


Abstract
10-((4-Hydroxypiperidin-1-yl)methyl)chromeno[4,3,2-de]phthalazin-3(2H)-one (E7016), an inhibitor of poly(ADP-ribose) polymerase, is being developed for anticancer therapy. One of the major metabolites identified in preclinical animal studies was the product of an apparent oxidation and ring opening of the 4-hydroxypiperidine. In vitro, this oxidized metabolite could not be generated by incubating E7016 with animal or human liver microsomes. Further studies revealed the formation of this unique metabolite in hepatocytes. In a NAD(P)+-dependent manner, this metabolite was also generated by liver S9 fractions and recombinant human flavin-containing monooxygenase (FMO) 5 that was fortified with liver cytosol fractions. In animal and human liver S9, this metabolic pathway could be inhibited by 4-methylpyrazole, bis-p-nitrophenylphosphate (BNPP), or a brief heat treatment at 50°C. Based on these results, the overall metabolic pathway was believed to involve a two-step oxidation process: dehydrogenation of the secondary alcohol in liver cytosol followed by an FMO5-mediated Baeyer-Villiger oxidation in liver microsomes. The two oxidation steps were coupled via regeneration of NAD(P)+ and NAD(P)H. To further confirm this mechanism, the proposed ketone intermediate was independently synthesized. In an NAD(P)H-dependent manner, the synthetic ketone intermediate was metabolized to the same ring-opened metabolite in animal and human liver microsomes. This metabolic reaction was also inhibited by BNPP or a brief heat treatment at 50°C. Methimazole, the substrate/inhibitor of FMO1 and FMO3, did not inhibit this reaction. The specificity of FMO5 toward catalyzing this Baeyer-Villiger oxidation was further demonstrated by incubating the synthetic ketone intermediate in recombinant enzymes.
Introduction
Flavin-containing monooxygenases (FMOs) comprise a group of flavin adenine dinucleotide-containing enzymes that are widely present in nature. They generally catalyze oxidation of compounds containing soft nucleophilic groups. Typical FMO-catalyzed reactions are monooxygenation of heteroatoms such as nitrogen, sulfur, and phosphorus (Hines et al., 1994; Cashman 1995; Ziegler 2002; Lang and Kalgutkar 2003). Although NADPH is apparently the preferred cofactor of FMOs, the enzymes can use either NADPH or NADH as the electron supplier to complete their catalytic cycle (Hvattum et al., 1991; Sausen et al., 1993). The key intermediate of the FMO catalytic cycle is a reactive peroxide intermediate C(4α)-hydroperoxyflavin. In some cases, the reactive C(4α)-hydroperoxyflavin can act as an “enzymatic peroxide” to catalyze a Baeyer-Villiger oxidation of a carbonyl compound, such as a ketone or an aldehyde. Such reactions are well known in microbials and plants but have not been widely recognized in mammalian species (Wong and Whitesides 1994). Using enzyme isolated from pig liver microsomes, salicylaldehyde was reported to undergo a Baeyer-Villiger oxidation catalyzed by FMO1 (Chen et al., 1995). The major metabolite of a keto progestagen (17α)-17-hydroxy-11-methylene-19-norpregna-4,15-dien-20-yn-3-one (Org 30659), was reported to be a ring-opened hydroxyl carboxylic acid. The mechanism of its formation was speculated to be an FMO-mediated Baeyer-Villiger oxidation. However, there was no evidence to support the involvement of a specific FMO isoform in this biotransformation (Verhoeven et al., 1998).
As an inhibitor of poly(ADP-ribose) polymerase, 10-((4-hydroxypiperidin-1-yl)methyl)chromeno[4,3,2-de]phthalazin-3(2H)-one (E7016) (Fig. 1) is currently being investigated in the clinic as a potential anticancer agent. In preclinical studies, a ring-opened hydroxyl carboxylic acid, 3-((2-hydroxyethyl)((3-oxo-2,3-dihydrochromeno[4,3,2-de]phthalazin-10-yl)methyl)amino)propanoic acid (ER-879123), was identified as one of the major metabolites of E7016 in rats and dogs in vivo. In dogs, the metabolite was isolated from the feces. The structure of this metabolite was identified by proton NMR and mass spectrometry and confirmed by comparison with a chemically synthesized standard. However, the mechanism for the formation of ER-879123 was unknown. Furthermore, this metabolite was not detected by incubating E7016 with liver microsomes. In the current in vitro study, we report that the mechanism leading to the formation of this unique metabolite involves an FMO5-mediated Baeyer-Villiger oxidation of a ketone intermediate.

Structures of E7016 and its Baeyer-Villiger metabolite, ER-879123, with synthetic conditions for the ketone intermediate, ER-879819, and ER-879123. (COCl)2, oxalyl chloride; DCE, 1,2-dichloroethane; Et3N, triethylamine; KOH, potassium hydroxide; THF, tetrahydrofuran; AcOH, acetic acid; MeOH, methanol; RT, room temperature.
Materials and Methods
Materials.
E7016, its hydroxyl carboxylate metabolite ER-879123, and the ketone intermediate 10-((4-oxopiperidin-1-yl)methyl)chromeno[4,3,2-de] phthalazin-3(2H)-one (ER-879819) were synthesized by the Process Research Department, Eisai Inc. (Andover, MA). NAD+ and NADP+ were purchased from Calbiochem (San Diego, CA). NADH and NADPH were purchased from Sigma-Aldrich (St. Louis, MO). Potassium phosphate buffer was obtained from BD Biosciences (San Jose, CA). All solvents used were of high-performance liquid chromatography (HPLC) grade. Acetonitrile was purchased from Mallinckrodt Baker, Inc. (Phillipsburg, NJ), and other solvents were purchased from EMD Science (San Diego, CA).
Recombinant FMO Supersomes, pooled liver S9 fractions, pooled human liver cytosolic fractions, and liver microsomes were purchased from BD Biosciences. Pooled dog and rat liver cytosolic fractions were purchased from XenoTech, LLC (Lenexa, KS). Pooled human and dog cryopreserved hepatocytes were purchased from In Vitro Technologies (Baltimore, MD), and pooled rat cryopreserved hepatocytes were purchased from CellzDirect (Durham, NC). Human materials comprised a mixture of both male and female donors; dog and rat materials were from male animals only.
Analytical Methods.
