in vivo.
Materials and Methods
Chemicals.
3-Cyano-7-ethoxycoumarin was obtained from Molecular Probes (Eugene, OR). 7-Methoxy-4-(aminomethyl)-coumarin, and 7-benzyloxy-4-trifluoromethylcoumarin were from GENTEST Corporation (Woburn, MA). Ticlopidine was obtained from ICN Biomedicals Inc. (Aurora, OH). Glucose 6-phosphate, β-nicotinamide, adenine dinucleotide phosphate, reduced NADPH, diclofenac, 7-methoxy-4-trifluoromethylcoumarin (MFC), α-naphthoflavone, quinidine, niclosamide, tinidazole, pyrimethamine, albendazole, thiabendazole, suramin, pyrantel, sulfaphenazole, diethylcarbamazine, and quinine were purchased from Sigma Chemical Co. (St. Louis, MO). 4-OH-Diclofenac, bufuralol, 1′-OH-bufuralol,S-mephenytoin, and 4-OH-mephenytoin were from Ultrafine (Manchester, UK). 4-Acetamidophenol and phenacetin were purchased from Sigma-Aldrich (Steinheim, Germany). 4-Aminophenyl sulfone (dapsone) and artemisinin were obtained from Aldrich Chemical Co. (Milwaukee, WI). Amodiaquine, (+)-chloroquine, melarsoprol, (−)-chloroquine, desethylchloroquine, 4-chlorophenylbiguanide, proguanil, and cycloguanil were generous gifts from Prof. Anders Björkman and Prof. Lars Gustafsson (Karolinska Institute, Stockholm, Sweden). Dr. Michael Ashton (Uppsala University, Uppsala, Sweden) kindly provided dihydroartemisinin and artesunate. All other reagents used were of analytical or HPLC grade.
Recombinant Enzymes and Human Liver Microsomes.
Microsomes from yeast expressing human CYP isoforms -1A2, -2C9, -2C19, -2D6, and -3A4 were produced as previously described (Masimirembwa et al., 1999). Human liver microsomes were prepared from a pooled set of liver pieces of patients undergoing liver resections according to the method of Pearce et al. (1996).
Fluorometric Assays.
Fluorometric assays (Crespi et al., 1997) were done in black Costar 96-well plates (Corning Incorporated, Corning, NY) under experimental conditions as shown in Table 1. Addition of reagents to the 96-well plates was done by hand-pipetting. Each reaction mixture consisted of the appropriate concentration of enzyme, 1 mM NADPH, substrate, and inhibitor in the appropriate concentration of potassium phosphate buffer (pH 7.4) as described by GENTEST (www.gentest.com). Twelve data points were used to generate the Km andVmax. Test compounds and positive control inhibitors were dissolved in water, methanol, acetonitrile, or dimethylformamide to give 10 mM stock solutions. The final solvent concentrations, causing less than 20% inhibition in incubation mixtures, were between 1 and 3%. Due to problems of solubility, only 1 mM solutions of niclosamide, primaquine, and albendazole could be made. Dimethyl sulfoxide was avoided as a solvent due to its high capacity to inactivate CYPs. All compounds were tested for fluorescence or metabolism to fluorescent metabolites at the different excitation and emission wavelengths for the assays. Primaquine was very fluorescent under all the assay conditions and so could not be studied with these assays. The inhibitors were serially diluted to give final concentrations, in duplicate, ranging from 0.09 to 200 μM for 10 mM stock solutions and 0.009 to 20 μM for 1 mM stock solutions. The following were used as positive controls: α-naphthoflavone for CYP1A2, sulfaphenazole for CYP2C9, ticlopidine for CYP2C19, quinidine for CYP2D6, and ketoconazole for CYP3A4. The reaction mixtures were prewarmed for 10 min at 35°C, and NADPH was added to start the reactions. The incubation times used are shown in Table 1, and 75 μl of 20% 0.5 M Tris and 80% acetonitrile was used to stop the reactions. For the CYP2D6 assay, an NADPH-regenerating system (0.4 mM glucose 6-phosphate, 0.4 mM MgCl2, 0.5 M sodium citrate, and 0.4 U/ml glucose-6-phosphate dehydrogenase) was used. The fluorescence was measured using a Wallac 1420 Victor
Experimental conditions and Michaelis-Menten kinetics for different rCYP fluorescence assays
HPLC Assays.
