


Abstract
The metabolism of apigenin, a weak estrogenic flavonoid phytochemical, was investigated in the rat. After a single oral administration of radiolabeled apigenin, 51.0% of radioactivity was recovered in urine, 12.0% in feces, 1.2% in the blood, 0.4% in the kidneys, 9.4% in the intestine, 1.2% in the liver, and 24.8% in the rest of the body within 10 days. Sex differences appear with regard to the nature of compounds eliminated via the urinary route: immature male and female rats, like mature female rats, excreted a higher percentage of the mono-glucuronoconjugate of apigenin than the mono-sulfoconjugate of apigenin (10.0-31.6% versus 2.0-3.6%, respectively). Mature male rats excreted the same compounds in an inverse ratio (4.9% and 13.9%, respectively). Radioactivity appeared in the blood only 24 h after oral administration. Blood kinetics showed a high elimination half-time (91.8 h), a distribution volume of 259 ml, and a plasmatic clearance of 1.95 ml/h. All of the parameters calculated from these experiments suggested a slow metabolism of apigenin, with a slow absorption and a slow elimination phase. Thus, a possible accumulation of this flavonoid in the body can be hypothesized.
Flavonoids are polyphenolic compounds that occur ubiquitously in plants and that are present in variable amounts in the diet. Beneficial health properties of flavonoids have been reported and much attention has been paid to their antioxidant activities that affect oxygen free radicals and lipid peroxidation, both of which are involved in many diseases (Di Carlo et al., 1999). Cancer prevention by flavonoids has also been observed (Le Marchand, 2002). Several mechanisms, such as modulation of the xenobiotic metabolizing enzymes involved in the bioactivation of carcinogens, or apoptosis induction, could explain such an effect (Wang et al., 1999, Middleton et al., 2000).
Some of the flavonoids have additional properties: several isoflavones including genistein are well known as potent phytoestrogens and could act on hormonal cancers, coronary heart disease, strokes, osteoporosis, and menopausal symptoms. Apigenin (Fig. 1) is a flavone present in the western diet: in aromatic plants (camomilla, rosemary, parsley) but also in celery, apple, honey, fennel, and wheat germ (Hertog et al., 1992; Karakaya and Nehir, 1999). Even though apigenin is less active than its homologous isoflavone (genistein), its estrogenic properties have already been described. In vitro, apigenin has estrogenic activity on growth of transfected cells that are estrogen-dependent and has additive effects with 17β-estradiol (Le Bail et al., 1998). In vivo, apigenin decreases the endogenous estrogen receptor levels in mouse uterus (Breinholt et al., 2000), enhances the estrogenicity of low doses of estradiol in immature rats (Stroheker et al., 2003), and has a protective effect on skin tumorigenesis, a hormonal-dependent cancer (Birt et al., 2001). Antifertility properties have also been observed: apigenin is an active constituent of Striga orobanchioides, a medicinal plant used for its contraceptive properties (Hiremath et al., 2000). Many mechanisms of action have been proposed to explain these (anti-) estrogenic and (anti-) carcinogenic properties: interaction with estrogen receptors (Miksicek, 1995; Le Bail et al., 1998), modulation of the biosynthesis and the metabolism of steroid hormones (Dai et al., 1997; Ibrahim and Abul-Hajj, 1990), enhancement of gap-junction intercellular communication, and apoptosis induction (Suschetet et al., 1998; Wang et al., 1999).

Structure of apigenin.
