


Abstract
Clinical reports indicate that cardiotoxicity due to donepezil can occur after coadministration with cilostazol. We speculated that the concentration of donepezil in heart tissue might be increased as a result of interaction with cilostazol at efflux transporters such as P-glycoprotein (P-gp, ABCB1) and breast cancer resistance protein (BCRP, ABCG2), which are expressed in many tissues including the heart, and our study tested this hypothesis. First, donepezil was confirmed to be a substrate of both BCRP and P-glycoprotein in transporter-transfected cells in vitro. Cilostazol inhibited BCRP and P-glycoprotein with half-inhibitory concentrations of 130 nM and 12.7 μM, respectively. Considering the clinically achievable unbound plasma concentration of cilostazol (about 200 nM), it is plausible that BCRP-mediated transport of donepezil would be affected by cilostazol in vivo. Indeed, in an in vivo rat study, we found that coadministration of cilostazol significantly increased the concentrations of donepezil in the heart and brain, where BCRP functions as a part of the blood–tissue barrier, whereas the plasma concentration of donepezil was unaffected. In addition, in vitro accumulation of donepezil in heart tissue slices of rats was significantly increased in the presence of cilostazol. These results indicate that donepezil-cilostazol interaction at BCRP may be clinically relevant in heart and brain tissues. In other words, the tissue distribution of drugs can be influenced by drug-drug interaction (DDI) at efflux transporters in certain tissues (local DDI) without any apparent change in plasma concentration (systemic DDI).
Introduction
Donepezil, a selective, reversible inhibitor of acetylcholinesterase, is used for treatment of Alzheimer’s disease. Although donepezil is generally well tolerated, there are some reports of side effects associated with cholinergic effects. In addition, it has been reported that donepezil may cause cardiac toxicity, such as QT prolongation (Takaya et al., 2009). There are also case reports of cardiac toxicity of donepezil in patients concurrently receiving cilostazol, benidipine, or memantine (Tanaka et al., 2009; Shinozaki, 2012; Igeta et al., 2013). However, the mechanism of the putative drug–drug interaction (DDI) has not yet been clarified, even though donepezil is often administered with other drugs, as it is frequently prescribed for elderly people. On the other hand, it has been proposed that coadministration of donepezil with cilostazol is more effective in patients with mild dementia (Ihara et al., 2014), and this has been supported by findings in an animal model (Lee et al., 2007). Accordingly, it is important to examine whether interaction of donepezil with cilostazol might occur, and if it does, whether it might have clinical relevance.
Pharmacologic activity usually depends on the unbound drug concentration at the site of action. Because donepezil shows high bioavailability after oral administration (more than 90% in rats) (Matsui et al., 1999), it is unlikely that coadministered cilostazol increases intestinal absorption of donepezil. Clearance of donepezil is mainly due to metabolism via CYP3A4 and partly via CYP2D6, so interaction of the two drugs at drug-metabolizing enzymes could contribute to increased systemic exposure to donepezil; however, the increase of plasma concentration of donepezil by ketoconazole, a typical inhibitor of cytochrome P450 3A4 (CYP3A4), was reported to be mild, amounting to at most 30% (Tiseo et al., 1998a). Another possibility is an increase in distribution to heart tissue.
Transporters are involved in drug absorption, tissue distribution and excretion, and DDI at transporters can cause significant changes in plasma concentrations of affected drugs; on the other hand, DDI at transporters involved in tissue distribution, such as P-glycoprotein (ABCB1) and breast cancer resistance protein (BCRP, ABCG2), may alter the tissue distribution without any apparent change in plasma concentration (Sadeque et al., 2000; Sasongko et al., 2005). Accordingly, we considered that, if the distribution of donepezil to heart tissue is regulated by efflux transporters, and if the efflux process is inhibited by cilostazol, accumulation of donepezil in the tissue might be increased.
P-glycoprotein is expressed in endothelium of heart tissue and could serve to limit drug distribution to the heart (Meissner et al., 2002). A significant increase in the distribution of vinblastine to heart tissue was observed in Mdr1a gene knockout mice, demonstrating that P-glycoprotein is functional in heart tissue to exclude substrate drugs (Schinkel et al., 1994). In addition, QT prolongation by romidepsin has been suggested to be influenced by allelic variation of the ABCB1 gene, suggesting that P-glycoprotein contributes to the blood–heart barrier (Sissung et al., 2011).
Previous reports on the transport of donepezil by P-glycoprotein are conflicting: values of efflux ratio ranging from unity to 2.3 have been found in P-glycoprotein–expressing Madin-Darby canine kidney (MDCK) cells (Summerfield et al., 2007; Lili et al., 2013). An in vivo study in mice suggested that donepezil distribution to brain is controlled by P-glycoprotein, based on the increase of accumulation by cyclosporine A (Ishiwata et al., 2007). Furthermore, we have previously shown that cilostazol efflux transport is mediated by P-glycoprotein (Toyobuku et al., 2003). Accordingly, it is possible that inhibition of P-glycoprotein by cilostazol would lead to increased accumulation of donepezil in the heart if donepezil is clearly shown to be a substrate of P-glycoprotein.
In addition to P-glycoprotein, BCRP is also expressed in heart, mainly in capillary endothelial cells and cardiomyocytes (Meissner et al., 2006; Solbach et al., 2008; Higashikuni et al., 2010; Emmert et al., 2013), so BCRP is also a potential site of DDI. However, it has not been reported whether donepezil is a substrate of BCRP.
Our study examined the hypothesis that cardiotoxicity associated with coadministration of donepezil and cilostazol is a result of interaction of the two drugs at efflux transporters such as P-glycoprotein and BCRP in heart tissue (local DDI). In other words, we examined whether concomitantly administered cilostazol inhibits efflux transport of donepezil, resulting in increased accumulation of donepezil in the heart without any apparent change of systemic exposure.
Materials and Methods
Chemicals and Animals.
Donepezil and inulin carboxyl, [14C]inulin (2.5 mCi/g) were purchased from Tokyo Chemical Industry (Tokyo, Japan) and ARC (St. Louis, MO), respectively. Cilostazol and tetraethylammonium (TEA) were purchased from Wako Pure Chemical Industries (Osaka, Japan) and Sigma-Aldrich (St. Louis, MO), respectively. All other reagents were purchased from Sigma-Aldrich, Wako Pure Chemical Industries, Thermo Fisher Scientific (Waltham, MA), Kanto Chemical (Tokyo, Japan), and Nacalai Tesque (Kyoto, Japan).