To compare fluorescence and HPLC assays, the effect of 20 μM solutions of all the compounds on enzyme activities was determined using HPLC assays. Complete IC50 curves were done for compounds identified as potent inhibitors. Incubation conditions for HPLC assays were done as described by Masimirembwa et al. (1999)and the phenacetin O-deethylation for CYP1A2 as described byKobayashi et al. (1998). Experimental conditions are shown in Table2.
Michaelis-Menten kinetics for different CYP marker reactions
Data Analysis.
The parameters Km,Vmax, and IC50 were determined by nonlinear least-squares regression analysis using GraFit version 3.0 (Erithacus Software Limited, Middlesex, UK). The inhibitor constant (Ki) values were calculated from IC50 values, assuming competitive inhibition, according to the following relationship: IC50 =Ki(1 + S/Km), thus when S = Km,Ki = IC50/2.
For compounds identified as potent inhibitors (Table 4), the mechanism of inhibition was determined by fitting the data to different inhibition models by nonlinear regression using GraFit version 4.0. The predicted percentage of inhibition of a specific CYP in vivo was then calculated using the Ki values generated from human liver microsomes and the reportedCmax of the drugs using the following relationships:
Inhibition of human liver microsomal CYP1A2 phenacetin O-deethylation and CYP2D6 bufuralol 1′-hydroxylation by antiparasitic drugs
Results
Michaelis-Menten Kinetics.
The Michaelis-Menten kinetic parameters for the fluorescence assays using recombinant enzymes are shown in Table 1 with Table 2 showing those for HPLC-based assays using both recombinant enzymes and human liver microsomes. Although data for bufuralol 1′-hydroxylation displayed two Km values of 9.3 and 277 μM, data for the other CYPs displayed singleKm Michaelis-Menten kinetics in the substrate ranges studied (data not shown).
Inhibition Kinetics.
The inhibitory effects of the antiparasitic drugs and some of their metabolites on the five CYP isoforms are shown in Table 4 with results showing that CYP1A2 and -2D6 were affected the most. TheKi values were calculated from IC50 values determined at [S] =Km using fluorescence-based assays and typical graphs obtained are shown in Fig.1. Known potent inhibitors of the respective CYPs were used as positive controls givingKi values in the same range as those reported in the literature: α-naphthoflavone for CYP1A2 (Ki = 0.003 μM), sulfaphenazole for CYP2C9 (Ki = 0.67 μM), ticlopidine for CYP2C19 (Ki = 0.77 μM), quinidine for CYP2D6 (Ki = 0.009 μM), and ketoconazole for CYP3A4 (Ki = 0.008 μM).

Inhibition of the recombinant CYP1A2-catalyzed fluorometric reaction by antiparasitic drugs.
○, α-naphthoflavone, Ki = 0.003 μM; ●, niclosamide, Ki = 1.19 μM; ■, thiabendazole, Ki = 0.61 μM; ▪, artemisinin, Ki = 2.97 μM.
There was a general agreement between the data from fluorescence-based and that from the HPLC-based assays using recombinant enzymes (Table3) in that compounds that were not inhibitors with the fluorescence assays showed reduced or no effect on enzyme activity using the HPLC-based assays. The compounds with lowKi values in the fluorescence-based assays caused, as expected, a great reduction in activity at 20 μM using HPLC-based assays.
Comparison of FLUO assays and HPLC assays in determination of the inhibitory effects of antiparasitic drugs on the five major drug-metabolizing rCYPs
Apparent Activation of CYP Activity.