Analyses were performed on an AB Sciex QSTAR Elite mass spectrometer and an AB Sciex API4000 QTrap mass spectrometer (AB Sciex, Framingham, MA). Both mass spectrometers were interfaced with HPLC systems including LC-10AD binary pumps and SIL-HTC autosamplers (Shimadzu, Kyoto, Japan). The HPLC column used was a Synergi Fusion-RP column (150 × 2.00 mm, 4 μ; Phenomenex, Torrance, CA). HPLC resolution was achieved with a gradient consisting of solvent A (water-acetonitrile-acetic acid, 95:5:0.05) and solvent B (water-acetonitrile-acetic acid, 5:95:0.05). The gradient consisted of the following steps: hold 99% solvent A for 1 min, then a linear gradient from 99 to 95% solvent A between 1 and 5 min, from 95 to 80% solvent A between 5 and 10 min, and from 80 to 5% solvent A between 10 and 18 min, hold 5% solvent A between 18 and 20 min before returning to 99% solvent A, and hold for an additional 4 min to reequilibrate the column. The flow rate was 350 μl/min, and the injection volume was 10 μl. Both mass spectrometers were operated in positive ion scan mode using the TurboSprayIon source. The ionization spray voltage was set at 5500 V, and source temperature was kept at 500°C. The QSTAR Elite mass spectrometer was operated in the product ion scan mode and was calibrated externally and daily using the parent ion and product ions of E7016 (m/z 350.1499, m/z 249.0659, and m/z 100.0757). The product ion scan of m/z 382.10 was used for monitoring formation of ER-879123. After data collection, the mass spectra were internally recalibrated using the parent ion of ER-879123 (m/z 382.1397). Data processing was performed using AB Sciex Analyst QS 2.0 software. The API4000 QTrap mass spectrometer was used to monitor the formation of ER-879123 in the selected reaction-monitoring (SRM) mode with the transition of m/z 382 to m/z 249. The formation of a potential lactone intermediate was also monitored in some samples using the SRM transition of m/z 364 to m/z 249. Data processing was performed using AB Sciex Analyst 1.4 software.
To characterize the chemically synthesized ketone intermediate ER-879819 and ER-879123, both 1H and 13C NMR spectra were obtained on a Unity Inova NMR spectrometer (Varian, Palo Alto, CA) at 400 MHz for 1H spectra and at 100 MHz for 13C spectra. 2H6-DMSO was used as the solvent for both compounds. Chemical shifts are reported as δ values relative to internal DMSO (1H δ 2.50 ppm, 13C δ 40.6 ppm). High resolution mass spectrometry data on the compounds were obtained on a Micromass Q-TOF Micro mass spectrometer (Waters, Milford, MA) using electrospray ionization (ESI) in the positive ion scan mode.
Synthesis of ER-879123.
To a solution of tert-butyl 3-((2-hydroxyethyl)((3-oxo-2,3-dihydrochromeno[4,3,2-de]phthalazin-10-yl)methyl)amino)propanoate (ER-886830) (1.0 g, 2.28 mmol, 1.0 Eq) (Fig. 1) in tetrahydrofuran (15 ml) and water (5 ml) was added solid potassium hydroxide (663 mg, 11.8 mmol, 5.2 Eq). The resulting mixture was allowed to stir for 60 h, after which it was neutralized with acetic acid (0.62 ml, 11.8 mmol, 5.2 Eq), resulting in the precipitation of a gummy solid. Methanol (4.5 ml) was added, resulting in a free-flowing solid. This slurry was allowed to stir for 3 h, after which it was filtered and washed with 1:1 methanol/water (one 4-ml wash) and water (one 6-ml wash). The isolated solid was allowed to dry under vacuum to afford ER-879123 (744 mg, 85% yield) as a white solid.
Synthesis of the Ketone Intermediate ER-879819.
To a stirred solution of oxalyl chloride (0.775 ml, 9.16 mmol, 3.2 Eq) in 1,2-dichloroethane (20 ml) at 0°C DMSO was added dropwise (5 ml, 70 mmol, 25 Eq), resulting in gas evolution. This mixture was stirred for 30 min at 0°C, after which a slurry of E7016 (1.0 g, 2.86 mmol, 1.0 Eq) in DMSO (5 ml) was added in one portion. The resulting mixture was stirred for 30 min, after which triethylamine (3.2 ml, 22.9 mmol, 8.0 Eq) was added. The mixture was allowed to warm to room temperature with stirring overnight. The mixture was added to water (250 ml) and stirred for 30 min. Stirring was halted, and the mixture was allowed to settle. The supernatant solution was decanted, and the remaining lower gummy material was slurried in methanol (30 ml) for 2 h to solidify. The solids were filtered and washed with methanol (two 1-ml washes) and allowed to dry under vacuum for 6 h. The mother liquors were concentrated to dryness, and the residue was slurried in methanol (20 ml) to afford a second crop of solid. In total, 400 mg (40% yield) of ER-879819 was obtained as a white solid.
Incubation of E7016 in Liver Microsomes.
To a final volume of 250 μl of potassium phosphate buffer (0.1 M, pH 7.4), E7016 in acetonitrile stock solution was added at a final concentration of 50 μM (final acetonitrile 1%). The stock solution of cofactor was added to make the final NADPH concentration of 2.5 mM. For control samples, potassium phosphate buffer was added in the place of cofactor. The mixtures were preincubated at 37°C for 3 min. The reactions were initiated by addition of ice-cold dog liver microsomes at a final protein concentration of 0.5 mg/ml, and the incubations were continued at 37°C for 60 min. The liver microsomes were kept on ice before the initiation of reactions. At the end of the incubation, an equal volume (250 μl) of methanol-acetonitrile (v/v, 1:1) was added to quench the reactions. After centrifugation at 2000g for 10 min on a Beckman Allegra 6R centrifuge (Beckman Coulter, Fullerton, CA), the supernatants were analyzed for potential formation of ER-879123 with the AB Sciex QSTAR Elite LC-MS/MS system using the product ion scan of m/z 382.1 in the positive ion scan mode. The results were compared with the HPLC retention time and MS/MS fragment pattern of the synthetic ER-879123 standard. Incubations and analyses were also conducted in a similar manner in rat and human liver microsomes at the same final protein concentration.
Incubation of E7016 with Hepatocytes.
To a final volume of 250 μl of Dulbecco's potassium phosphate-buffered saline (0.1 M, pH 7.4), E7016 in acetonitrile stock solution was added at a final concentration of 50 μM (final acetonitrile 1%). The mixtures were preincubated at 37°C for 3 min. Reactions were initiated by adding ice-cold rat, dog, or human hepatocytes (1 × 106 cells/ml). The hepatocytes were kept on ice before the initiation of reactions. The incubations were continued at 37°C for 120 min. At the end of the incubation, an equal volume (250 μl) of methanol-acetonitrile (v/v, 1:1) was added to quench the reactions. After centrifugation at 2000g for 10 min on a Beckman Allegra 6R centrifuge, the supernatants were analyzed for potential formation of ER-879123 with the AB Sciex QSTAR Elite LC-MS/MS system using a product ion scan of m/z 382.1 in the positive ion scan mode. The results were compared with the HPLC retention time and MS/MS fragment pattern of the synthetic standard of ER-879123.
Incubation of E7016 in Liver S9 Fractions.
To a final volume of 250 μl of potassium phosphate buffer (0.1 M, pH 7.4), E7016 in acetonitrile stock solution was added at a final concentration of 50 μM (final acetonitrile 1%). A stock solution of NAD+ or NADP+ was also added at a final concentration of 2.5 mM. For control incubations, potassium phosphate buffer was added in place of the cofactor. The mixtures were preincubated at 37°C for 3 min. The reactions were initiated by adding ice-cold S9 fractions of dog or human liver at the final protein concentration of 2 mg/ml, and the incubations were continued at 37°C for 60 min. The liver S9 fractions were kept on ice before the initiation of reactions.