All the animal studies were approved by the Committee of Kanazawa University for the Care and Use of Laboratory Animals and were performed in accordance with its guidelines for the care and use of laboratory animals (AP-143148). Seven-week-old Wistar male and female rats were purchased from Sankyo Labo Service (Tokyo, Japan).
Transport Study in MDCK Cells.
MDCK cells overexpressing BCRP or P-glycoprotein were obtained from the Netherlands Cancer Institute (Amsterdam). MDCK cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum, 100 units/ml benzylpenicillin, and 100 µg/ml streptomycin at 37°C under an atmosphere of 5% CO2 in air. For bidirectional transport studies, MDCK cells were cultured on Transwell filter membrane inserts (Becton Dickinson, Franklin Lakes, NJ), surface area 0.9 cm2 and pore size 3 µm, at a density of 2.0×105 cells/cm2 for 5 days before use for each experiment. The confluent cells were preincubated with transport medium (5.4 mM KCl, 136.9 mM NaCl, 0.34 mM Na2HPO4・7H2O, 0.44 mM KH2PO4, 4.2 mM NaHCO3, 1.3 mM CaCl2, 0.49 mM MgCl2・6H2O, 0.41 mM MgSO4・7H2O, 19.5 mM glucose, 10 mM HEPES/Tris, pH 7.4), containing TEA (1 mM) and bovine serum albumin (0.1%, v/v). Transport measurement was initiated by adding transport medium containing donepezil. Inhibitors of BCRP and P-glycoprotein were added to both the apical and basolateral sides during preincubation and incubation. When the transport reaction was initiated, 0.5-ml and 1.5-ml aliquots of transport medium were added to the apical and basolateral sides of the Transwell insert membrane, respectively. Transport of donepezil was measured in both directions, apical (AP)-to-basolateral (BL) and basolateral-to-apical side. The receiver side solution (100 µL) was withdrawn for measurement of transport and replaced with an equal volume of transport medium at 15, 30, 45, and 60 minutes.
All the drugs used were quantitated by high-performance liquid chromatography (HPLC) analysis. Before the HPLC analysis, the sample was mixed vigorously with 100 µl of acetonitrile for 1 minute, and then centrifuged at 15,000 rpm and 4°C for 15 minutes. The supernatant was transferred to a 1.5-ml polypropylene microcentrifuge tube and evaporated to dryness. The residue was reconstituted in 100 µl of the mobile phase used for HPLC analysis.
The apparent permeability (Papp, cm/s) of donepezil across the cell monolayer was calculated using the following equation: where Q is the amount of donepezil transported over time t, C is the initial concentration in the donor side, and A is the surface area of the membrane used.
where Q is the amount of donepezil transported over time t, C is the initial concentration in the donor side, and A is the surface area of the membrane used.
Kinetic parameters were estimated by means of nonlinear least-squares analysis using the MULTI program (Yamaoka et al., 1981). The inhibitory effect of cilostazol on donepezil or quinidine transport was expressed as a percentage of the control, and the inhibitor concentration giving half-maximum inhibition (IC50) was obtained by applying the following equation: where [I] is the inhibitor concentration.
where [I] is the inhibitor concentration.
The affinity of donepezil for BCRP (Michaelis constant Km), the maximal rate of BCRP-mediated donepezil transport (Vmax), and the apparently nonsaturable component (kd), which may represent diffusion, paracellular transport, and/or transport via a low-affinity transporter that is distinct from BCRP in MDCK cells, were obtained by fitting the data to the following equation: where v and C are transport rate and initial concentration of donepezil, respectively.
where v and C are transport rate and initial concentration of donepezil, respectively.
In Vitro Tissue Accumulation Study Using Rat Heart Slices.
The accumulation study of donepezil in heart slices from male Wistar rats was conducted as described elsewhere, with some modifications (Iwata et al., 2008). Briefly, rats were anesthetized with ether and exsanguinated from the descending aorta. The heart was immediately excised and rinsed with ice-cold saline. Atrium and connective tissue were removed carefully, and the heart was sectioned at 350 µm with a microslicer (Zero 1; Dosaka EM, Kyoto, Japan). The heart slices were preincubated with ice-cold test solution containing 120 mM NaCl, 16.2 mM KCl, 1 mM CaCl2, 1.2 mM MgSO4, 10 mM NaH2PO4/Na2HPO4 (pH 7.5) and oxygenated by bubbling with 99.5% O2 gas. After preincubation, the heart slices were incubated with donepezil at 37°C for 30 minutes with bubbling of 99.5% O2. Inhibitors used were present during both preincubation and incubation. At appropriate times, slices were picked up from the solution containing donepezil, and rinsed with ice-cold saline. They were blotted to dryness, weighed, and homogenized with 2 ml of water using a homogenizer (Ultra Turrax T25; IKA Werke, Staufen im Breisgau, Germany). The obtained homogenate (500 µL) was transferred to a 1.5-ml polypropylene microcentrifuge tube, and 500 µl of acetonitrile was added to it. The samples were vortexed for 1 minute and centrifuged at 15,000 rpm and 4°C for 15 minutes. The supernatant was evaporated to dryness, and the resultant residue was reconstituted in 100 µl of the mobile phase used for HPLC analysis.
Extracellular adhered water space was evaluated in terms of apparent uptake of [14C]inulin. After incubation with [14C]inulin, tissue slices were blotted to dryness, weighed, and solubilized with 150 µl of 1 N NaOH. Then the sample was neutralized with 30 µl of 5 N HCl and mixed with scintillation liquid (Clear-sol I; Nacalai Tesque, Kyoto, Japan). Radioactivity was quantitated with a liquid scintillation counter (LCS-6100; Hitachi Aloka Medical, Tokyo, Japan). All the accumulation values were expressed as slice-to-medium concentration ratio, obtained by dividing the accumulated amount in the heart slices by the final concentration in the medium and the weight of tissue slices.
In Vivo Animal Study.
Female Wistar rats were used in the in vivo animal study. For preparation of dosing solution, donepezil was dissolved in saline, and cilostazol was suspended in methylcellulose solution (0.5%, w/v). Cilostazol (100 or 500 mg/kg body weight) was administered to rats orally 30 minutes before intravenous administration of donepezil. When donepezil (1 mg/kg body weight) and elacridar (1 mg/kg body weight) were coadministered to rats intravenously, donepezil was dissolved in a mixture of saline and polyethylene glycol 600 (3:1, v/v). The volumes of oral and intravenous administration were 3 and 1 ml/kg body weight, respectively. Elacridar was dissolved in dimethylsulfoxide (DMSO) and diluted in a mixture of saline and polyethylene glycol 600. The blood was collected from the jugular vein at 0.5, 1, 2, and 3 hours after intravenous administration of donepezil to rats. The plasma was obtained by centrifugation of the blood at 5000 rpm and 4°C for 10 minutes.