Some compounds showed an apparent activation of the activity of some of the CYPs (Table 3). An increase in activity of more than 20% compared with the control activity was considered to be apparent activation. Activation was observed with atovaquone on CYP1A2, niclosamide on CYP2C9, and diethylcarbamazine on CYP2C19. This apparent activation of enzyme activities observed from fluorescence-based assays was, however, not seen with the HPLC-based assays using recombinant enzymes (Table3). Instead, niclosamide inhibited CYP2C9-catalyzed diclofenac 4-hydroxylation (Ki = 6.00 μM). The apparent activation of CYP2C9 by niclosamide was further studied (Fig.2). In the presence of niclosamide, the CYP2C9 activity was increased by about 400%. Interestingly, coincubation of niclosamide with sulfaphenazole, a potent inhibitor of CYP2C9, resulted in inhibition of enzyme activity by 30% (i.e., abolishing the activation by niclosamide and reducing the inhibitory effect of sulfaphenazole). Further experiments showed that the apparent activation was not due to fluorescence by a complex of niclosamide and the fluorescent metabolite, or a complex of an unknown metabolite of niclosamide and the substrate, or a complex of an unknown metabolite of niclosamide and the fluorescent metabolite (7-hydroxy-4-trifluoromethylcoumarin).

Apparent activation of recombinant CYP2C9 activity by niclosamide.
○, sulfaphenazole, Ki = 0.67 μM; ●, niclosamide; ■, sulfaphenazole and niclosamide.
Comparison of Inhibition Data in the Different Systems.
For the potent inhibitors of CYP1A2 and -2D6, theKi values obtained using the fluorescence assays and those obtained using the HPLC assays showed a fairly good agreement; r2 = 0.81 (Fig.3A) using recombinant CYPs. There was also good agreement between Ki values obtained using recombinant CYPs and using human liver microsomes on the same HPLC marker reactions, r2 = 0.825 (Fig. 3B). Importantly, there was good correlation,r2 = 0.724, betweenKi values obtained using the fluorescence assays with recombinant CYPs and those obtained using HPLC-based assays with human liver microsomes (Fig. 3C). Quinine and dihydroartemisinin had Ki values of 2.40 and 16.10 μM using recombinant CYP2D6 and -1A2 but had relatively different values, 15.51 and 3.67 μM, using human liver microsomes on the same CYP marker reactions, respectively.