To test the involvement of the thermally unstable FMO in the oxidation, dog or human liver S9 fractions were preincubated at 50°C for 5 min in the absence of cofactors and then cooled on ice. To determine the effects of prototypical inhibitors of alcohol dehydrogenases, FMOs, and esterases on this metabolic pathway, 4-methylpyrazole (0.5 mM final concentration), methimazole (0.5 mM final concentration), or bis(p-nitrophenyl)phosphate (BNPP) (0.5 mM final concentration), respectively, was added.
At the end of incubations, an equal volume (250 μl) of methanol-acetonitrile (v/v, 1:1) was added to quench the reactions. After centrifugation at 2000g for 10 min on a Beckman Allegra 6R centrifuge, the formation of ER-879123 was assessed on the AB Sciex API4000 QTrap LC-MS/MS system in the SRM mode with the transition of m/z 382 to m/z 249.
Incubation of E7016 in Liver Cytosol-Fortified Recombinant FMOs.
To a final volume of 250 μl of potassium phosphate buffer (0.1 M, pH 7.4), E7016 in acetonitrile stock solution was added at a final concentration of 50 μM (final acetonitrile 1%). A stock solution of NAD+ and a stock solution of rat, dog, or human liver cytosol were also added at the final concentrations of 2.5 mM NAD+ and 1 mg/ml cytosolic protein, respectively. For control incubations, potassium phosphate buffer was added in place of either NAD+ or liver cytosol or both. The mixtures were preincubated at 37°C for 3 min, and the reactions were initiated by addition of ice-cold recombinant enzymes of FMO1, FMO3, or FMO5 Supersomes at final protein concentrations of 400 μg/ml. The recombinant enzymes were kept on ice before the initiation of reactions. The incubations were conducted at 37°C for 60 min. At the end of the incubation, an equal volume (250 μl) of methanol-acetonitrile (v/v, 1:1) was added to quench the reactions. After centrifugation at 2000g for 10 min on a Beckman Allegra 6R centrifuge, the supernatants were analyzed for potential formation of ER-879123 on the AB Sciex QSTAR Elite LC-MS/MS system using a product ion scan of m/z 382.1 in the positive ion scan mode. The results were compared with the HPLC retention time and MS/MS fragment pattern of the ER-879123 synthetic standard.
Incubation of ER-879819 in Liver Microsomes.
To a final volume of 250 μl of potassium phosphate buffer (0.1 M, pH 7.4), ER-879819 in acetonitrile stock solution was added at a final concentration of 10 μM (final acetonitrile 1%). A stock solution of NADH was also added at a final concentration of 2.5 mM. For control incubations, potassium phosphate buffer was added in the place of NADH. The mixtures were preincubated at 37°C for 3 min. The reactions were initiated by addition of ice-cold dog or human liver microsomes at a final protein concentration of 0.5 mg/ml, and the incubations were continued at 37°C for 60 min. The liver microsomes were kept on ice before the initiation of reactions. The effect of heating the liver microsomes and enzymatic inhibitors was tested as before.
At the end of incubations, an equal volume (250 μl) of methanol-acetonitrile (v/v, 1:1) was added to quench the reactions. After centrifugation at 2000g for 10 min on a Beckman Allegra 6R centrifuge, the formation of ER-879123 was assessed on the AB Sciex API4000 QTrap LC-MS/MS system using the SRM mode with the transition of m/z 382 to m/z 249. The formation of a potential lactone intermediate was also monitored by using the SRM transition of m/z 364 to m/z 249.
Incubation of ER-879819 in Recombinant Enzymes.
To avoid thermal inactivation of FMO enzymes, the incubation procedure recommended by BD Biosciences was followed (FMO BD-Supersome Guidelines for Use, 2010, http://www.bdbiosciences.com/external_files/dl/doc/manuals/live/web_enabled/adme_supersomes_fmo_enzymes.pdf). To a final volume of 250 μl of potassium phosphate buffer (0.1 M, pH 7.4), ER-879819 in acetonitrile stock solution was added at a final concentration of 10 μM (final acetonitrile 1%). A stock solution of NADH or NADPH was added at the final concentration of 2.5 mM. For control incubations, potassium phosphate buffer was added in the place of the stock solution of the cofactors. The mixtures were preincubated at 37°C for 3 min, and the reactions were initiated by adding ice-cold recombinant enzymes of FMO5 Supersomes at final protein concentrations of 100 μg/ml. The recombinant enzymes were kept on ice before the initiation of reactions. The incubations were conducted at 37°C for 2, 10, 30, and 60 min. At the end of each time point, an equal volume (250 μl) of methanol-acetonitrile (v/v, 1:1) was added to quench the reactions. After centrifugation at 2000g for 10 min on a Beckman Allegra 6R centrifuge, the formation of ER-879123 was assessed on the AB Sciex API4000 QTrap LC-MS/MS system using the SRM mode with the transition of m/z 382 to m/z 249. The formation of a potential lactone intermediate was also monitored using the SRM transition of m/z 364 to m/z 249.
In the presence of either NADH or NADPH, the formation of ER-879123 was also compared in the incubations of ER-879819 (final concentration 10 μM) with recombinant FMO1, FMO3, or FMO5 at a final protein concentration of 100 μg/ml. The incubations were conducted at 37°C for 60 min. The same sample workup procedure and analytical methods as described above were followed to assess the formation of ER-879123.
Results
Synthesis and Structural Confirmation of ER-879123 and ER-879819.
The structures of E7016 and ER-879123 are shown in Fig. 1. During preclinical development of E7016 as a potential anticancer agent, ER-879123 was identified as one of the major metabolites in animal pharmacokinetic models. In dogs, this metabolite was isolated from the feces and characterized by NMR spectroscopy. Its structure was confirmed by independent chemical synthesis of a standard. The last step of its synthesis is shown in Fig. 1. 1H NMR of ER-879123: δ 12.5 (br s, 1H), 7.75 (s, 1H), 7.66 (m, 2H), 7.42 (dd, J = 2.0, 6.8 Hz, 1H), 7.28 (d, J = 8.4 Hz, 1H), 7.05 (d, J = 8.0 Hz), 3.47 (s, 2H), 3.34 (t, J = 6.2 Hz, 2H), 2.60 (t, J = 7.0 Hz, 2H), 2.36 (t, J = 6.2 Hz, 2H), 2.19 (t, J = 7.0 Hz, 2H). 13C NMR: δ 176.0, 159.7, 151.3, 151.2, 137.3, 135.4, 134.4, 133.2, 128.3, 123.2, 119.8, 119.6, 118.3, 118.1, 117.1, 60.3, 58.5, 56.5, 51.2, 34.8. The exact mass of MH+ determined by high-resolution mass spectrometry was m/z 382.1437, which supported the elemental assignment of C20H20N3O5+ (calculated as 382.1397). The major MS/MS product ion was m/z 249.1, suggesting double oxidation of the 4-hydroxypiperidine moiety of E7016 (Fig. 2A). The 1H NMR spectra of ER-879123 revealed two pairs of methylene protons between δ 2.1 and 3.4 ppm. Both sets of methylene protons were triplets with coupling constants of 6.2 and 7.0 Hz, respectively, resulting from vicinal coupling with the adjacent methylene group.