Rats were sacrificed 3 hours after administration of the donepezil by anesthesia with ether followed by exsanguination. The whole heart and brain tissues were immediately excised and frozen at −30°C until analysis. Frozen heart and brain samples were thawed, weighed, and homogenized with 2 ml of water using a homogenizer. Before the HPLC analysis, 500 µl of homogenate was transferred to a 1.5-ml disposable microcentrifuge tube, and 500 µl of acetonitrile was added to deproteinize the tissue. The samples were vigorously mixed for 1 minute and centrifuged at 15,000 rpm and 4°C for 15 minutes. The resultant supernatant was evaporated to dryness, and the residue was reconstituted in 100 µl of the mobile phase used for HPLC analysis.
Pharmacokinetic analysis of the plasma profiles after intravenous administration was performed with the MULTI program using a one-compartment model according to the following equation: where Cp is the plasma concentration of donepezil, Vd is the volume of distribution, ke is a first-order elimination rate constant, and t is time after administration. CLtot (total clearance) was obtained by applying the following equation:
where Cp is the plasma concentration of donepezil, Vd is the volume of distribution, ke is a first-order elimination rate constant, and t is time after administration. CLtot (total clearance) was obtained by applying the following equation: where AUC0–∞ is the area under the plasma concentration–time curve extrapolated to infinity.
where AUC0–∞ is the area under the plasma concentration–time curve extrapolated to infinity.
Analytic Methods.
The concentrations of drugs were quantified by HPLC. An HPLC system (Waters Corporation, Milford, MA) was equipped with a 2475 variable fluorescence detector, 2487 UV absorbance detector, and 2690 separation module. A Waters 2695 solvent delivery system was used to operate the isocratic flow of mobile phase. The analytical column was a Mightysil RP-18 GP column (4.6 mm × 250 mm i.d., 5 µm particle size; Kanto Chemical, Tokyo, Japan). All the samples for HPLC analysis were filtered through membranes with a pore size of 0.45 μm.
For donepezil analysis, the mobile phase consisted of water, acetonitrile, and methanol (80:17:3, v/v/v) with 0.1% acetic acid (v/v) and separation was performed isocratically at 32°C and a flow rate of 1.0 ml/min with fluorescence detection (excitation 321 nm and emission 387 nm). For quinidine, the mobile phase consisted of 10 mM sodium acetate buffer (pH 5.0) and methanol (50:50, v/v). Separation was carried out isocratically at 35°C and a flow rate of 0.6 ml/min, with fluorescence detection (excitation 350 nm and emission 450 nm). For cilostazol, the mobile phase consisted of 20 mM ammonium acetate buffer (pH 5.0) and acetonitrile (55:45, v/v), and separation was performed isocratically at ambient temperature at a flow rate of 1.0 ml/min with UV detection at 254 nm. For elacridar, the mobile phase was a mixture of 50 mM ammonium acetate buffer (pH 4.2) and acetonitrile (65:35, v/v), and separation was performed isocratically at 30°C at a flow rate of 1.0 ml/min with fluorescence detection (excitation 260 nm and emission 460 nm). The injected volume of samples into HPLC was 50 µL.
All the data are presented as the mean ± S.E.M. or S.D. Statistical significance was evaluated by means of Student’s t test, with P < 0.05 considered statistically significant.
Results
Transcellular Transport of Donepezil in MDCK Cells.
To examine whether donepezil is a substrate of efflux transporters, transcellular transport of donepezil by MDCK cells expressing BCRP or P-glycoprotein was compared with that in Mock cells. Figure 1A shows the time course of donepezil (3 μM) transport in the apical (AP)-to-basolateral (BL) and basolateral-to-apical directions in the three types of MDCK cells cultured on Transwell inserts. Figure 1B shows the apparent permeability of donepezil obtained from the slope of the results shown in Fig. 1A. In Mock cells, there was no difference between the basolateral-to-apical and apical-to-basolateral transport of donepezil. However, basolateral-to-apical transport in MDCK cells expressing BCRP or P-glycoprotein was significantly higher than that in the opposite direction. These results demonstrate that donepezil is a substrate of both BCRP and P-glycoprotein.

Transcellular transport of donepezil by MDCK cells expressing BCRP or P-glycoprotein and Mock cells. (A) Time course of permeated amount of donepezil (3 µM) across monolayers of P-glycoprotein-expressing (triangle), BCRP-expressing (square), and Mock (circle) cells in the apical (AP)-to-basolateral (BL) direction (open symbols) and the basolateral (BL)-to-apical (AP) direction (closed symbols) were evaluated for 60 minutes at 37°C and pH 7.4. Data represent the mean ± S.E.M. (n = 3). (B) Permeability of donepezil (obtained from the slope of Fig. 1A) is shown in the apical (AP)-to-basolateral (BL) (open columns) and basolateral (BL)-to-apical (AP) (closed columns) directions. *Statistically significant difference (P < 0.05) versus the value in the apical (AP)-to-basolateral (BL) direction by Student’s t test. Data represent the mean ± S.E.M. (n = 3).
Inhibitory Effect of Cilostazol on Transport Activity of P-gp and BCRP.
We first examined whether cilostazol is an inhibitor of P-glycoprotein and/or BCRP, using MDCK cells in the presence of increasing concentrations of cilostazol. The results are shown in Fig. 2. In the case of P-glycoprotein, donepezil transport was insufficient to enable quantitative analysis of the inhibitory effect of cilostazol, so quinidine was used as the substrate of P-glycoprotein. In the case of BCRP, donepezil was used as the substrate. Cilostazol inhibited P-glycoprotein-mediated transport of quinidine (5 μM) with the IC50 of 12.7 ± 0.22 µM. The obtained IC50 is much higher than the observed unbound plasma concentration of cilostazol (143∼203 nM) at clinically used dose levels (Suri et al., 1998). Cilostazol also inhibited BCRP-mediated transport of 1 μM donepezil in a concentration-dependent manner with an IC50 of 130 ± 31.7 nM. In this case, the IC50 is lower than the clinically relevant unbound cilostazol concentration. These results suggest that cilostazol may affect the transport activity of BCRP, but not P-glycoprotein, at clinically achievable plasma unbound concentrations.

Inhibitory effect of cilostazol on BCRP and P-glycoprotein. Concentration dependence of cilostazol in inhibiting BCRP (○) and P-glycoprotein (●) was evaluated by measuring transcellular transport of donepezil (1 µM) and quinidine (5 µM), respectively, from the basolateral to apical side across MDCK cells for 60 minutes at 37°C and pH 7.4. The results are shown as a percentage of control, which was measured in the absence of cilostazol. Each point represents the mean ± S.E.M. (n = 3).