Even though the biological and health properties of apigenin have been extensively studied, our knowledge concerning its metabolism and its pharmacokinetics needs to be extended. In vivo metabolism studies published in 1972 showed that maximal excretion of apigenin and its metabolites occurred between 48 and 72 h after oral administration and that derivatives were all conjugated compounds (primary metabolites were not detected) (Griffiths and Smith, 1972). More recently, apigenin elimination in human urine after parsley consumption has been studied (Nielsen et al., 1999). Apigenin appeared in urine after 24 h, the fraction of apigenin intake excreted being 0.58%. In vitro studies have already shown that apigenin undergoes transformation into luteolin and conjugated derivatives (glucurono- and sulfoconjugates). Recently, using rat hepatic microsomes, we have identified two new hydroxylated derivatives, namely, scutellarein and iso-scutellarein. Moreover, phase II apigenin biotransformation leads to the formation of three mono-glucuronated, and one mono-sulfated, compounds. Its major phase I metabolite, luteolin, can also be conjugated to four different mono-glucuronated compounds, two sulfated compounds, and one methylated compound (namely, diosmetin). Despite the diversity of possible phase I and II metabolites that can be formed, only phase II derivatives were recovered from a perfused rat liver ex vivo system and one metabolite, mono-glucuronated and mono-sulfated, was detected only in these conditions (Gradolatto et al., 2004). But this work needs to be completed with the in vivo apigenin metabolism study, to provide new elements concerning the bioavailability and kinetic parameters of this compound.
Thus, the aim of the present work was to investigate in vivo apigenin metabolism in rats. The intragastric route was chosen because this kind of compound is provided by food consumption and must be metabolized and pass through the intestinal barrier to join systemic circulation. First, the elimination of radioactivity was measured in urine and feces after a single oral administration of [3H]apigenin to male and female Wistar rats. Potential storage tissues were also analyzed (blood, liver, kidney, intestines, and body residues), and specific time-point samples were further analyzed to investigate potential sex differences. Second, blood kinetics was investigated to determine kinetic parameters. Finally, a bile excretion experiment was carried out to check whether, like other flavonoids, apigenin metabolism can occur via an enterohepatic pathway.
Materials and Methods
Chemicals. Apigenin (CAS number 520-36-5), luteolin (CAS number 491-70-3), and diosmetin (CAS number: 520-34-3) were purchased from Extrasynthèse (Genay, France; purity >90%). [3H]Apigenin (specific activity 15-20 Ci/mmol, radiochemical purity above 95%) was provided by SibTech, Inc. (Newington, CT). Tritiation is randomly distributed on the phenolic skeleton. Sulfatase (type VI) and β-glucuronidase (type H-3AF) were purchased from Sigma-Aldrich Chimie (Saint-Quentin Fallavier, France). Soluene-350 and the scintillation fluid Ultima Gold were purchased from PerkinElmer Life and Analytical Sciences (Rungis, France). Isofluran was provided by Baxter (Maurepas, France). All other chemicals were provided by standard commercial sources and were of the highest quality available.
Animals and Diets. Mature and immature SPF Wistar rats were obtained from Janvier (Le Genest Saint-Isle, France). They were housed in wire metabolic cages in a room maintained at 22°C with a 12-h light/dark period. The animals were fed ad libitum on a purified semiliquid phytoestrogen-free diet (synthesized at Institut National de la Recherche Agronomique, Jouy-en-Josas, France). They were maintained in accordance with the French Ministry of Agriculture guidelines for care and use of laboratory animals. Animals were fasted for 16 h before and 2 h after drug administration; water was provided ad libitum.
Apigenin Elimination. Ten female and 10 male rats (8 weeks old) weighing 200 ± 30 g received, via intragastric route, one single dose of 10 mg of unlabeled apigenin coupled with 10 μCi/kg b.wt. [3H]apigenin (specific activity of the administered dose, 1 μCi/mg) dissolved in corn oil (dosing volume, 5 ml/kg b.wt.). One male and one female control rat received only corn oil. The same experiment was repeated with immature male and female animals (3 weeks old) (n = 6) for the first 24 h of the experiment (55.6 ± 1.0 g).
Urine and feces were collected daily for 10 days in precooled flasks. The volume of urine samples was measured and adjusted to pH 4 with HCl. Urine samples corresponding to the maximum elimination of radioactivity were analyzed using the LC-MS technique. Feces were directly frozen after weighing and stored at -80°C for further analysis.
At the end of sample collection, aorta blood collection was carried out under isofluran anesthesia. The blood was immediately treated for radioactivity counting. The livers, kidneys, and intestines were frozen at -80°C for further analysis. The rest of the body was crushed with a mixer and frozen at -80°C for further analysis.