Lack of formation of ER-879123 on incubation of E7016 with dog liver microsomes. LC-MS/MS chromatograms and product ion spectra of m/z 382.1 for the ER-879123 standard (A) and for samples obtained from 60-min incubations of E7016 in dog liver microsomes (B) are shown.
To probe the mechanism of ER-879123 formation, the ketone intermediate ER-879819 was synthesized from E7016. Figure 1 also shows the synthetic conditions. 1H NMR of ER-879819: δ 12.6 (br s, 1H), 8.07 (d, J = 2 Hz, 1H), 7.90 (comp m, 2 H), 7.70 (dd, J = 2.0, 7.2 Hz, 1H), 7.54 (dd, J = 2.0, 8.4 Hz, 1H), 7.39 (d, J = 8.4 Hz, 1H), 3.69 (s, 2H), 2.73 (t, J = 6.0 Hz, 4H), 2.37 (t, J = 6.0 Hz, 4H). 13C NMR: δ 209.4, 159.8, 151.7, 131.4, 136.2, 135.5, 134.7, 133.4, 128.5, 123.7, 120.0, 119.8, 118.7, 118.5, 117.3, 61.0, 53.4, 41.8. High-resolution mass spectrometry of the MH+ gave m/z of 348.1378, which supported the elemental assignment of C20H18N3O3+ (calculated as 348.1343).
Lack of ER-879123 Formation in Incubations of E7016 with Liver Microsomes.
After a 60-min incubation of E7016 with dog liver microsomes in the presence of NADPH, several double oxidation metabolites were detected using the product ion scan at m/z 382.1. Two of them had retention times similar to that of the ER-879123 synthetic standard. However, both metabolites had a major MS/MS fragment of m/z 265.1, suggesting addition of a single oxygen to the 4-hydroxypiperidine moiety and another single oxygen on the left-hand side chromeno[4,3,2-de]phthalazin-3(2H)-one moiety (Fig. 2B). Metabolism of E7016 was also studied in rat and human liver microsomes. Similar to the observation in dog liver microsomes, no formation of ER-879123 was detected (data not shown).
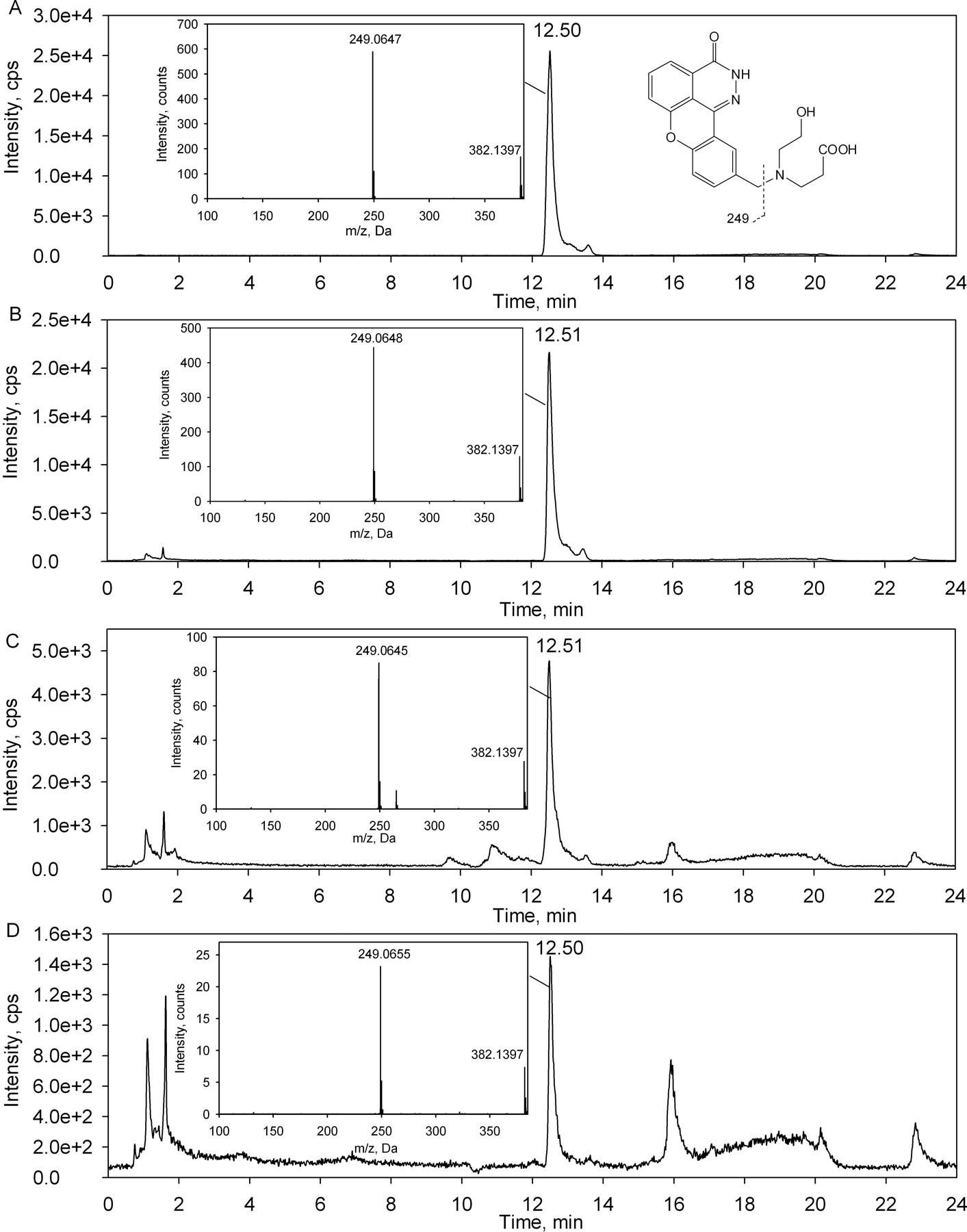
Formation of ER-879123 in Incubations of E7016 with Hepatocytes.
After a 120-min incubation of E7016 with suspensions of rat, dog, or human hepatocytes, formation of ER-879123 was detected in all three species (Fig. 3). The ring-opened hydroxyl carboxylate metabolite generated in each species matched both the HPLC retention time and MS/MS fragment of the synthetic standard. The major MS/MS fragment of m/z 249.1 indicated a double oxidation on the 4-hydroxypiperidine ring. The relative amount of this metabolite formed after a 120-min incubation was found to be most abundant in the rat followed by dog and human under the same incubation conditions (Fig. 3).

Formation of ER-879123 on incubation of E7016 with hepatocytes. LC-MS/MS chromatograms and product ion spectra of m/z 382.1 for ER-879123 standard (A) and for samples obtained from 120-min incubations of E7016 in hepatocytes of rats (B), dogs (C), and humans (D) are shown.
