


Abstract
The in vitro metabolism of ropinirole was investigated with the aim of identifying the cytochrome P450 enzymes responsible for its biotransformation. The pathways of metabolism after incubation of ropinirole with human liver microsomes wereN-despropylation and hydroxylation. Enzyme kinetics demonstrated the involvement of at least two enzymes contributing to each pathway. A high affinity component with a KMof 5–87 μM and a low affinity component with aKM of approximately two orders of magnitude greater were evident. The high affinity component could be abolished by pre-incubation of the microsomes with furafylline. Additionally, incubation of ropinirole with microsomes derived from CYP1A2 transfected cells readily produced the N-despropyl and hydroxy metabolites. Some inhibition of ropinirole metabolism was also observed with ketoconazole, indicating a minor contribution by CYP3A. Multivariate correlation data were consistent with the involvement of the cytochrome P450 enzymes 1A2 and 3A in the metabolism of ropinirole. Thus, it could be concluded that the major P450 enzyme responsible for ropinirole metabolism at lower (clinically relevant) concentrations is CYP1A2 with a contribution from CYP3A, particularly at higher concentrations.
Ropinirole is a potent dopamine D2-receptor agonist currently inT development as a treatment for Parkinson’s disease (1). Previous studies have indicated that this compound is extensively metabolized by the cytochrome P450 mono-oxygenase system producing a number of oxidative metabolites. After an oral or iv dose to man approximately 10% of the dose is excreted unchanged, the remainder is metabolizedvia N-despropylation or hydroxylation pathways (fig.1, data on file at SB Pharmaceuticals).
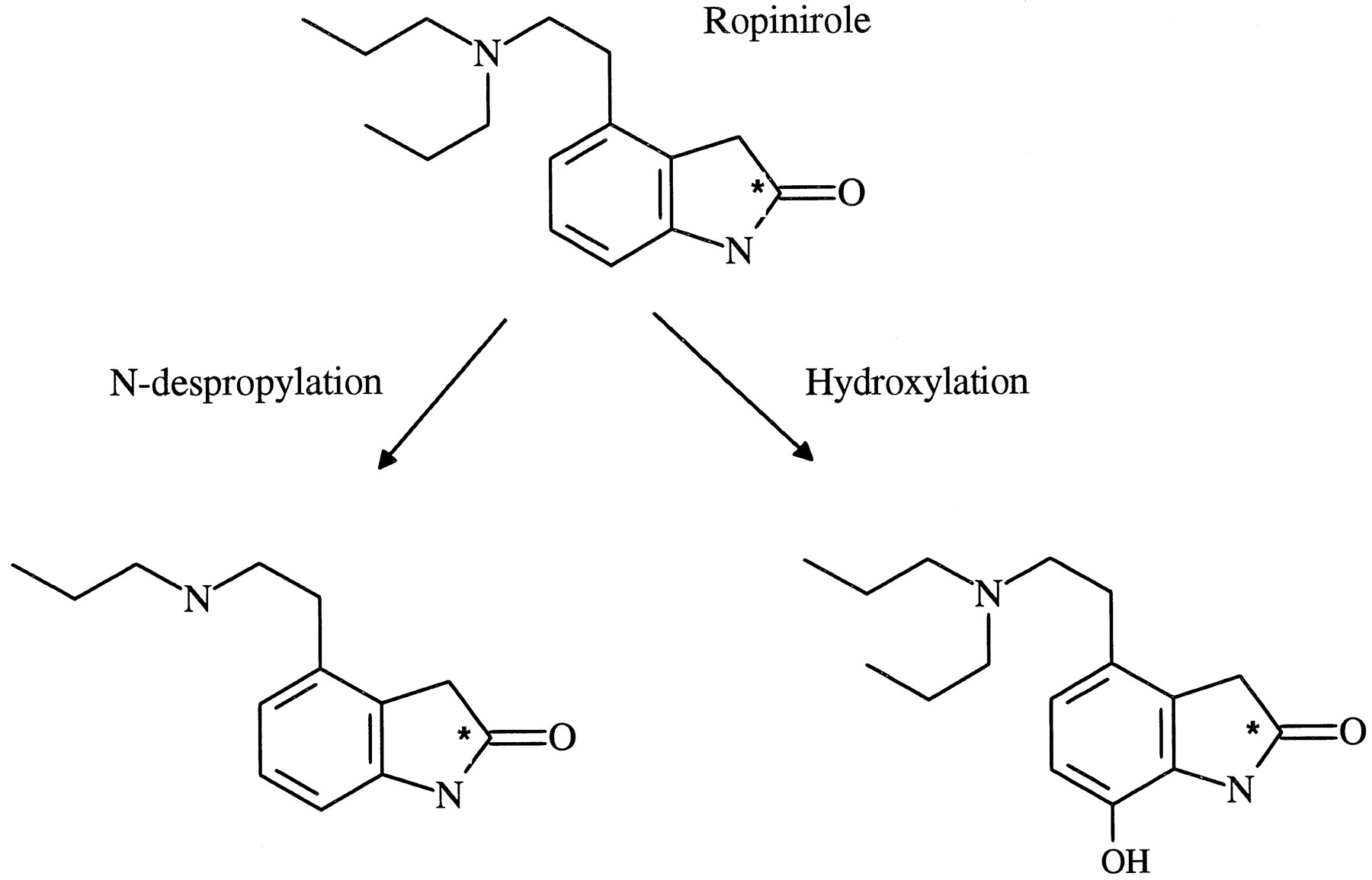
Metabolic pathways of ropinirole.
* Indicates the position of radiolabel.