Concentration Dependence of BCRP-Mediated Donepezil Transport.
To elucidate whether BCRP-mediated donepezil transport is clinically important, we examined the affinity of donepezil for BCRP. Figure 3 shows the concentration dependence of in vitro donepezil transport by BCRP. The transport consisted of both saturable and apparently nonsaturable components with Michaelis constant (Km), maximum transport rate (Vmax), and nonsaturable first-order rate constant (kd) values of 4.03 ± 0.45 μM, 10.9 ± 1.15 pmol/min/cm2, and 1.16 ± 0.097 μl/min/cm2, respectively.

Concentration dependence of BCRP-mediated transport of donepezil. Transport of donepezil from the basolateral to apical side across BCRP-expressing MDCK cells was measured at concentrations from 0.1 to 40 µM for 60 minutes at 37°C and pH 7.4. Solid, dotted, and broken lines represent the total, BCRP-mediated, and apparently nonsaturable transport components predicted from the estimated kinetic parameters, Km, Vmax, and kd, respectively. Data represent the mean ± S.E.M. (n = 3).
Because the obtained Km value is higher than maximum concentration of donepezil in plasma (70 nM) at clinically used dose levels (Tiseo et al., 1998b), BCRP-mediated transport of donepezil would not be saturated under these conditions.
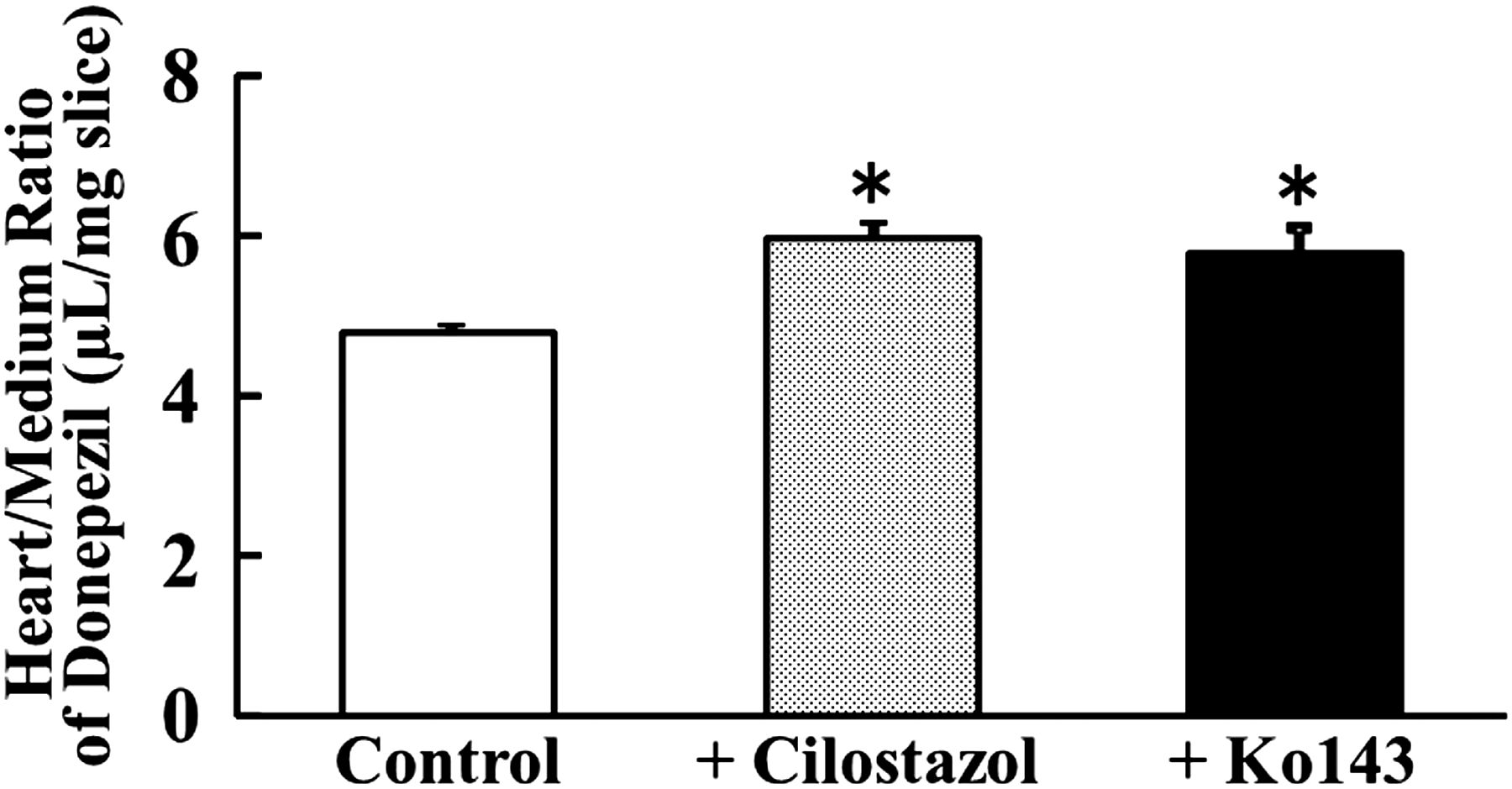
Effect of Cilostazol and Ko143 on In Vitro Accumulation of Donepezil in Heart Tissue Slices.
To examine the effect of cilostazol on donepezil distribution to the heart, we measured accumulation of donepezil in rat heart tissue slices. In this study, BCRP inhibitor Ko143 [(3S,6S,12aS)-1,2,3,4,6,7,12,12a-octahydro-9-methoxy-6-(2-methylpropyl)-1,4-dioxopyrazino[1ʹ,2ʹ:1,6]pyrido[3,4-b]indole-3-propanoic acid 1,1-dimethylethyl ester] (0.1 μM) and cilostazol (5 μM) were used. Figure 4 shows the accumulation of donepezil at 0.1 μM in heart tissue slices at 30 minutes. When heart tissue slices were incubated with donepezil in the presence of cilostazol, the accumulation (evaluated in terms of slice-to-medium concentration ratio) of donepezil was significantly increased as compared with the control (from 4.78 to 5.95). Similarly, when accumulation of donepezil in heart slices was examined in the presence of Ko143 as a BCRP inhibitor, a significant increase was again observed (from 4.78 to 5.78). These results suggest that the increase of donepezil accumulation in heart can be accounted for by the inhibition of BCRP by cilostazol.