Formation of ER-879123 in Incubations of E7016 with Liver S9.
Formation of ER-879123 was observed after a 60-min incubation of E7016 with dog or human liver S9 in the presence of NAD+ or NADP+ (Table 1). The reaction was clearly cofactor-dependent. NADP+ seemed to be a less favorable cofactor in these incubations. In dog and human S9, the extent of ER-879123 formation with NADP+ as the cofactor was approximately 40 and 65%, respectively, of that observed with NAD+ as the cofactor. The alcohol dehydrogenase inhibitor, 4-methylpyrazole, decreased the formation of ER-879123 by approximately 30% in both dog and human. The carboxylesterase inhibitor, BNPP, decreased the formation of this metabolite by approximately 70% in dog and by 25% in human. The FMO1 and FMO3 substrate/inhibitor methimazole did not inhibit this biotransformation. However, a brief 50°C treatment of liver S9 in the absence of cofactor decreased the formation of ER-879123 by more than 80% in dog S9 and by approximately 20% in human S9.
Formation of ER-879123 after 60-min incubations of E7016 with dog and human liver S9 fractions under various incubation conditions
Data are means ± S.D. n = 3.
Formation of ER-879123 in Incubations of E7016 with Liver Cytosol-Fortified FMOs.
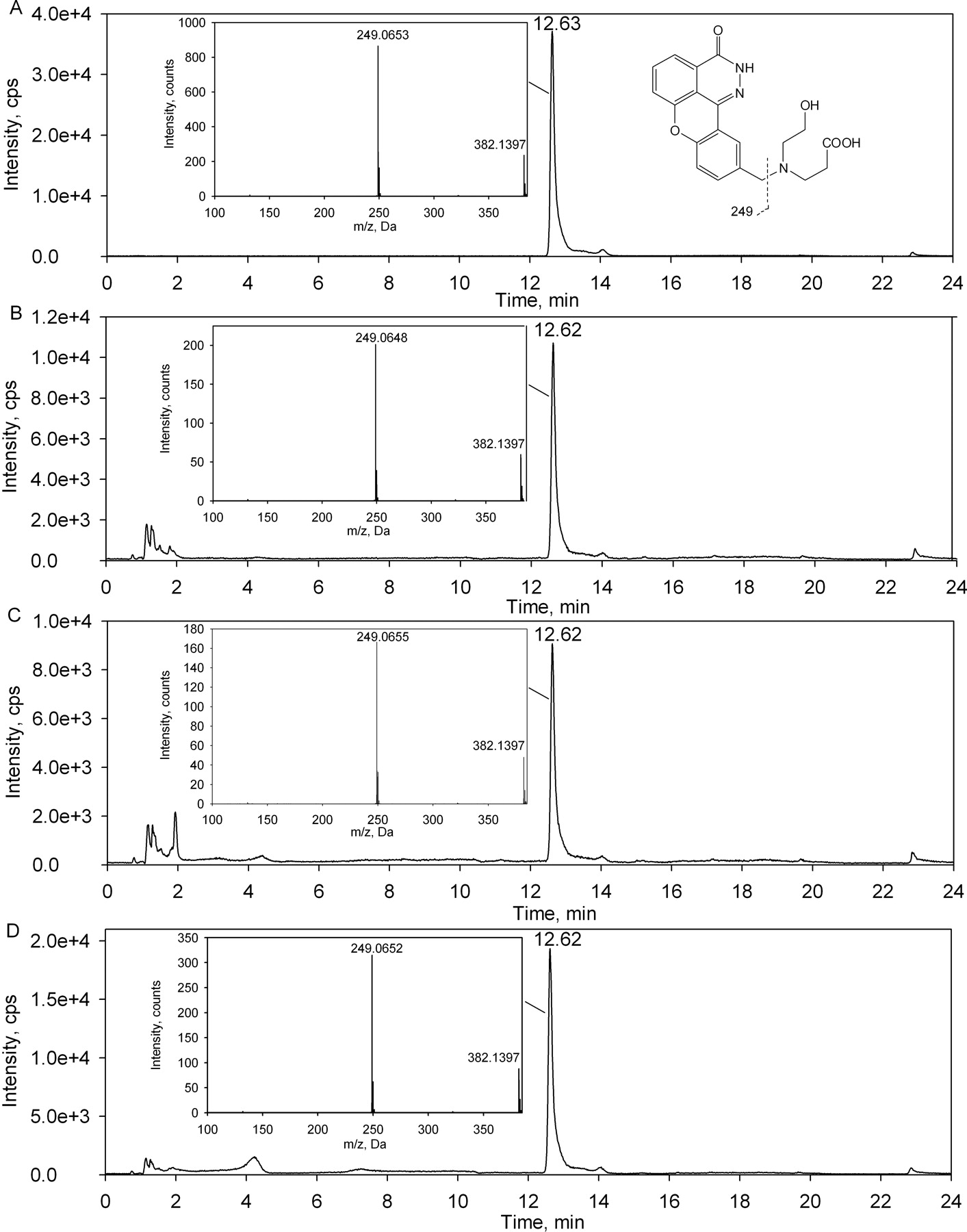
In the presence of NAD+, after a 60-min incubation of E7016 with Supersomes of recombinant FMO5 fortified with liver cytosolic fractions, a significant amount of ER-879123 was detected. The HPLC retention time and MS/MS fragmentation of the double oxidation metabolite (MH+ m/z 382.1) found in the incubations matched those of the synthetic standard of ER-879123 (Fig. 4). No such metabolite could be detected in the incubations without either liver cytosolic fractions or cofactor.

LC-MS/MS chromatograms for a product ion scan of m/z 382.1 for ER-879123 (A) and for samples obtained from 60-min incubations of E7016 with Supersomes of recombinant FMO5 fortified with liver cytosolic fractions of rats (B), dogs (C), and humans (D) are shown. The incubations were conducted in the presence of NAD+.
With recombinant human FMO1 and FMO3 fortified with dog liver cytosol, no formation of ER-879123 was detected after incubations of E7016 in the presence of NAD+. In the incubation with FMO3, there was no metabolite detected with a molecular ion of m/z 382.1. In the incubation with FMO1, a metabolite was detected with the molecular ion of m/z 382.1 and the major MS/MS fragment of m/z 249.1. However, the HPLC retention time of this metabolite was at 10.7 min, which was much shorter than that of the ER-879123 standard. Of interest, the formation of this unknown metabolite in cytosol-fortified recombinant FMO1 was also dependent on the presence of both NAD+ and liver cytosol.
Formation of ER-879123 in Incubations of ER-879819 with Liver Microsomes.
Formation of ER-879123 was observed after a 60-min incubation of ER-879819 with dog or human liver microsomes in the presence of NADH (Table 2). The reaction was again cofactor-dependent. The amount of this metabolite formed in human liver microsomes, under the same incubation conditions, was much less than that observed in dog liver microsomes. The carboxylesterase inhibitor, BNPP, decreased the formation of this metabolite by approximately 60% in both dog and human microsomes. When the formation of ER-879123 decreased with the presence of BNPP, a possible lactone intermediate with molecular ion MH+ of m/z 364 and a major MS/MS fragment of m/z 249 was detected. Consistent with the incubations of E7016 in liver S9 fractions, the FMO1 and FMO3 substrate/inhibitor methimazole showed no inhibitory effect toward this biotransformation. However, a brief 50°C treatment of liver microsomes in the absence of cofactor decreased the formation of ER-879123 by nearly 90% in dog microsomes and by approximately 40% in human microsomes.