Blood Kinetics. Four female rats (8 weeks old) weighing 225 ± 25 g received one single dose of 10 mg coupled with 10 μCi of apigenin/kg b.wt. dissolved in corn oil (dosing volume, 5 ml/kg b.wt.). One female control rat received only corn oil.
Blood was withdrawn under heparin from the orbital vein at 8, 24, 48, 72, 120, 170, 216, 242, 264, 288 and 360 h after intragastric administration. At the end of the experiment, carbon dioxide was used for euthanasia of the animals.
Data were analyzed using noncompartmental methods with Kinetica software (InnaPhase, Champs sur Marne, France). The AUC was computed using the Log Linear Method, trapezoidal when Cn > Cn-1. The elimination half-life (t1/2) was calculated as t1/2 = ln2/Lz where Lz is the elimination rate constant. The plasma clearance (Clp) was calculated as Clp = dose/AUC.
Bile collection. The bile ducts of four female rats (8 weeks old) weighing 253 ± 4 g were cannulated and the bile was collected with a system allowing the animals to move freely (Harvard Apparatus, Les Ulis, France). After 16 h of fasting, the animals received one single dose of 10 mg coupled with 10 μCi of apigenin / kg BW dissolved in corn oil via the intragastric route (dosing volume: 5 ml / kg b.wt.). One control female rat received only corn oil. Bile was collected with an autosampler hour by hour during 48 h. At the end of the experiment, carbon dioxide was used for euthanasia of the animals.
Radioactivity Measurement. Samples from the control rats were counted to determine the background count rate for each matrix. The measurements were made by a model TRI CARB 2000 (United Technologies Packard, France) liquid scintillation counter. The counts per minute were converted to disintegrations per minute based on a quench curve stored in the instrument's microprocessor. The yield of counting was determined by a standard curve. Quenching was corrected by external standardization. The yield of counting was 91.57 ± 0.73% for blood, 100.94 ± 3.20% for carcasses, 91.41 ± 1.66% for feces, 103.37 ± 2.54% for intestine, 119.38 ± 14.73% for kidneys, 95.18 ± 0.84% for livers, and 88.12 ± 2.85 for urine.
Aliquots of urine (1 ml) and bile (100 μl) were directly counted in 10 and 5 ml of scintillation fluid, respectively. Aliquots of blood (150 μl) were digested by Soluene-350 in the presence of isopropanol and bleached by H2O2 30% for 1 h at 50°C before the addition of 10 ml of scintillation fluid for counting.
The kidneys, livers, and intestines were homogenized with ultrapure water (1 g:1 ml) by Ultra-Turrax. The feces were first hydrated with ultrapure water (1 g:4 ml) for 2 h before homogenization with Ultra-Turrax. Aliquots (400 μl) were digested by Soluene-350 in the presence of isopropanol and bleached by H2O2 30%for 2 h at 50°C before the addition of 10 ml of scintillation fluid for counting.
LC-MS Analysis. Analyses of 24-h urine samples were performed after high-performance liquid chromatographic separation as previously described (Gradolatto et al., 2004). The interface was realized between a Waters Alliance system (Waters, Milford, MA) including a model 2690 separation module, a 996 photodiode array detector, and a single quadrupole Waters LCZ Platform equipped with an electrospray ionization source operated in a negative ionization mode. The following conditions, as described elsewhere (Gradolatto et al., 2004), were used: electrospray ionization capillary voltage, 3.50 kV; temperature desolvation, 150°C; desolvation gas, 400 l/h; fragmentor voltage, 20 V.
Statistical Analysis. Data from the apigenin elimination experiment (percentage of cumulative and final radioactivity counting) and data from urine and blood LC-MS analysis (percentage of recovered compounds) were submitted to analysis of variance (p ≤ 0.05). Data from the LC-MS urine analysis of mature and immature male and female rats were submitted to the Kruskal-Wallis test (α ≤ 0.05). Calculations were made with Stat Box Pro software.