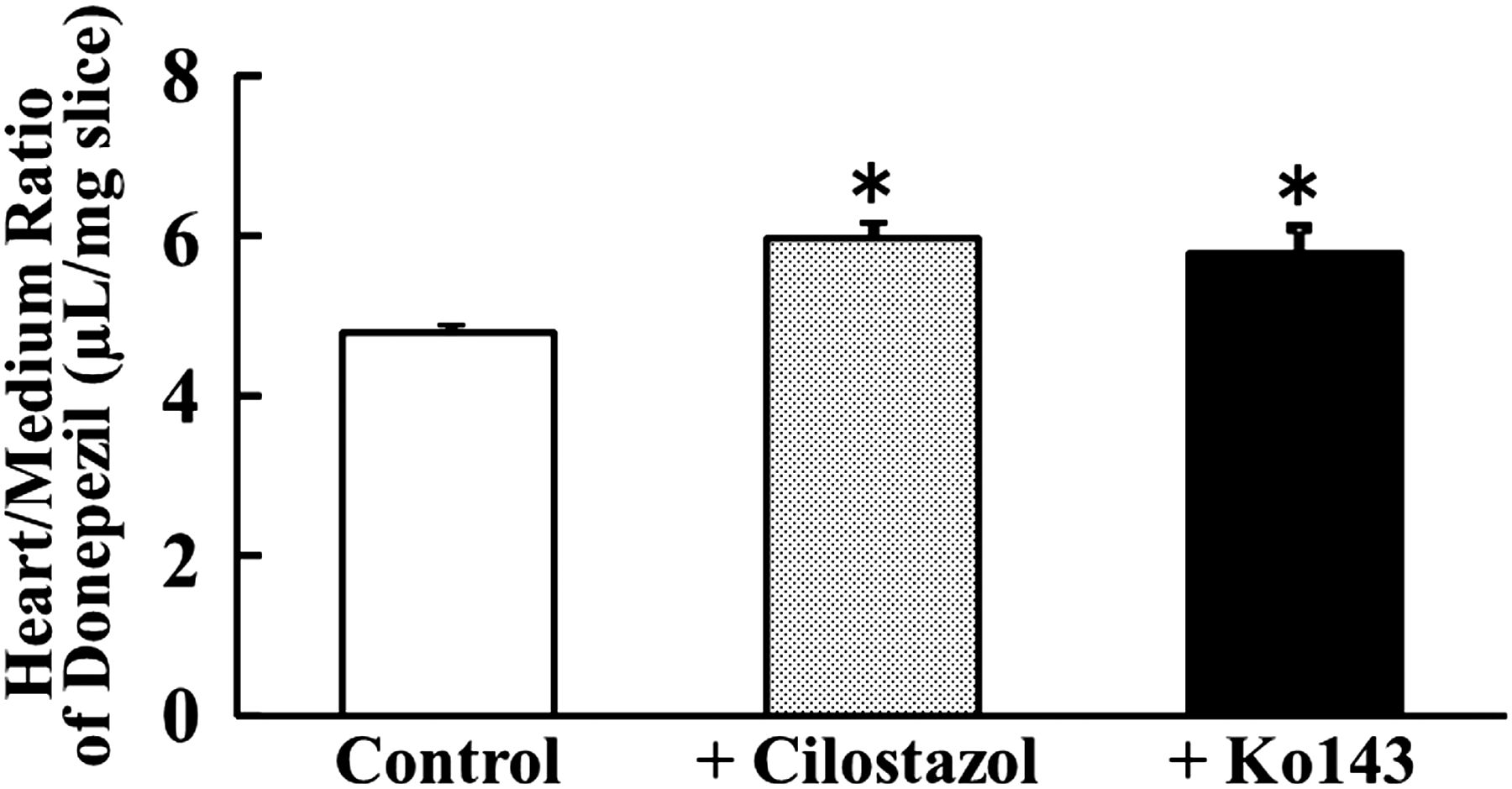
Effect of cilostazol and Ko143 on in vitro accumulation of donepezil in rat heart slices. Accumulation of donepezil (0.1 µM) in rat heart tissue slices was measured for 30 minutes at 37°C without (open column) or with cilostazol (5 µM, dotted column) or Ko143 (0.1 µM, closed column). An asterisk indicates a statistically significant difference (P < 0.05) versus the control by Student’s t test. Data represent the mean ± S.E.M (n = 3).
Effects of Cilostazol and Elacridar on Disposition of Donepezil in Rats.
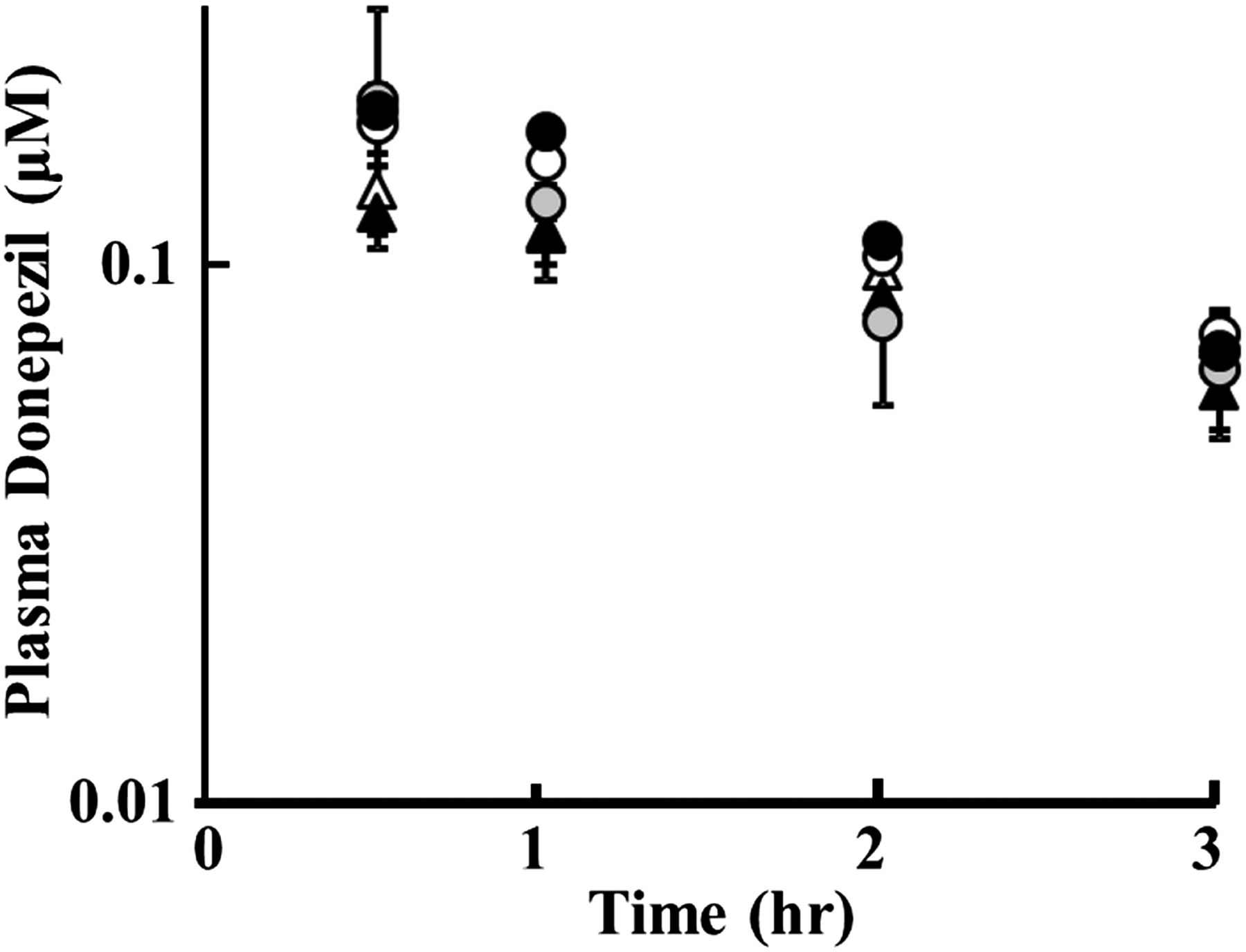
The in vivo effect of cilostazol on the disposition of donepezil was examined in rats. Figure 5 shows the plasma concentration-time curve of donepezil in female rats after bolus intravenous administration of 1 mg/kg with and without cilostazol (100 mg/kg or 500 mg/kg) or elacridar (1 mg/kg) as an inhibitor of both BCRP and P-glycoprotein. Table 1 summarizes the pharmacokinetic parameters of donepezil obtained by one-compartment model analysis.
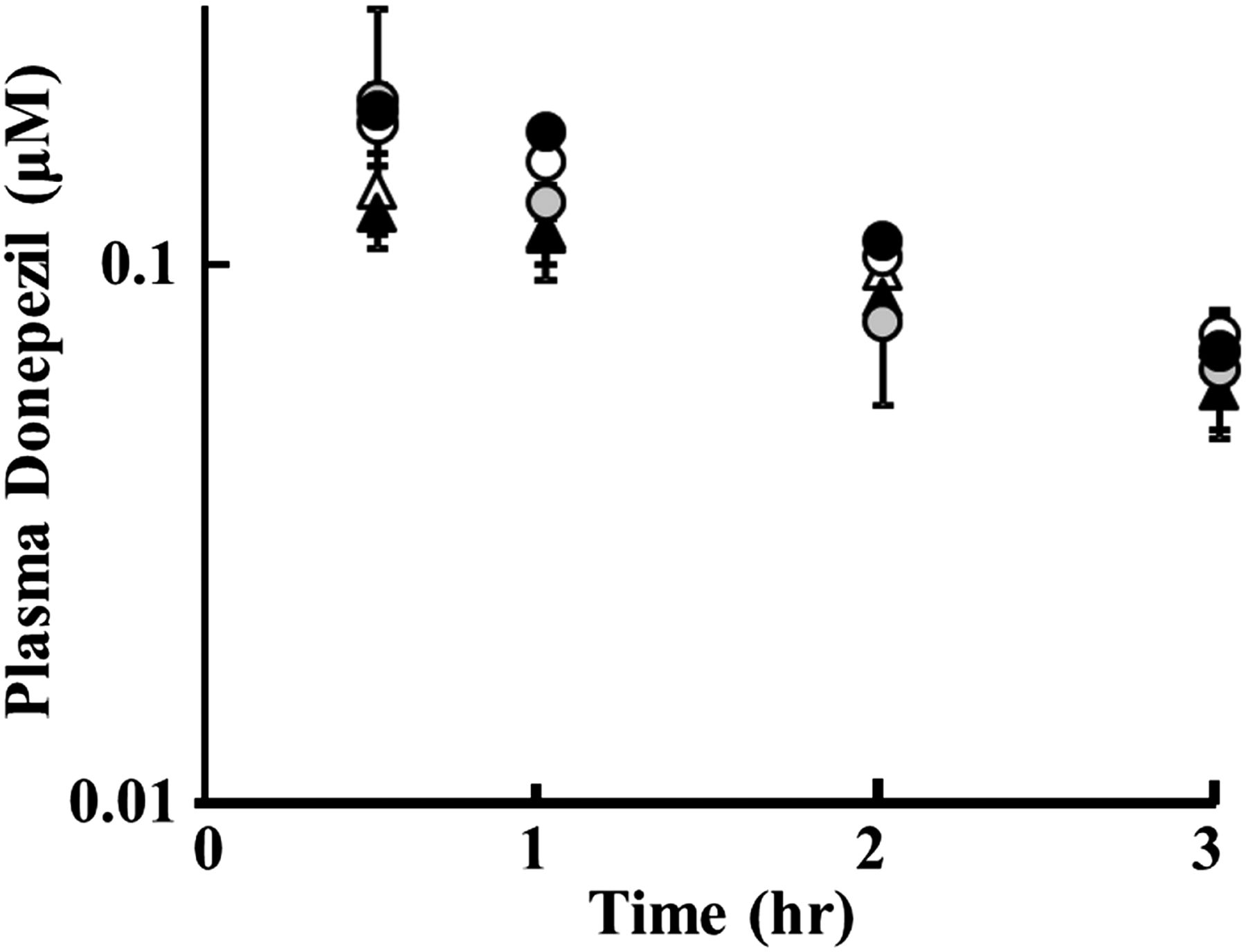
Effect of cilostazol and elacridar on plasma concentration of intravenously administered donepezil in rats. Plasma concentrations of donepezil after intravenous administration of donepezil alone at 1.0 mg/kg (○ as the control for cilostazol study/▵ with DMSO as the control for elacridar study) or concurrently administered with cilostazol at 100 mg/kg (gray circles) or 500 mg/kg (●) orally, or elacridar at 1.0 mg/kg (▴) intravenously. Data represent the mean ± S.D. (n = 3).
Pharmacokinetic parameters of donepezil after intravenous bolus administration with and without cilostazol or elacridar in female rats
Cilostazol (100 mg/kg or 500 mg/kg) dissolved in 0.5% methylcellulose was administered orally to female rats 30 minutes before intravenous administration of donepezil (1 mg/kg). Elacridar (1 mg/kg) dissolved in 5% DMSO was simultaneously administered with donepezil intravenously, and DMSO was also similarly administered in control experiments. Each experiment was repeated 3 times, and the mean values of pharmacokinetic parameters are shown with the S.D. (n = 3).