Formation of ER-879123 after 60-min incubations of ER-879819 with dog and human liver microsomes under various incubation conditions
Data are means ± S.D. n = 3.
Formation of ER-879123 in Incubations of ER-879819 with Recombinant Enzymes.
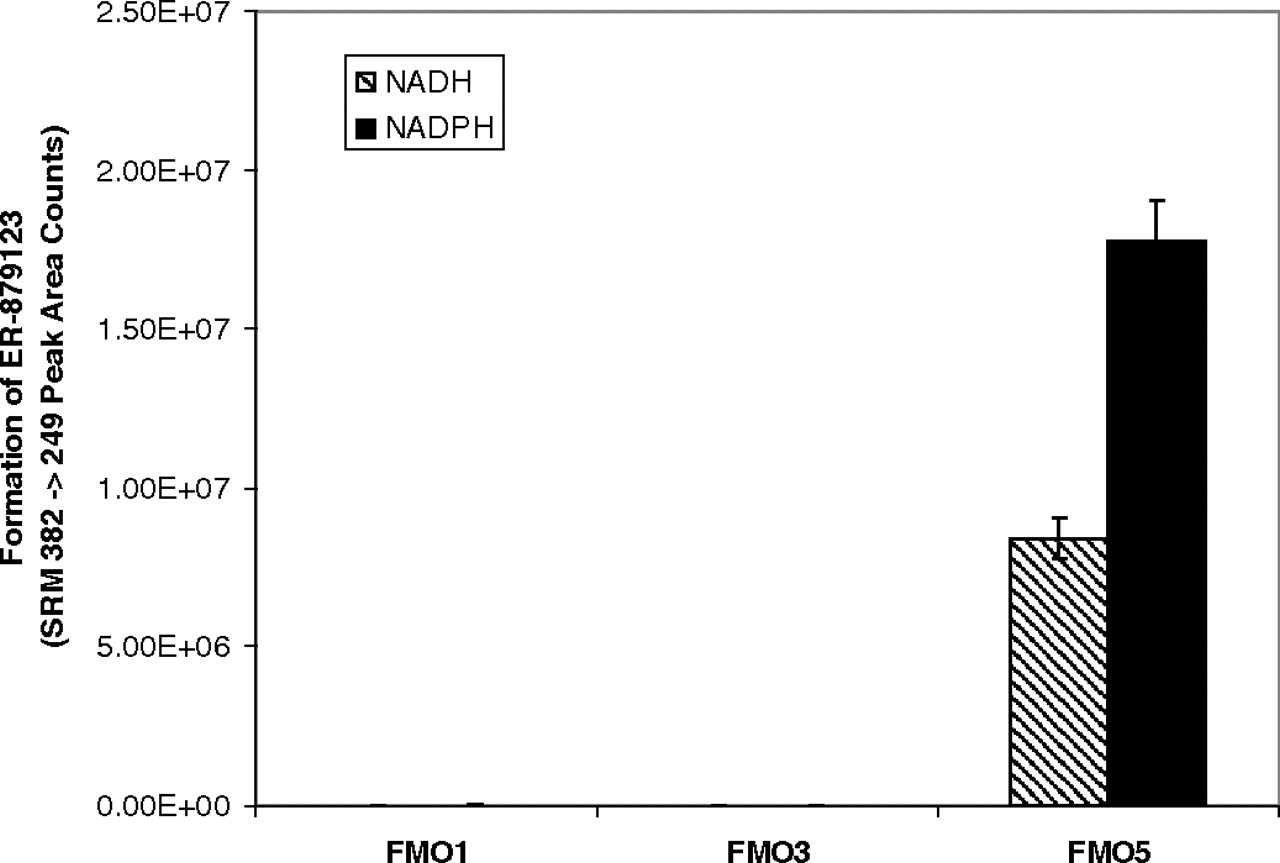
In the presence of either NADH or NADPH, a significant and time-dependent formation of ER-879123 (m/z 382 → m/z 249) was detected after incubation of the ketone ER-879819 with recombinant FMO5 (Fig. 5A). A time-dependent formation of the potential lactone intermediate (m/z 364 → m/z 249) was also detected (Fig. 5B). Insignificant amounts of the metabolites were detected in the incubations without cofactors. For either metabolite, a slightly higher turnover was observed using NADPH as the cofactor, rather than NADH. This observation was in contrast with the observation of the incubations of E7016 with liver S9 fractions for which NAD+ was clearly the preferred cofactor for the formation of ER-879123. The result implies that the cofactor preference of the overall metabolic pathway of E7016 may be more related to the first oxidoreductase-mediated step than to the second FMO5-mediated step. After incubation of ER-879819 with recombinant enzymes of major FMO isoforms, the formation of ER-879123 was detected only in the FMO5 incubation (Fig. 6).
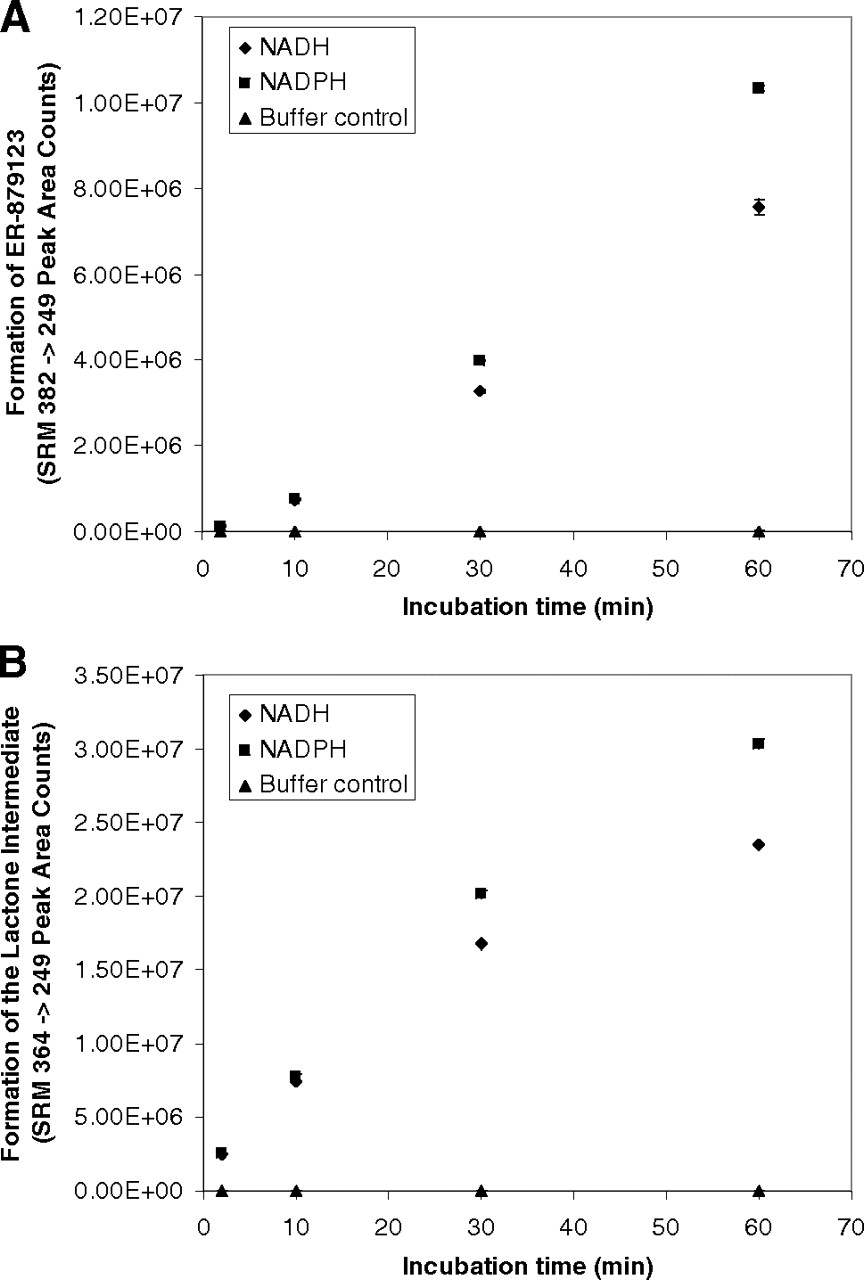
Time courses for the formation of ER-879123 (A) and the putative lactone intermediate (B) in incubations of the ketone intermediate ER-879819 with recombinant FMO5 (n = 3; mean ± S.D.).