Results
Apigenin Elimination. At the end of the experiment, 51% of radioactivity was recovered in urine, 12% in feces, 1.18% in blood, 0.32% in kidneys, 9.41% in intestines, and 1.18% in livers with a significant difference between males and females (average values from male and female data; details are given in Table 1). Approximately a quarter of the recovered radioactivity (24.8%) was still present in the rest of the body after 10 days post-ingestion.
Spread of the radioactivity after single oral administration of radiolabeled apigenin.
Values are mean percentages ± S.E.M. (n = 10) of the recovered radioactivity after administration of one single dose of 10 mg of unlabeled apigenin coupled with 10 μCi of [3H]apigenin/kg b.wt. (specific activity, 1 μCi/mg) to male and female mature Wistar rats. Data for urine and feces correspond to the cumulative radioactivity detected for 10 days. Data for the other tissues correspond to the recovered radioactivity on the 10th day.
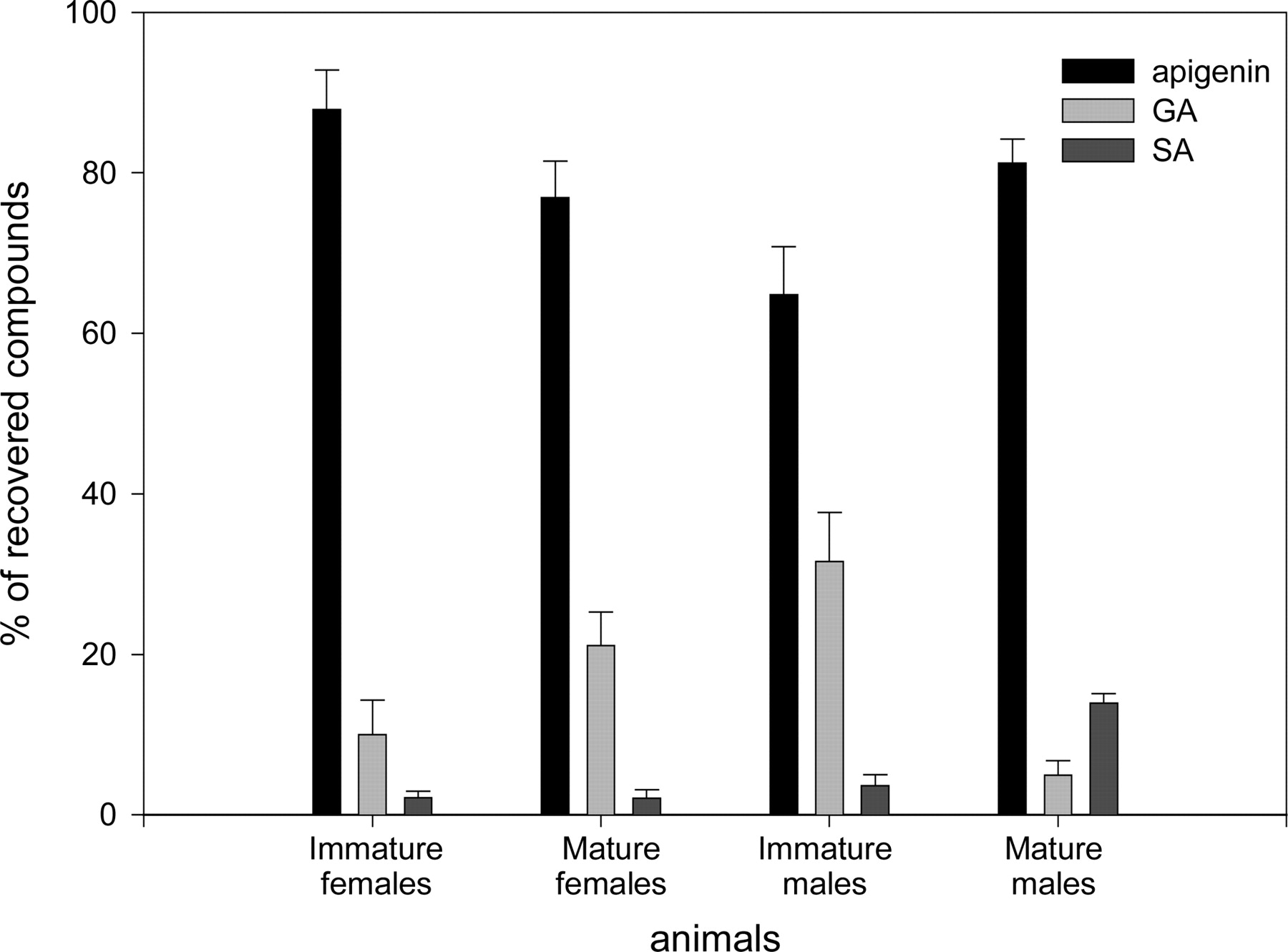
A single oral dose of [3H]apigenin administered to male and female rats led to a major elimination of radioactivity via the urine pathway and, to a lesser extent, via feces (51% and 12% of the ingested dose at the end of the experiment). This excretion mainly occurred during the first 24 h post-ingestion. No significant difference between male and female rats was observed when considering radioactivity recovery (Tables 2 and 3). LC-MS analysis of 24-h urine samples of mature and immature animals revealed that both rat groups excreted a high proportion of unchanged apigenin (64.8-87.8%; m/z 269). In contrast, the proportion of conjugated derivatives highlighted a strong difference related to sex and maturity of animals (Fig. 2). Analysis of urine samples collected from immature males and females revealed the presence of two compounds eluted at 25 and 30 min (m/z of 445 and 349, respectively). These products correspond to glucuronated and sulfated derivatives of apigenin (10.0 versus 31.6% for female and male glucuronoconjugates and 2.2 versus 3.6% for female and male sulfoconjugates, respectively). In mature female samples, these metabolites were in the same proportion as those observed for immature animals (21.1 and 2.0% for glucurono and sulfo derivatives, respectively). In contrast, LC-MS analysis of mature male samples revealed the presence of the same compounds in an inverse proportion: 4.9 versus 13.9% for glucuronated and sulfated derivatives, respectively.