To date there is no information available concerning which specific P4501 enzymes are involved in the metabolism of ropinirole. This knowledge can help predict drug interactions and genetically based inter-individual variations of drug metabolism and can prove invaluable when directing drug interaction studies in the clinic. The aim of this study was therefore to investigate the in vitro metabolism of ropinirole and to identify the specific cytochrome P450 enzymes involved. Ropinirole was incubated with human liver microsomes and microsomes from cells transfected with CYP1A2 and the products of metabolism identified. The kinetics of metabolite formation were investigated and chemical inhibitors selective for individual P450 enzymes were used to indicate the enzyme(s) responsible. A multivariate correlation method was used to provide further evidence for the cytochrome P450 enzymes involved in the metabolism of ropinirole.
Materials and Methods
Chemicals.
14C-Ropinirole (radiochemical purity 98.3%, specific activity 22 mCi/mmole), non-radiolabeled ropinirole,N-despropyl ropinirole, and hydroxy ropinirole were synthesized in the laboratories of SmithKline Beecham Pharmaceuticals (Harlow, UK). Furafylline, 4-hydroxy mephenytoin, 1′-hydroxy bufuralol, 6-hydroxy chlorzoxazone, and hydroxy tolbutamide were obtained from Salford Ultrafine Chemicals and Research Ltd. (Manchester, UK). Ethoxyresorufin and resorufin were obtained from Pierce and Warriner (Chester, UK). (4-14C) Testosterone and (1-14C) lauric acid were purchased from Amersham Ltd. (Bucks, UK). Testosterone metabolite standards were obtained from Steraloids Ltd. (Croydon, UK). (±) Bufuralol and S-mephenytoin were obtained from Chemical Development, SmithKline Beecham Pharmaceuticals. Coumarin, 7-hydroxy coumarin, tolbutamide, chlorzoxazone, testosterone, ketoconazole, and quinidine sulfate were all obtained from Sigma Chemical Co. Ltd. (Dorset, UK). All other reagents were purchased from commercial sources and were of the purest grade available.
Human Liver Samples and Preparation of Human Liver Microsomes.