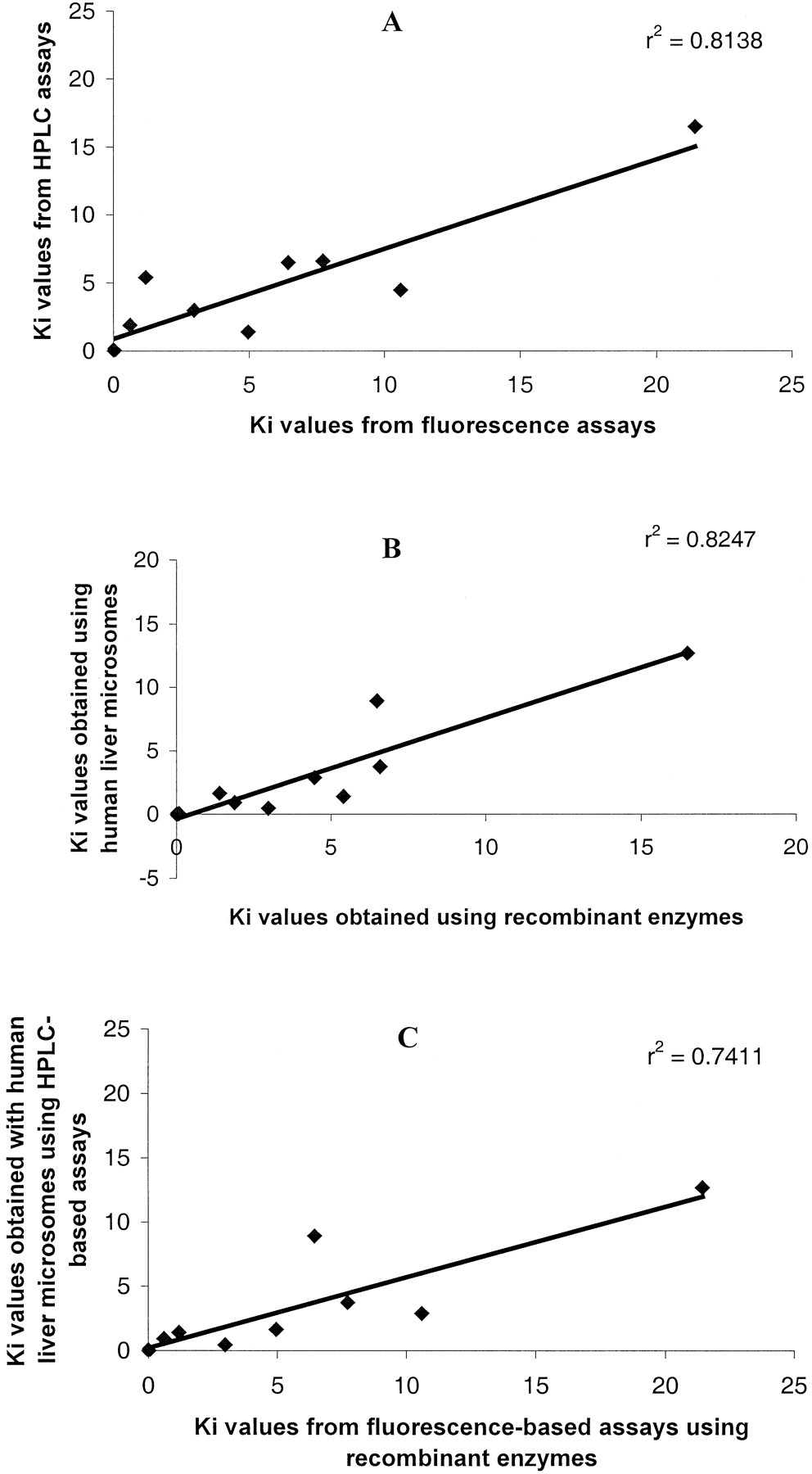
Correlations of Ki values obtained for 10 inhibitors of CYP1A2 and CYP2D6 using different systems.
A, data using the fluorescence assays with recombinant CYPs against data using HPLC assays with recombinant enzymes. B, data using HPLC assays with recombinant CYPs against data using HPLC assays with human liver microsomes. C, data using fluorescence assays with recombinant CYPs against data using HPLC assays with human liver microsomes.
Prediction of in Vivo Inhibitory Effects from in Vitro Data.
The antiparasitic drugs (Table 4) affected CYP1A2 and -2D6 the most. Potent inhibitors of CYP1A2 included primaquine (Ki = 0.22 μM), thiabendazole (Ki = 1.54 μM), artemisinin (Ki = 0.43 μM), dihydroartemisinin (Ki = 3.67 μM), and niclosamide (Ki = 2.70 μM). Proguanil, cycloguanil, amodiaquine, desethylamodiaquine, chloroquine, and quinine inhibited CYP2D6 with Ki values of 6.76, 5.97, 2.1, 4.13, 12.68, and 15.51 μM, respectively. The maximum plasma concentration (Cmax) the drugs attain in vivo at doses used for treatment were used to calculate the percentage of inhibition of the enzymes in vivo (Table 4). Thiabendazole, primaquine, and artemisinin are expected to significantly inhibit CYP1A2 in vivo by 98, 67, and 76%, respectively. Amodiaquine and its metabolite desethylamodiaquine are expected to inhibit about 97 and 99% of CYP2D6 activity in vivo, respectively.
Discussion
The results of our study show a reasonable agreement between inhibition data obtained using the fluorescence-based assays and the HPLC-based assays and between recombinant CYPs and human liver microsomes. A number of antiparasitic drugs have been shown to inhibit some CYPs with potential clinical consequences.
Methodological Issues.