There were no significant differences in plasma concentration between the control rats and cilostazol- or elacridar-treated rats at each measured time (Fig. 5). In addition, no significant change in total clearance (CLtot), volume of distribution (Vd), or plasma AUC was observed in the case of coadministration of cilostazol or elacridar (Table 1). Here, plasma total concentrations of cilostazol at 100 and 500 mg/kg doses were 8.54 ± 0.67 and 16.7 ± 3.85 μM, respectively, at 3.5 hours after oral administration, and that of elacridar was 9.05 ± 1.05 μM at 30 minutes after intravenous administration.
Table 2 shows the concentration and tissue-to-plasma concentration ratio (Kp) in heart and brain tissues at 3 hours after intravenous administration with and without oral cilostazol or intravenous elacridar. The brain was measured as a reference because BCRP and P-glycoprotein both function as a part of the blood–brain barrier. As mentioned earlier (Fig. 5), the treatments caused no significant changes in plasma concentration of donepezil. However, cilostazol caused a concentration-dependent increase in Kp in both brain and heart. At the high dose of cilostazol, a statistically significant increase in Kp was observed in the heart from 6.32 to 7.82 (+24%), and in the brain from 7.67 to 9.47 (+23%). Similarly, elacridar significantly increased the Kp of donepezil from 5.5 to 7.92 (+44%) in the heart and from 6.78 to 10.3 (+52%) in the brain.
Plasma and tissue (brain and heart) concentrations and Kp of donepezil at 3 hours after intravenous administration with and without cilostazol or elacridar in rats
Donepezil concentrations in plasma, heart, and brain were measured at 3 hours after intravenous administration as described in Table 1. The results are shown as mean and S.D. (n = 3). Kp was obtained by dividing the tissue concentration by the plasma concentration. In the case of elacridar, the control rats were administered DMSO under the same conditions as used for the rats treated with elacridar.
In this study, observed plasma unbound concentrations of cilostazol at 3.5 hours after oral administration were 85.4 ± 6.7 nM and 167 ± 39 nM at doses of 100 and 500 mg/kg, respectively, using the reported plasma unbound fraction of 0.01 in rats (Kamada et al., 2011), and that of elacridar at 30 minutes after 1 mg/kg intravenous administration was 181 ± 0.02 nM, using the plasma unbound fraction of 0.02 (Kallem et al., 2012). The IC50 values of cilostazol for BCRP and P-glycoprotein were 130 nM and 12.7 μM, respectively (this study), and those of elacridar were 252 nM and 4 nM (Kwak et al., 2010), respectively.
Accordingly, based on the plasma unbound concentrations and the IC50 values for each transporter, cilostazol (167 nM concentration at 500 mg/kg dose) may be able to inhibit BCRP (IC50 130 nM) but not P-glycoprotein (IC50 12.7 μM), and elacridar (181 nM) is considered to inhibit BCRP (IC50 252 nM) partly and P-glycoprotein (4 nM) completely, assuming that the affinity for rat P-glycoprotein and BCRP is similar to that in humans. These results suggest that cilostazol may affect local tissue concentrations (in brain and heart) without causing any significant change of plasma concentration of donepezil by inhibiting BCRP, which serves as a tissue distribution barrier to substrate drugs.
Discussion
DDIs at efflux transporters such as P-glycoprotein and BCRP have been well documented to influence intestinal absorption and urinary and biliary elimination, but clinically relevant DDIs relating to the tissue distribution process are poorly understood. In the present study, we focused on the interaction of donepezil and cilostazol at efflux transporters as a potential explanation of the clinically reported cardiotoxicity of donepezil when coadministered with cilostazol (Tanaka et al., 2009; Shinozaki, 2012; Igeta et al., 2013). This is an important issue because simultaneous use of donepezil and cilostazol has been proposed to improve the pharmacologic outcome (Lee et al., 2007; Ihara et al., 2014).
We first confirmed that donepezil is a substrate of BCRP. It was also a substrate of P-glycoprotein, though there are conflicting reports as to the contribution of P-glycoprotein to donepezil transport (Summerfield et al., 2007; Lili et al., 2013). Here, we modified the transport study method by adding TEA and serum albumin to the medium to minimize the effect of nonspecific binding and/or transport by other transporters natively expressed in host cells because donepezil is a substrate of several organic cation transporters (Kim et al., 2010) that might be functional in MDCK cells and thus might disturb quantitative analysis of donepezil transport by P-glycoprotein and/or BCRP. In the presence of TEA and serum albumin, unidirectional transport by P-glycoprotein and BCRP was clearly demonstrated (Fig. 1).
We used this modified method to examine the inhibitory potential of cilostazol on donepezil transport by P-glycoprotein and BCRP. Interestingly, both P-glycoprotein and BCRP were inhibited by cilostazol, though the IC50 for P-glycoprotein (12.7 μM) was much higher than that for BCRP (130 nM). Consideration of the relationship between IC50 value and plasma concentration is essential to evaluate the clinical relevance of in vivo DDIs. In the Guidance for Industry: Drug Interaction Studies issued by the U.S. Food and Drug Administration in 2012 (CDER 2012), the criterion for evaluation of potential risk of efflux transporter inhibition is [I]1/IC50 ≥ 0.1, where [I]1 is defined as the mean steady-state total (bound and unbound) maximum plasma concentration (Cmax). Here, if we take [I]1 as the clinically evaluated pharmacologically minimum effective concentration of cilostazol, which is about 1.3 μM (interview form of cilostazol), the [I]1/IC50 ratio at BCRP is 10 (1.3/0.13), which is much higher than the critical value of 0.1. However, in the case of P-glycoprotein, the ratio is 0.102, which is a borderline value, suggesting a low risk of DDI at P-glycoprotein. If the total concentration of cilostazol is replaced with the unbound concentration, the [I]1/IC50 ratio at BCRP is 0.5 based on plasma protein binding of cilostazol of about 95% in humans (Suri et al., 1998), and this is still within the range of drug interaction risk. In addition, a total Cmax of 3.32 μM (unbound Cmax 0.166 μM) was reported in certain populations of patients who take cilostazol (Suri et al., 1998), and in this case the [I]1/IC50 ratio is higher than unity. Thus, we cannot rule out interaction of cilostazol at BCRP clinically.