Cumulative excretion (percentage of the recovered dose) of radioactivity in urine and feces after single oral administration of [3H]apigenin to male and female rats
Values are mean percentages ± S.E.M. (n = 10 rats) of the recovered radioactivity. Data of urine and feces correspond to the cumulative radioactivity recovered each day for 10 days.
Single dose pharmacokinetic parameters for total radioactivity in female rats (n = 4)

Percentage of compounds recovered from urine samples collected during the 24 h after one single intragastric administration of 10 mg of apigenin/kg b.wt. to male and female, mature and immature rats. Values are mean ± standard error of the mean (n = 4), and asterisks indicate significant difference from other animal groups (Kruskal-Wallis test, p < 0.05).
Blood kinetics. Preliminary experiments were first realized to determine which time point could be analyzed during this kinetic.
During the first 8 h after apigenin administration, no radioactivity was detected. It appeared at 9 h post-administration time and increased to reach a maximum at 24 h post-ingestion time. Then, it slowly decreased in a monophasic manner, suggesting a slow distribution phase (Fig. 3). The calculated pharmacokinetic parameters showed that blood radioactivity decreased with a high elimination half-life of 91.8 ± 5.6 h and with a Tmax occurring at 24 h. The volume of distribution was quite high (259.1 ± 25.8 ml) and plasmatic clearance was about 2 ml/h.

Milligram equivalents of apigenin per milliliter recovered in the whole blood of female rats following intragastric administration of 10 mg and 10 μCi of [3H]apigenin/kg b.wt.
Bile Excretion. Radioactivity in bile samples appeared during the second hour after [3H]apigenin ingestion and increased during the third hour to a dose equivalent to 0.59 ± 0.2 mg of apigenin/ml (Fig. 4). Radioactivity decreased during the fourth hour and increased once again during the fifth and sixth hours to a dose equivalent to 0.35 ± 0.03 apigenin/ml. Then it slowly decreased, and baseline (0.12 mg Eq of apigenin/ml ± 0.05) was recovered approximately 15 h after ingestion of [3H]apigenin.