Human liver tissue from eleven individuals (coded H6, H27, H30, H32, H33, H35, H37, H99, H100, H102, and H103) was obtained from either Vitron Inc.(Tuscon, AZ) or the International Institute for the Advancement of Medicine (Exron, PA). All samples were from otherwise healthy donors and in all cases the cause of death did not result from any known biochemical deficiency in the liver. Liver samples were stored at approximately −80°C until use. Human liver microsomal fractions were prepared by differential ultracentrifugation. After homogenization in 50 mM Tris-HCl buffer, pH 7.4, containing 0.25 M sucrose, the microsomal fraction was isolated from the supernatant of a 20 min, 10,000 gav. spin by ultracentrifugation at 100,000 gav. for 60 min. The microsomal pellet was resuspended in the Tris-HCl buffer and recentrifuged at 100,000 gav. for 60 min. The final pellet was resuspended in 50 mM potassium phosphate buffer, pH 7.4, and stored at approximately −80°C until required. Microsomal protein concentrations were determined by the method of Smith et al.(2) using the Pierce BCA (bicinchoninic acid) Protein Assay Reagent. None of the human livers used in this study was deficient in any of the cytochrome P450 activities measured, including the polymorphically expressed P450 activities bufuralol 1′-hydroxylase (CYP2D6) andS-mephenytoin hydroxylase (CYP2C19). Microsomes from human lymphoblastoid cells transfected with CYP1A2 cDNA were purchased from Gentest Corporation (Woburn, MA).
Investigation of Ropinirole Metabolism in Human Liver Microsomes.
14C-Ropinirole (final concentration 1–500 μM, added in Milli-Q water), was incubated with an NADPH generating system (final concentration 0.5 mM NADP, 5 mM glucose-6-phosphate, and 1 U/ml glucose-6-phosphate dehydrogenase in 2% (w/v) sodium hydrogen carbonate) with microsomal protein (0.8–3.8 mg/ml incubation) in 50 mM potassium phosphate buffer, pH 7.4, to give a final incubation volume of 250 μl. Incubations were initiated by the addition of a NADPH generating system after a 3-min preincubation period and were continued at approximately 37°C for 30 min in a shaking water bath. Initial velocity conditions were established which were linear with respect to both protein and time of incubation for each human liver. The reaction was terminated by adding 50 μl 5% (w/v) trichloroacetic acid. Extraction efficiencies after protein precipitation were >91%. After centrifugation, supernatants were analyzed directly by HPLC.
HPLC of incubates was performed using a Merck Hitachi L62000 system (Merck Ltd., Lutterworth, Leicestershire, UK) and a Supelcosil LC-ABZ column (5 μm, 4.6 mm × 15 cm, from Supelco UK, Ltd., Poole, Dorsef, UK) maintained at a temperature of approximately 40°C with a flow rate of 1.0 ml/min. Elution was obtained with 0.1 M ammonium acetate, pH 4 (solvent A) with a gradient of acetonitrile (solvent B). The elution conditions were a linear gradient of 0–15% solvent B over 10 min followed by a linear gradient to 100% solvent B by 12 min and isocratic 100% solvent B by 15 min. A Ramona-5 (Lablogic, Inc., Sheffield, Yorkshire, UK) was used for radiodetection and a Jasco 875 for UV absorbance at 250 nm. The radioactive peaks of interest on the chromatogram were integrated and the area under each peak expressed as a percentage of the total integrated area using Lablogic Rachel software. Rates of formation of ropinirole metabolites were evaluated from the fractional conversion of ropinirole apparent from the radiochromatogram. Peaks were identified using retention time comparison with authentic ropinirole, N-despropyl ropinirole, and hydroxy ropinirole and confirmed using LC/MS.
For analysis of enzyme kinetics, the production of metabolites in incubates containing ropinirole at concentrations from 1 to 8000 μM was investigated in duplicate under initial rate conditions in microsomes from three human livers (H32, H99, and H102). Ropinirole was incubated with approximately 1 mg/ml microsomal protein for 30 min. The apparent Vmax and KMvalues for the formation of N-despropyl and hydroxy ropinirole were determined.
The rates of ropinirole metabolism in microsomes from nine human livers were investigated by incubating 75 μM ropinirole with 0.8–3.8 mg/ml microsomal protein for 30 min. The rates of ropiniroleN-despropylation and hydroxylation were then correlated with the rates of specific cytochrome P450 activities.
Inhibition Experiments.