The good correlation of Ki values obtained using rCYPs with the fluorometric assays and the HPLC assays means that there will not be much loss in data quality in the move to the HTS assays. The best correlation was between inhibition data from rCYP and HLMs using the same HPLC marker reactions. This further validates the use of easily available and cheaper rCYP (Masimirembwa et al., 1999) in our HTS assays. A better correlation was observed between inhibition data obtained using the fluorometric assays with rCYP and the HPLC assays with human liver microsomes compared with correlations reported for some inhibitors on different CYP3A4 marker substrates (Kenworthy et al., 1999). In this study, only quinine and dihydroartemisinin, among the potent inhibitors, showed very differentKi values when using rCYP and HLM, results for which we currently have no explanation. Using the fluorometric assays, there were 3 of 29 cases of apparent activation of CYP activity (Table 3). Using the HPLC marker reactions, the apparent activators were either noninhibitors (atovaquone and diethylcarbamazine) or potent inhibitors (niclosamide). For primary screens, the benefits of the HTS fluorescence assays far outweigh the few discordant cases. Besides the huge save in costs (with respect to reagents, equipment, and human resources), the saving in time is tremendous.
Cases of Apparent CYP Activation.
In our study, we observed apparent activation of CYP1A2 by atovaquone and CYP2C19 by diethylcarbamazine. We also observed activation of CYP2C9 by niclosamide of up to 400%. In the presence of the potent CYP2C9 inhibitor sulfaphenazole, niclosamide's activation was abolished (Fig. 2), indicating a role of the enzyme in the increase in fluorescence. Further experiments, however, showed that the activation was not due to the fluorescence of niclosamide, its possible metabolite, or complexes of niclosamide or its metabolite with 7-MFC or 7-hydroxy-4-trifluoromethylcoumarin. We, therefore, cannot explain the cause of the apparent activation. Recently, acetonitrile has been shown to cause substrate-dependent effects on CYP2C9 activity (Tang et al., 2000). The solvent increased diclofenac 4-hydroxylase and tolbutamide methyl hydroxylase activities, but decreased celecoxib methyl hydroxylase activity and had no effect on phenytoin hydroxylase.
Prediction of in Vivo Drug-Drug Interactions.
The inhibition parameters, Ki and IC50, generated in vitro only become useful when they can successfully predict in vivo effects. For inhibition-based interactions, one of the major difficulties is estimating the actual concentration of the inhibitor available to the enzyme. With respect to antiparasitic drugs, some of which accumulate in special tissues (e.g., chloroquine in the liver and melanin-containing tissues), calculation of the concentration of drug reaching the enzyme is even more difficult (Abdi et al., 1995). In addition, the fraction of the dose metabolized by a particular pathway is also important because it determines the magnitude of an interaction (Rodrigues and Wong, 1997). In this study, we have used the maximum plasma concentration (Cmax) as the concentration of drug available to the enzyme and no attempt was made to estimate the concentration of inhibitor in liver or to correct for protein binding.
Most of the antiparasitic drugs are predicted not to pose any risk for important drug-CYP interactions. Some of the drugs, however, could have clinically important interactions with CYP1A2 and CYP2D6. Potent inhibitors of CYP1A2 included artemisinin, dihydroartemisinin, thiabendazole, niclosamide, and primaquine. Cycloguanil, proguanil, amodiaquine, desethylamodiaquine, chloroquine, and quinine were potent inhibitors of CYP2D6. Niclosamide potently inhibited CYP3A4.
Thiabendazole, a broad-spectrum anthelminthic drug (Edwards and Breckenridge, 1988) has been shown to increase theophylline serum levels, resulting in serious side effects (Lew et al., 1989; Schneider et al., 1990). The Ki for thiabendazole's inhibition of CYP1A2 obtained with human liver microsomes (1.54 μM) is far below the plasma concentration it achieves (89 μM). Thiabendazole is, therefore, predicted to inhibit 98% of CYP1A2 activity in vivo (Tables 3 and 4). Given that theophylline is metabolized by CYP1A2 (Tjia et al., 1996) and has a narrow therapeutic index, our results suggest that the interaction between thiabendazole and theophylline may be due to the inhibition of CYP1A2 by thiabendazole.