Milligram equivalents of apigenin per milliliter of ingested radioactivity recovered in the cumulative bile samples of female rats following intragastric administration of 10 mg and 10 μCi of [3H]apigenin/kg b.wt. Mean ± standard error of the mean (n = 4).
Discussion
To our knowledge, no information concerning the pharmacokinetics of apigenin in rat were available. Our data show that apigenin excretion occurred mainly via the urinary pathway, with 16% of ingested radioactivity recovered from male urine samples within the first 24 h, and 18% for female. After this time point, radioactivity excretion decreased and 40% was recovered within 5 days. Because glucuronoconjugates of apigenin (GA) only represent a part of this percentage (Fig. 2), we can assume that within 5 days, the proportion of GA excreted is less than 40% of the ingested dose. Griffiths and Smith (1972) observed GA excretion after administration of one single dose of apigenin to rats. But in this previous work, GA excretion was not maximal after 24 h and increased until the second day post-ingestion, and finally, 75% of GA was recovered within 5 days. The differences observed between these two studies could be explained by the initial dose administration, since a 100 mg/kg b.wt. dose was administered by Griffiths and Smith (1972), i.e., 10-fold higher than the one delivered in our study. Even though the kinetics of elimination are not exactly the same, both works show that urinary excretion of apigenin is an important route.
These results are very different from those observed in human volunteers after parsley ingestion: within 24 h after ingestion, only 0.58% of apigenin was recovered in urine samples (Nielsen and Dragsted, 1998). Moreover, even though the authors highlighted the importance of individual differences in apigenin absorption, they could not detect any differences between male and female subjects after the enzymatic digestion of derivatives. This was also the case in our study concerning radioactivity recovery alone. Nevertheless, LC-MS analysis quickly invalidated this observation, by pointing out the different evolution of sex maturity resulting in different proportions of metabolism products. The application of a detection method allowing estimation of metabolite proportion (Basly et al., 2003) showed that the ratio of GA to sulfated metabolites of apigenin (SA) in immature rats (male and female) and mature females was the inverse ratio in mature male rats (10-31:2.0-3.6% in immature rats and mature female rats, versus 4.9:13.9% in mature males for GA and SA, respectively). To our knowledge, this is the first time that such a difference has been observed, and this finding could be explained by differences in enzyme activities related to sex maturation of animals. It is well known that phase I enzyme metabolism and, more specifically, cytochromes P450, is regulated by growth hormone in a gender-specific manner (Shapiro et al., 1995 and Pampori et al., 2001). This is also the case for some phase II metabolism enzymes (Mugford and Kedderis, 1998). Some UDP-glucuronosyl- and sulfotransferases can be regulated via endogenous steroid hormones and are consequently sex- and age-regulated, thus explaining discrepancies between metabolites obtained from immature and mature males and females (Mugford and Kedderis, 1998). Moreover, some aryl sulfotransferase activities in mature male rat livers are 2- to 3-fold greater than those in mature female livers, thus explaining why SA are predominant in urine samples of mature males (Mulder, 1986).
Apigenin kinetics appear to differ from that of other flavonoids like quercetin. When administered to rats, only 20% of this flavonol was absorbed through the intestinal barrier, 30% was excreted in expired CO2, and 30% was recovered unchanged in the feces (Ueno et al., 1983). In the present work, 63% of radioactivity was excreted via the urinary and fecal route (only 12% for the latter), suggesting a high absorption of apigenin. After 10 days, 24.8% of radioactivity was still present in the rest of the body, suggesting an accumulation of this compound in the body and slow elimination.
Moreover, it should be noted that none of the phase I derivatives of apigenin were recovered at any time in the experiment. Previous work has shown that in vitro apigenin phase I metabolism (hepatic rat microsomes) mainly led to the formation of luteolin, whereas only phase II conjugates were recovered in ex vivo experiments (Nielsen and Dragsted, 1998; Gradolatto et al., 2004). The current work has confirmed the fact that phase I metabolites are compounds obtained only in vitro.