The effect of specific inhibitors of individual cytochrome P450 enzymes on the metabolism of ropinirole was investigated. The specific inhibitors furafylline (10 μM), sulfaphenazole (10 μM), quinidine (1 μM), and ketoconazole (1 μM) were added to incubates containing ropinirole (10 μM) to investigate the involvement of CYP1A2 (3, 4), CYP2C9 (5, 6), CYP2D6 (7), and CYP3A (8, 9), respectively. The effect of furafylline (10 μM) was also determined with a range of ropinirole concentrations (10–8000 μM). Furafylline, sulfaphenazole, and ketoconazole were added to incubates in methanol (final concentration 2% v/v); therefore, methanol (final concentration 2% v/v) was also added to the control incubations for these experiments. Quinidine was dissolved in 50 mM phosphate buffer, pH 7.4. Furafylline was preincubated with microsomes (final protein concentration 0.8 mg/ml incubate) and NADPH generating system for 10 min before adding ropinirole to initiate the reaction. In all other assays, reactions were initiated by the addition of the NADPH generating system. The effect of inhibitors on ropinirole metabolism was investigated in microsomes from two human livers and all incubations were performed in duplicate.
Determination of Cytochrome P450 Activities.
Ethoxyresorufin O-deethylase (EROD), coumarin 7-hydroxylase, tolbutamide hydroxylase, S-mephenytoin 4-hydroxylase, bufuralol 1′-hydroxylase, chlorzoxazone 6-hydroxylase, testosterone 6β-hydroxylase and lauric ω-hydroxylase were used as probe activities for CYP1A2 (10, 11), CYP2A6 (12), CYP2C9/8 (6), CYP2C19 (13), CYP2D6 (14), CYP2E1 (15), CYP3A (16), and CYP4A11 (17), respectively.
All microsomal assays were carried out under similar conditions at approximately 37°C in potassium phosphate buffer, pH 7.4, using a NADPH generating system consisting of NADP, glucose 6-phosphate, and glucose 6-phosphate dehydrogenase. Assays of EROD with a substrate concentration of 2 μM (18) and coumarin 7-hydroxylase using 100 μM coumarin (19) were carried out as reported by these authors. Tolbutamide hydroxylase, S-mephenytoin 4-hydroxylase, bufuralol 1′-hydroxylase, chlorzoxazone 6-hydroxylase, testosterone 6β-hydroxylase, and lauric ω-hydroxylase were determined as described previously (20), using 500 μM tolbutamide, 100 μM mephenytoin, 10 μM bufuralol, 100 μM chlorzoxazone, 250 μM testosterone and 100 μM lauric acid, respectively.
Analysis of Results.
The Michaelis-Menten parameters were determined from a nonlinear least squares regression analysis for two-enzyme model, using Grafit v 3.0 (21). Inhibition of enzyme activities were expressed as a per cent of control enzyme activities.
A multiple linear regression analysis, using SAS/INSIGHT Version 6 (22), enabled objective identification of the cytochrome P450 enzyme(s) explaining the variation in each ropinirole metabolite. To identify the enzymes involved, a model was fitted initially including all possible cytochrome P450 enzyme activities. Then, statistical tests (i.e., F- or, in this case, equivalentlyt tests) were used to test for the exclusion ofeach enzyme from the model: the cytochrome P450 enzyme activity corresponding to the lowest value of the test statistic was omitted if its p-value was greater than the 10% significance level and the change in the adjusted R2 value indicated that little explanatory power would be lost by its omission. The F- or t-test statistics were then calculated for the new model and again the explanatory variable corresponding to the lowest value of the statistic was excluded if itsp-value was greater than the 10% significance level and little explanatory power would be lost by its exclusion. These steps were repeated until no other cytochrome P450 enzyme activity could be omitted. This multiple regression procedure used to select those enzymes involved in each metabolic pathway of ropinirole is called the “backward elimination” procedure (23).
Results
In Vitro Metabolism of Ropinirole
Two major products of metabolism were evident following incubation of ropinirole with human liver microsomes and an NADPH generating system. Using co-chromatography with reference materials and mass spectrometry, these metabolites were identified asN-despropyl and hydroxy ropinirole. No metabolites were evident following incubation of ropinirole with human liver microsomes in the absence of NADPH. Metabolism of ropinirole (75 μM concentration) varied in the human liver microsomes investigated with the N-despropylation and hydroxylation pathways ranging from 1.6 to 23.5 nmol/hr/mg protein and 0.4 to 5.8 nmol/hr/mg protein, respectively (fig. 2). In the human livers investigated, the rate of N-despropylation exceeded the rate of hydroxylation by an average of 4-fold.