In our study, primaquine is predicted to inhibit 67% of CYP1A2 activity in vivo (Table 4). Primaquine has been shown to decrease the clearance of antipyrine, resulting in an increase in its half-life (Back et al., 1983). Antipyrine has been shown to be metabolized by many CYPs with CYP1A2 significantly contributing to its metabolism (Sharer and Wrighton, 1996). Our results, therefore, show that the interaction between antipyrine and primaquine may be due to inhibition of CYP1A2 by primaquine. Recently, primaquine has been recommended for use as prophylactic for malaria (Schwartz and Regev-Yochay, 1999). There is, therefore, a high risk of it interacting with drugs that are substrates of CYP1A2 because the drug will be taken for up to 3 weeks.
Thiabendazole and primaquine show selective inhibition of CYP1A2 with respect to the five CYP isoforms in this study. They could, therefore, be used as selective positive control inhibitors instead of the commonly used mechanism-based inhibitor furafylline (Clarke et al., 1994), the toxic α-naphthoflavone (Liehr et al., 1991), or the unselective fluvoxamine (Venkatakrishnan et al., 1999).
Artemisinin and dihydroartemisinin are very important antimalarials because they are effective against multidrug-resistant strains of malaria parasites (Dhingra et al., 2000). The drugs are usually used in combination with other antimalarials (Na-Bangchang et al., 1999). Careful drug and patient monitoring should be done when artemisinin is coadministered with drugs that are CYP1A2 substrates because it is predicted to inhibit about 76% of CYP1A2 activity in vivo. Although niclosamide is a potent inhibitor of CYP1A2 and -3A4, it is not significantly absorbed (Abdi et al., 1995) and is, therefore, not expected to inhibit these enzymes in vivo.
Amodiaquine, an antimalarial, is converted in the body to the active metabolite desethylamodiaquine, which has a much longer elimination half-life than the parent drug. Amodiaquine and desethylamodiaquine are predicted to inhibit 97 and 99% of CYP2D6 activity in vivo, respectively. This means that an individual's metabolic status could be changed from an extensive metabolizer to a poor metabolizer status with respect to CYP2D6 activity after administration of amodiaquine. Knowledge of these potential interactions with CYP2D6 is important when doing phenotyping studies (Masimirembwa and Hasler, 1997). CYP2D6 has been shown to be responsible for metabolism of many antipsychotic drugs, most of which have narrow therapeutic indices. Our results show that amodiaquine and its metabolite might interact with coadministered antipsychotic drugs metabolized by CYP2D6 with possible clinically significant consequences.
Conclusions
This study has evaluated the reliability of using data obtained using HTS fluorescence assays with rCYPs. Data from these assays are of sufficient quality to use in the drug discovery process. Assessment of the potential of antiparasitic drugs to inhibit CYPs showed that some of them could result in undesirable interactions with CYP1A2 and -2D6. Our results also suggest inhibition of CYP1A2 as the mechanism behind the observed thiabendazole/theophylline and primaquine/antipyrine interactions in vivo.
Acknowledgments
We thank Drs. Richard Thompson, Tommy B. Andersson, and Scott Boyer for useful discussions and Katarina Rubin for technical assistance.
Footnotes
-
Send reprint requests to: Dr. Collen Mutowembwa Masimirembwa, Department of DMPK and Bioanalytical Chemistry, AstraZeneca, R & D Mölndal, S-431 83 Mölndal, Sweden. E-mail: collen.masimirembwa{at}astrazeneca.com
-
Tashinga Bapiro is a recipient of a fellowship from International Programme in Chemical Sciences, Uppsala University, Sweden.
- Abbreviations used are::
- CYP cytochrome P450
- HPLC, high-performance liquid chromatography
- HTS
- high throughput screening
- rCYP
- recombinant cytochrome P450
- FLUO
- fluorescence
- HLM
- human liver microsome
- MFC
- 7-methoxy-4-trifluoromethylcoumarin
- Received June 30, 2000.
- Accepted October 17, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}