On the other hand, the clinically relevant plasma concentration of donepezil (70 nM, Tiseo et al., 1998b) is much lower than the Km of BCRP-mediated transport of donepezil (4.03 μM, Fig. 3), which means that BCRP is not saturated in the clinical setting and can efficiently transport donepezil. These results indicate that in vivo DDI of donepezil and cilostazol at BCRP could be clinically significant in humans.
We then conducted an animal study to confirm the influence of cilostazol on the tissue distribution of donepezil. First, in vitro accumulation of donepezil in heart tissue slices at the steady state was increased in the presence of cilostazol (5 μM) as well as Ko143 (0.1 μM), a selective BCRP inhibitor (Fig. 4). Although the extent of the increase in Kp value is not so high (24% of the control), the increment of Kp is more than 1.1 (from 4.78 to 5.95), which can be quantitatively considered as a significant increase.
BCRP is localized at endothelial cells and cardiomyocytes in heart (Meissner et al., 2006; Solbach et al., 2008). It is not clear whether this localization of BCRP in heart tissue is well reflected in the currently used tissue-slice method because the tissue surface might be exposed to the drug solution directly, and BCRP may not be able to exclude donepezil as efficiently as it would in its role as a tissue barrier transporter in intact tissue. However, the observation of significantly increased accumulation of donepezil in the heart in the presence of BCRP inhibitor Ko143, to an extent comparable with the effect of cilostazol, indicates that BCRP is at least partly functional under the conditions of our experimental method. Accordingly, it is reasonable to conclude that cilostazol increases donepezil accumulation in heart tissue by inhibiting BCRP.
The in vivo pharmacokinetic study indicated an increase in Kp of donepezil in both heart and brain in the presence of cilostazol. Here, as the control inhibitor, we used elacridar, which inhibits both BCRP (IC50 252 nM) and P-glycoprotein (IC50 4 nM) depending on the concentration (Kwak et al., 2010); a previous study showed clear in vivo inhibition of P-glycoprotein at the blood–brain barrier after intravenous administration (Kallem et al., 2012). The increments of Kp of donepezil in heart and brain by elacridar were 44% and 52%, respectively (Table 2). Because the observed plasma unbound concentration of elacridar was 181 nM in the present study at 30 minutes after administration, the observed concentration was considered sufficiently high to inhibit P-glycoprotein completely and BCRP partially.
The effect of cilostazol was dose-dependent with significant increases of 24% and 23% in heart and brain, respectively, at the high dose of 500 mg/kg, though the increment was not statistically significant at the low dose of 100 mg/kg. Plasma total concentrations of cilostazol at the low and high doses were 8.54 and 16.7 μM, respectively, and the unbound fraction in rat plasma was about 1% (Kamada et al., 2011), so these doses are comparable with the unbound concentrations achievable at clinical doses in humans (1.35 to 2.7 μM, Interview form of cilostazol).
Considering the IC50 values of cilostazol for BCRP and P-glycoprotein, 0.13 and 12.7 μM, respectively, the plasma cilostazol concentration should be high enough to interact with BCRP at the high dose but not with P-glycoprotein, as discussed earlier. Furthermore, higher tissue accumulation of donepezil than shown in Table 2 is possible, because the measured time point at 3 hours after administration may not be at steady state for tissue distribution of donepezil.
Although there may be species differences in the affinities of cilostazol for BCRP and P-glycoprotein, these results show that cilostazol could interact with these efflux transporters and influence the tissue accumulation of substrate drugs, including donepezil, at clinically achievable concentrations in vivo. In the present study, we used female rats because there is a sex difference in the pharmacokinetics of cilostazol; females show higher plasma concentrations with a longer half-life (Kamada et al., 2011), which is useful for the current study.
Another important point is an absence of any significant change in the pharmacokinetic parameters derived from plasma concentrations, despite the significant increase of Kp in both heart and brain. Although donepezil is transported by BCRP and P-glycoprotein, their contributions to the overall elimination of donepezil are likely to be small because metabolism by CYP3A4 and CYP2D6 dominates in elimination. Cilostazol is eliminated mainly through metabolism by CYP3A4 and CYP3A5, but the Km values (from 1.6 to 5.6 μM) are higher than the clinically achievable plasma total concentration of cilostazol (Hiratsuka et al., 2007). Accordingly, cilostazol is expected to have only a minimal effect on metabolism of donepezil, which is consistent with the results of the present study in rats.
The reason why no change in plasma concentration was observed in spite of the increased accumulation in tissues such as brain and heart would be that the amounts of donepezil distributed to these tissues are too small to affect the plasma concentration, due to the small contribution of these tissues to the total distribution volume. Accordingly, this type of DDI at efflux transporters involved in the tissue distribution process should be carefully considered because local tissue concentration can be significantly increased without any apparent change in systemic exposure.
In conclusion, donepezil was found to be a substrate of both P-glycoprotein and BCRP. Cilostazol inhibits BCRP potently and P-glycoprotein weakly. The IC50 of cilostazol for BCRP lies within the range of the expected plasma concentration at clinically used doses. Therefore, the increased accumulation of donepezil in the heart and brain upon concurrent administration of cilostazol can be explained in terms of DDI at efflux transporters in brain and heart (local DDI), despite the apparent absence of any change in plasma concentration (absence of systemic DDI). Because ABCG2 shows genetic polymorphisms, pharmacogenetic study of the relationship between genotype and the incidence of donepezil-induced cardiotoxicity are warranted for the proposed local DDI.
Authorship Contributions
Participated in research design: Shinozaki, Takeuchi, Tamai.
Conducted experiments: Takeuchi, Nakanishi.
Performed data analysis: Takeuchi, Tamai.
Contributed to the writing of the manuscript: Takeuchi, Shinozaki, Nakanishi, Tamai.
Footnotes
- Received August 8, 2015.
- Accepted October 8, 2015.
This study was supported by a Grant-in-Aid for Challenging Exploratory Research from the Japan Society for the Promotion of Science.
Abbreviations
- AUC
- area under the plasma concentration–time curve
- BCRP
- breast cancer resistance protein
- CLtot
- total clearance
- DDI
- drug–drug interaction
- DMSO
- dimethylsulfoxide
- HPLC
- high-performance liquid chromatography
- Ko143
- (3S,6S,12aS)-1,2,3,4,6,7,12,12a-octahydro-9-methoxy-6-(2-methylpropyl)-1,4-dioxopyrazino[1ʹ,2ʹ:1,6]pyrido[3,4-b]indole-3-propanoic acid 1,1-dimethylethyl ester
- Kp
- tissue-to-plasma concentration ratio
- MDCK
- Madin-Darby canine kidney
- TEA
- tetraethylammonium
- Vd
- volume of distribution
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}