Variation of ropinirole N-despropylation and hydroxylation in microsomes from human livers (N = 11).
Identical pathways of metabolism were observed following incubation of ropinirole with microsomes derived from human lymphoblastoid cells transfected with CYP1A2 cDNA. The two metabolites produced, identified using co-chromatography with reference materials, wereN-despropyl and hydroxy ropinirole. As observed in the human liver microsomes, the N-despropyl metabolite exceeded the hydroxy metabolite by approximately 4-fold (fig. 3).

Radiochromatogram illustrating the metabolism of ropinirole with microsomes from human lymphoblastoid cell lines expressing CYP1A2 cDNA.
Kinetic analysis of ropinirole metabolism
Analysis of ropinirole N-despropylation and hydroxylation in the three human livers investigated demonstrated biphasic enzyme kinetics with respect to substrate concentration (fig.4a). The Michaelis-Menten parameters for the components involved are shown in table 1. For ropiniroleN-despropylation a relatively high affinity component, with a KM of 5–51 μM, was evident; the other contributing enzyme(s) had a KM of approximately two orders of magnitude higher, indicating a lower affinity enzyme. For ropinirole hydroxylation a relatively high affinity enzyme component was evident, with a KM of 8–87 μM, the lower affinity enzyme having a KM of approximately two orders of magnitude higher. Although the standard errors for the calculated parameters were large, it is clear from the kinetic plots that at least two enzymes, with KM values of two orders of magnitude difference, contribute to the metabolism of ropinirole. The errors in determining the Michaelis-Menten parameters may be attributed to the low affinity component(s) which could not be saturated despite the high concentrations of ropinirole used.

The effect of furafylline on Eadie-Hofstee plots for the N-despropylation and hydroxylation of ropinirole in human liver (H32) microsomes.
Apparent Michaelis-Menten parameters (± SE of determination) for theN-despropylation and hydroxylation of ropinirole in microsomes from three human livers
Correlation Experiments
Rates of ropinirole N-despropylation and hydroxylation were correlated with specific cytochrome P450 enzyme activities (table2). In the human livers investigated, statistically significant correlations were observed between rates of ropiniroleN-despropylation with EROD (1A2; p < 0.001) and testosterone 6β-hydroxylase (3A; p < 0.05). Ropinirole hydroxylation correlated with S-mephenytoin 4-hydroxylase (2C19, p < 0.05). Rates of ropiniroleN-despropylation and hydroxylation correlated with each other (t = 7.4, p < 0.001) andS-mephenytoin 4-hydroxylase correlated significantly with ethoxyresorufin O-deethylase (t = 4.3,p < 0.05) in the human livers investigated. No statistically significant or negative relationships were observed between ropinirole N-despropylation or hydroxylation and the other specific P450 enzyme activities investigated.
Correlation of ropinirole N-despropylation and hydroxylation with specific cytochrome P450 activities in human liver microsomes (n = 9)
Inhibition Experiments
The inhibition of ropinirole metabolism was investigated at substrate concentrations of 10 μM. At this concentration at least 50% of ropinirole N-despropylation activity was attributable to the high affinity component. The effects of selective P450 inhibitors on ropinirole metabolism are shown in table3. Furafylline demonstrated the most potent inhibitory effect on ropinirole metabolism, with both theN-despropylation and hydroxylation pathways being inhibited by approximately 50–80%. Some inhibition of ropinirole metabolism was also observed with ketoconazole, with 1 μM causing approximately 50% inhibition of each metabolic pathway in human liver H99. The microsomes from human liver H99 possessed higher CYP3A activity than human liver H32 (testosterone hydroxylase activity was 467 and 235 nmol/hr/mg protein in human livers H99 and H32, respectively). Little or no inhibition of ropinirole metabolism was observed with quinidine or sulfaphenazole.
The effect of selective P450 inhibitors on theN-despropylation and hydroxylation of ropinirole in human liver microsomes
The effect of furafylline inhibition was also determined in human liver microsomes incubated with a range of ropinirole concentrations (10–8000 μM). In the presence of furafylline, the high affinity enzyme was eliminated; kinetic analysis demonstrated low affinity enzyme(s) contributing to both the N-despropylation and hydroxylation pathways of ropinirole metabolism (fig. 4b).