Previous data from the literature concerning blood kinetics showed that, when diosmin was administered to humans, its half-life ranged from 26 to 43 h, with a peak occurring 2 h after administration (Cova et al., 1992), whereas diosmetin administered per os to rats appeared in blood after 6 h as unchanged and glucuronated compound (Boutin et al., 1993). Quercetin has been studied in different species and showed a half-time ranging from 0.7 to 2.4 h, a distribution volume of 6.21 up to 92.61 liters, and a total body clearance varying from 28.1 to 34.6 h-1 (Graefe et al., 1999). In pigs, after an oral dose of 50 mg/kg, only 17% of the quercetin administered was recovered in blood as free conjugate and derivative products within 8 h postadministration (Ader et al., 2000). In humans, the peak in blood occurred more rapidly, approximately 2.9 h after the flavonol administration (Hollman et al., 1995). In rats, flavonoids seemed to occur more rapidly. Luteolin, the in vitro phase I apigenin metabolite, given via gastric intubation, appeared in plasma after 15 min (Shimoi et al., 1998). Apigenin given to rats via the intraperitoneal pathway appeared in plasma 30 min after administration (Romanova et al., 2000).
Taken together, these results from the literature suggest a relatively rapid absorption of flavonoids, the presence in blood occurring within a few minutes to a few hours and half-times not exceeding 48 h. After 2 days, all of these compounds seem to be excreted from the body. In contrast, our results suggest a slow absorption of apigenin with the appearance of blood radioactivity 24 h after ingestion, with no traces before this time point. In addition, the half-time of 91.8 h and the fact that basal level is recovered 10 days after [3H]apigenin treatment confirm the hypothesis of the slow absorption and excretion of apigenin.
In conclusion, 1) urinary elimination seems to be the major elimination pathway of apigenin; 2) absorption seems to be a slow phenomenon since tritium was detected only after 24 h; 3) 48 h after ingestion, blood radioactivity is still high and corresponds to about 60% of the peak previously observed; and 4) the estimated distribution volume of radioactivity is 25 times higher than the theoretical blood volume (about 10 ml), suggesting accumulation in the tissues. Moreover, persistence of radioactivity in blood suggests an active enterohepatic pathway already described for other flavonoids (Hackett, 1986). These results corroborate the hypothesis of the slow metabolism and elimination of this compound.
Concerning biliary excretion, it was shown that flavonoid administration ([14C]quercetin or [14C]catechin) leads to a peak of radioactivity in bile after 2 h, containing the unchanged aglycone and its derivatives, with total recovery of baseline 48 h after administration (Shaw and Griffiths, 1980). In the case of genistein, 70 to 75% of radioactivity is recovered over a 4-h period (Sfakianos et al., 1997). In contrast, our results show that peak radioactivity is not detected before 3 to 6 h and that the basal level is recovered only 15 h after administration. Consequently, our results differ from results for other flavonoids and sustain the idea of slow apigenin pharmacokinetics. Therefore, biological effects of apigenin can be explained by its slow metabolism, its enterohepatic pathway leading to high persistence in blood, and, consequently, accumulation in the body.
Because apigenin is still present in the blood, detected 9 days after the administration of one single dose, accumulation seems possible and raises the question of the eventual consequences of chronic exposure to this compound, which is present in the daily western diet and which may affect animal and human health.
Acknowledgments
We thank Joëlle Chevalier and Lucien Guenot for invaluable advice and excellent technical assistance during this study.
Footnotes
-
This work was supported by grants from the Conseil Régional de Bourgogne, from the Ministère de l'Environnement, and from the Institut National de la Recherche Agronomique.
-
doi:10.1124/dmd.104.000893.
-
ABBREVIATIONS: LC-MS, liquid chromatography-mass spectrometry; GA, glucuronoconjugate(s) of apigenin; SA, sulfoconjugate(s) of apigenin.
- Received June 8, 2004.
- Accepted October 1, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}