Specificity of FMO5 for the formation of ER-879123 assessed by incubation of ER-879819 in recombinant enzymes (n = 3; mean ± S.D.).
Discussion
Oxygenation of an FMO substrate is mediated by the reactive –OOH moiety of the enzyme. During this process a water molecule is released (Eswaramoorthy et al., 2006). Because of this general mechanism, metabolites formed by FMOs are often the same as products of a chemical reaction involving a peroxyacid or peroxide (Ziegler 2002). Baeyer-Villiger oxidation involves insertion of an oxygen atom into the carbon–carbon bond next to a carbonyl group (aldehyde or ketone) to form an ester. Chemically, it is catalyzed by peroxyacid compounds (Renz and Meunier 1999). Biologically, it can be catalyzed by a group of flavin-containing enzymes called Baeyer-Villiger monooxygenases (BVMOs). Widely used in organic chemistry, BVMOs are mainly derived from microbial or plant sources (Wong and Whitesides 1994; Kroutil et al., 2004; Snajdrova et al., 2007; Kawamoto et al., 2008; Kołek et al., 2008). Some evidence suggests that mammalian FMOs may act as BVMOs in the metabolism of xenobiotics. For example, FMO1 from pig liver microsomes is known to catalyze the Baeyer-Villiger oxidation of salicylaldehyde to pyrocatechol (Chen et al., 1995). A cyclic ketone, Org 30659, is suspected to undergo FMO-catalyzed Baeyer-Villiger oxidation to a ring-opened hydroxyl carboxylate metabolite in rat, monkey, and human hepatocytes. However, in liver microsomes, reduction of the ketone became the predominant pathway, and the FMO responsible for Baeyer-Villiger oxidation could not be identified (Verhoeven et al., 1998). Nevertheless, the potential capability of mammalian FMOs to catalyze Baeyer-Villiger oxidations was recently highlighted by the existence of an almost identical sequence motif in the active sites of FMOs and BVMOs (Fraaije et al., 2002).
E7016 inhibits poly(ADP-ribose) polymerase by mimicking NAD+. Extensive metabolism studies were conducted during its preclinical development and revealed that the compound was metabolized at the 4-hydroxypiperidine moiety to ER-879123 as one of the major metabolic pathways in both rat and dog in vivo. The identity of this metabolite was conclusively established by comparing its mass and NMR spectra with those of the synthetic standard. However, in vitro ER-879123 could not be observed in liver microsomal incubations.
In contrast with results in liver microsomes, this ring-opened hydroxyl carboxylate was clearly generated in the incubations of hepatocytes across various species. It seems that liver microsomal enzymes alone were not sufficient to facilitate the biotransformation of E7016 to ER-879123, but hepatocytes provided the necessary enzyme systems to complete this metabolic pathway. As a secondary alcohol, we postulated that E7016 might be first oxidized at the 4-hydroxypiperidine to a ketone intermediate by liver cytosolic oxidoreductases and then oxidized via a Baeyer-Villiger reaction in liver microsomes to a lactone. This lactone then could be hydrolyzed to produce the ring-opened ER-879123 (Fig. 7). The biotransformation of E7016 to ER-879123 would take place via a unique pathway that involves oxidative enzymes in both cytosolic and microsomal fractions of the liver.

Overall metabolic pathway proposed for formation of ER-879123 from E7016.
To test this hypothesis, metabolism of E7016 was investigated in liver S9 fractions. The data indicated that the oxidized form of cofactor NAD(P)+ was required for the metabolism of E7016 to ER-879123. Many cytosolic oxidoreductases, such as alcohol dehydrogenases, are capable of oxidizing the secondary alcohol to the corresponding ketone using NAD(P)+ as a cofactor, which is concurrently converted to its reduced form NAD(P)H. The reduced cofactor NAD(P)H can then be used by the microsomal enzyme that catalyzes the subsequent Baeyer-Villiger oxidation, thus converting the cofactor back to its oxidized form of NAD(P)+ to complete the catalytic cycle. As a general inhibitor of alcohol dehydrogenases, 4-methylpyrazole inhibited the overall biotransformation. The carboxylesterase inhibitor BNPP also showed an inhibitory effect. These observations were consistent with the proposed overall metabolic pathway. With this information in hand, our efforts were focused on identifying the liver microsomal enzyme responsible for the second oxidation step. NAD+-dependent formation of ER-879123 in liver S9 could be significantly decreased by a brief heat treatment of the liver S9 at 50°C (Table 1). FMOs are well known for their thermal instability, and this observation provided circumstantial evidence for the involvement of an FMO in the second oxidation step. Of interest, methimazole at 0.5 mM showed no inhibitory effect toward the formation of ER-879123. Methimazole is well known as a substrate/inhibitor of FMO1 and FMO3, but it has very low affinity and inhibitory potency for FMO5 (Overby et al., 1995). This result suggested that an FMO isoform other than FMO1 and 3 was responsible for the secondary oxidative transformation. To identify the specific FMO isoform responsible for the second oxidation reaction, studies were conducted using recombinant enzymes. As shown in Fig. 4, the formation of ER-879123 was extensive in the incubations of E7016 with recombinant human FMO5, fortified with rat, dog, or human hepatic cytosol. Neither cytosol-fortified FMO1 nor FMO3 catalyzed the formation of this metabolite; thus, this metabolic reaction appeared to be specific to FMO5.