Discussion
This study has shown that the in vitro metabolism of ropinirole reflects the metabolism observed in vivo; that the two major pathways of ropinirole metabolism wereN-despropylation and hydroxylation. Rates ofN-despropylation exceeded those of hydroxylation by approximately 4-fold, similar to that observed following administration of ropinirole to man, where an average of 56% of an oral or iv dose of ropinirole is excreted by N-despropylation compared with 11% by hydroxylation (data on file at SB Pharmaceuticals).
The production of N-despropyl and hydroxy ropinirole in human liver microsomes was NADPH dependent, and kinetic analysis indicated the involvement of at least two cytochrome P450 enzymes in each pathway. For both pathways of ropinirole metabolism, a relatively high affinity enzyme and a low affinity enzyme were evident. As plasma concentrations of ropinirole are unlikely to exceed 0.01 μM after administration to man (24), the high affinity enzyme is most likely to be responsible for the metabolism of ropinirole in vivo. Due to analytical limitations it was not possible to perform these studies at these pharmacological concentrations. However, it was possible to identify the high affinity enzyme as CYP1A2.
Primary evidence for the involvement of CYP1A2 was provided by chemical inhibition studies. Furafylline, a well characterized inhibitor of CYP1A2 (4), inhibited both the N-despropylation and hydroxylation pathways but, more specifically, abolished the high affinity component of both these pathways. Further evidence for the involvement of CYP1A2 was provided using microsomes derived from cells transfected with CYP1A2 cDNA. Microsomes from these cells incubated with ropinirole produced the N-despropyl and hydroxy metabolites and in a similar ratio to that observed in human liver microsomes and in vivo.
At analytically practical concentrations it was not possible to arrive at conditions where only CYP1A2 made an appreciable contribution. For both the correlation analysis and the inhibition studies, the substrate concentration was such that both high and low affinity substrates could contribute, the relative contribution being dependent on the level of expression in an individual. For example, in an individual with high CYP3A activity (H99), ketoconazole and furafylline had similar effects (50% inhibition) on ropinirole hydroxylation. In another individual with more typical CYP3A activity (H32), furafylline caused about 75% inhibition but ketoconazole only caused about 10% inhibition. Equally, multivariate correlation analysis showed that ropiniroleN-despropylation was highly significantly correlated with CYP1A2 activity but also associated with CYP3A activity across a human liver bank.
With the exception of the correlation, the results for theN-despropylation and the hydroxylation are similar. Despite the lack of a correlation with CYP1A2 and CYP3A for the hydroxylation pathway, the two pathways correlate well with one another and theN-despropylation clearly correlates with these two activities. The correlation of the hydroxylation with CYP2C19 could not be supported in any other way, and it is of interest to note that CYP1A2 and CYP2C19 are coincidentally correlated in this liver bank.
This study has identified the cytochrome P450 enzymes involved in the metabolism of ropinirole. Although CYP3A may be capable of metabolizing ropinirole at high concentrations, CYP1A2 is the major P450 enzyme responsible at ropinirole concentrations equivalent to those observedin vivo. As a consequence the potential of ropinirole to interact with other substrates of CYP1A2 and with CYP1A2 inhibitors should be considered. CYP1A2 inhibitors, like ciprofloxacin (25) and fluvoxamine (26), would be expected to significantly decrease the clearance of ropinirole on co-administration. However, the potential of ropinirole to interfere with the elimination of other CYP1A2 substrates is likely to be limited by its low plasma levels in vivo and its comparatively low affinity for the CYP1A2 enzyme. This lack of interaction potential has been confirmed in vivo after co-administration of ropinirole and the CYP1A2 substrate theophylline (27).
Footnotes
-
Send reprint requests to: J. C. Bloomer, Department of Drug Metabolism and Pharmacokinetics, SmithKline Beecham Pharmaceuticals, The Frythe, Welwyn, AL6 9AR, UK
- Abbreviation used is::
- P450
- cytochrome P450. Nomenclature of individual P450 enzymes is as recommended by Nelson,et al. Pharmacogenetics 6, 1–42 (1996)
- Received December 14, 1996.
- Accepted March 17, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}