The intermediacy of a ketone was suggested by the proposed mechanism, but this species was not observed in the incubations with E7016. It is possible that the ketone intermediate may be in a rapid and unfavorable equilibrium with the alcohol and hence present in a very low concentration. Nevertheless, the small fraction of the ketone intermediate present would still be further consumed by FMO5, ultimately driving the oxidative process to completion. To confirm whether the ketone ER-879819 was a competent intermediate in the metabolic pathway, a standard was synthesized. By using this synthetic ketone intermediate directly, liver microsomes were able to catalyze the formation of ER-879123 without the presence of cytosolic fractions (Table 2). BNPP again showed a strong inhibitory effect toward the formation of ER-879123, implying the potential involvement of carboxylesterases in the final hydrolysis step. A brief heat treatment of the liver microsomes at 50°C significantly decreased the extent of this reaction, which again supported the potential role of FMOs. As observed in the incubations of E7016 with liver S9, the general substrate/inhibitor of FMO1 and FMO3, methimazole, showed no inhibitory effect. Consistent with the results using E7016, incubation of the ketone ER-879819 with Supersomes of FMO5 produced ER-879123 and the proposed lactone intermediate in a clear time-dependent manner (Fig. 5). Only minor differences were observed between NADH and NADPH as cofactor in this FMO5-catalyzed reaction. It has been reported that although NADPH is the preferred cofactor of FMOs, these enzymes can use either cofactor to complete their catalytic cycle (Hvattum et al., 1991; Sausen et al., 1993). This metabolic reaction appeared to be specific to FMO5, because recombinant FMO1 and FMO3 were not able to catalyze the reaction (Fig. 6). To our knowledge, this is the first report of such a reaction specifically mediated by FMO5.
Extensive knowledge on FMOs has accumulated since their discovery by Ziegler (Pettit et al., 1964). As a group of important drug-metabolizing enzymes, they catalyze oxidative reactions that are complementary to cytochrome P450-mediated biotransformations (Uetrecht and Trager, 2007). Among all the isoforms, FMO3 has been long recognized as the predominant isoform in adult human liver (Cashman 1995, Rettie and Fisher 1999). However, newer studies indicate that mRNA of FMO5 is nearly as abundant as that of FMO3 in adult human liver and is dominant in human small intestine (Zhang and Cashman 2006). Earlier studies reported that the FMO5 enzyme level is at least comparable to that of FMO3 in adult human liver microsomes (Overby et al., 1997). Although it is significantly expressed in liver and small intestine, FMO5 has been considered to be almost inactive against prototypical FMO substrates (Lang and Kalgutkar 2003). When its activity was assessed using traditional FMO substrates such as methimazole, ranitidine, and cimetidine, FMO5 showed very poor affinity and narrow selectivity (Overby et al., 1997). In addition, FMO5 also demonstrated atypical characteristics in terms of thermal stability and pH dependence in comparison with other FMO isoforms (Zhang et al., 2007). S-Oxidation of S-methyl-esonarimod was known to be mediated by FMO1, 3, and 5, but this reaction was not specific to FMO5 (Ohmi et al., 2003). Because of the lack of probe substrates, the potential physiological function and substrate specificity of FMO5 remain poorly understood. The low catalytic efficiency of FMO5 toward common FMO substrates was speculated to be due to its lack of the reactive C(4α)-hydroperoxyflavin (Ziegler 2002; Zhang et al., 2007). In this study, we report a unique probe reaction: a Baeyer-Villiger oxidation that is specifically catalyzed by FMO5. Because Baeyer-Villiger oxidations are catalyzed by peroxyacids and peroxides, this observation provides indirect evidence to support the existence of the peroxy intermediate in the catalytic cycle of FMO5. It has been suspected that FMO5 may play a role in certain physiological functions (Chen et al., 1995; Overby et al., 1995). However, because of the lack of understanding of FMO5 substrate specificity, it is very difficult to assess such potential roles. At this time, it is not known whether other compounds with moieties similar to those of E7016 and ER-879819 may also undergo an FMO5-mediated Baeyer-Villiger oxidation. Further studies will be focused on the mechanism of the FMO5-mediated oxidation step and the use of E7016 and ER-879819 as templates to explore the role of FMO5 toward metabolism of both xenobiotics and endogenous substrates.
Authorship Contributions
Participated in research design: Lai and Wong.
Conducted experiments: Lai, Farah, and Moniz.
Contributed new reagents or analytic tools: Moniz.
Performed data analysis: Lai and Farah.
Wrote or contributed to the writing of the manuscript: Lai, Farah, Moniz, and Wong.
Acknowledgments
We are deeply indebted to Robert Pelletier for his help in operations of the AB Sciex QSTAR mass spectrometer. We thank Dr. Carrie Liu for obtaining the high-resolution mass spectral data for synthetic ER-879819 and ER-879123. We are very grateful to Linda Buckley for her efforts in proofreading the manuscript and to Dr. Tsutomu Yoshimura and Dr. Jack Uetrecht for their suggestions on the article. We also thank Dr. Deepak Dalvie for an inspiring discussion regarding the possible mechanism and acknowledge Drs. Weizheng Xu and Jie Zhang for their contributions in the development of synthetic methods for E7016 and its metabolite.
Footnotes
- Received July 8, 2010.
- Accepted October 13, 2010.
Parts of this work were previously presented at the following conference: Lai WG, Farah N, Moniz GA, and Wong YN (2010) Baeyer-Villiger oxidation specifically catalyzed by human flavin-containing monooxygenase 5. 9th International Meeting of the International Society for the Study of Xenobiotics; 2010 Sept 4–8; Istanbul, Turkey. International Society for the Study of Xenobiotics, Washington, DC.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.035360.
ABBREVIATIONS:
- FMO
- flavin-containing monooxygenase
- Org 30659
- (17α)-17-hydroxy-11-methylene-19-norpregna-4,15-dien-20-yn-3-one
- E7016
- 10-((4-hydroxypiperidin-1-yl)methyl)chromeno[4,3,2-de]phthalazin-3(2H)-one
- ER-879123
- 3-((2-hydroxyethyl)((3-oxo-2,3-dihydrochromeno[4,3,2-de]phthalazin-10-yl)methyl)amino)propanoic acid
- ER-879819
- 10-((4-oxopiperidin-1-yl)methyl)chromeno[4,3,2-de]phthalazin-3(2H)-one
- HPLC
- high-performance liquid chromatography
- SRM
- selected reaction monitoring
- DMSO
- dimethyl sulfoxide
- ER-886830
- tert-butyl 3-((2-hydroxyethyl)((3-oxo-2,3-dihydrochromeno[4,3,2-de]phthalazin-10-yl)methyl)amino)propanoate
- LC
- liquid chromatography
- MS/MS
- tandem mass spectrometry
- HPLC
- high-performance liquid chromatography
- BNPP
- bis(p-nitrophenyl)phosphate
- BVMO
- Baeyer-Villiger monooxygenase.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}