


Abstract
PSC 833 has been used to overcome the phenomenon of multidrug resistance by inhibiting the P-glycoprotein (P-gp)-mediated efflux of antitumor drugs from tumor cells. Because P-gp expressed in several normal tissues may affect the disposition of its substrates, we examined the dose-dependent effect of PSC 833 on the disposition of vincristine (VCR) and digoxin (DGX) in rats. One-tenth milligram per kilogram PSC 833 was sufficient to significantly reduce the biliary excretion clearance of DGX from 3.0 ml/min/kg to 0.5 ml/min/kg, whereas 3 mg/kg PSC 833 was needed to significantly reduce the biliary excretion clearance of VCR from 36 ml/min/kg to 9 ml/min/kg. Three milligrams per kilogram PSC 833 significantly reduced the renal clearance of VCR by 30% but did not affect that of DGX significantly. The tissue-to-plasma DGX concentration ratio in the brain at 6 h after administration (0.34 versus 1.64), but not that of VCR at 2 h (1.07 versus 1.37), was significantly increased by PSC 833, 3 mg/kg. The differential effect of PSC 833 on the disposition of VCR and DGX may be ascribed to the different degree of contribution of P-gp to the disposition of these ligands.
One of the most important factors accounting for the acquisition of multidrug resistance (MDR)1 is the overexpression of P-glycoprotein (P-gp), an efflux pump for hydrophobic antitumor drugs, encoded by the MDR1 gene (Pastan and Gottesman, 1987). The pharmacological benefit of inhibiting the function of P-gp by many MDR modulators, including verapamil and cyclosporin A (CsA), has also been studied (Keller et al., 1992;Watanabe et al., 1995). Of the drugs investigated, PSC 833, a CsA analog, has been introduced clinically (Boote et al., 1996; Lum et al., 1993) because this compound is a potent inhibitor of transport via P-gp and is free of any immunosuppressant activity (Twentyman and Bleehen, 1991).
In addition to the overexpression of P-gp in tumor cells with acquired MDR, P-gp is also expressed in several normal tissues including renal tubular epithelial cells, hepatocytes, and the cerebral endothelial cells (Cordon-Cardo et al., 1990; Sugawara et al., 1988). P-gp expressed in these cells may play an important role in determining the disposition of its substrates within the body, e.g., the role of P-gp located on the bile canalicular membrane was demonstrated by Kamimoto et al. (1989), who reported the ATP-dependent uptake of daunorubicin into isolated membrane vesicles. We have also clarified the role of hepatic P-gp in the biliary excretion of vincristine (VCR) by demonstrating the inhibitory effect of verapamil in perfused rat liver (Watanabe et al., 1992) and by the finding that the induction of P-gp by phenothiazine results in increased biliary excretion of VCR (Watanabe et al., 1995). In the same manner, cumulative evidence suggests that renal excretion of digoxin (DGX), another P-gp substrate, is inhibited by MDR modulators such as verapamil and CsA (Charuk et al., 1994; de Lannoy et al., 1994; Hori et al., 1993).
The effect of P-gp on cerebral endothelial cells has been demonstrated both in vitro (Jette et al., 1995; Ohnishi et al., 1995) and in vivo (Sakata et al., 1994; Schinkel et al., 1994; Ohnishi et al., 1995).Schinkel et al. (1994) provided convincing evidence of the role of P-gp in determining the cerebral distribution of its substrates by demonstrating increased uptake of ivermectin and vinblastine inmdr1a knockout mice. Thus, it is plausible that the P-gp in the kidney and liver and in cerebral endothelial cells may be important in determining the plasma concentration of its substrates and their toxicity in the central nervous system, respectively. Based on such findings, preclinical studies have been performed to examine the effect of MDR modifiers on the disposition and toxicity of P-gp substrates (Colombo and Gonzalez, 1996; Didier and Loor, 1996).
In the present study, we examined the effect of PSC 833 on the disposition of P-gp substrates. We used VCR as a model compound because the biliary excretion mediated by P-gp may play an important role in determining its elimination (Watanabe et al., 1992, 1995). In addition, DGX was used because it might be possible to detect P-gp-mediated urinary excretion (de Lannoy et al., 1992; Ito et al., 1993).
Experimental Procedures
Materials.
[3H]DGX (15 Ci/mmol) and [3H]VCR (2–10 Ci/mmol) were purchased from New England Nuclear (Boston, MA) and Amersham Co. (Buckinghamshire, England), respectively. Unlabeled DGX and VCR were obtained from Wako Pure Chemicals Industries, Ltd. (Tokyo, Japan). PSC 833 was supplied by Novartis Pharma Ltd. (Basel, Switzerland). All other chemicals and reagents were commercial products of analytical grade.
Animals and Tumor Cells.
Ten-week-old female Sprague-Dawley (SD) rats weighing 220 to 270 g were purchased from Charles River Japan, Inc. (Tokyo, Japan). The animals had free access to water and food. The animal experiments were performed according to the guidelines provided by the Institutional Animal Care Committee (Graduate School of Pharmaceutical Sciences, The University of Tokyo). AH66 cells, rat ascites hepatoma cells that stably express rat P-gp on their surface (Miyamoto et al., 1992), were cultured in RPMI 1640 supplemented with 10% fetal calf serum in a humidified atmosphere with 5% CO2 at 37°C. The P-gp isoform was not determined in AH66 cells.
In Vivo Study.
The SD rats had their femoral vein and artery cannulated using polyethylene tubing (PE50; i.d. 0.58 mm; outside diameter 0.9655 mm, Becton Dickinson & Co., Parsippany, NJ). The bile duct was cannulated using polyethylene tubing (PE10; i.d. 0.28 mm; outside diameter 0.61 mm; Becton Dickinson & Co.) and the bladder was catheterized using polyethylene tubing (no. 8; outside diameter 2.33 mm; Hibiki Co., Tokyo, Japan) under light anesthesia with phenobarbital. The body temperature of the rats was maintained under suitable lighting. PSC 833, [3H]VCR, and [3H]DGX were dissolved in ethanol:PEG 200 (20:80 v/v), saline, and ethanol:saline (40:60 v/v), respectively. [3H]VCR (20 μCi, 0.5 mg/kg) and [3H]DGX (20 μCi, 0.25 mg/kg) were injected i.v. 30 min after an i.v. injection of PSC 833. Bile specimens were taken at 0 to 10, 10 to 30, 30 to 60, and 60 to 120 min for [3H]VCR and at 0 to 30, 30 to 60, 60 to 120, 120 to 240, and 240 to 360 min for [3H]DGX. Urine specimens were collected every 30 min for [3H]VCR and at 0 to 60, 60 to 120, 120 to 240, and 240 to 360 min for [3H]DGX. Blood was drawn at 1, 5, 10, 15, 30, 60, and 120 min for [3H]VCR and at 5, 10, 30, 60, 120, 240, and 360 min for [3H]DGX. At the end of the experiment, the tissue concentrations of [3H]VCR were determined by measuring the total radioactivity except for the liver and intestine. This was because our preliminary study had indicated that more than 80% of the radioactivity in tissues other than these two was associated with intact [3H]VCR. Concentrations of intact [3H]VCR and [3H]DGX in plasma, bile, urine, and tissues were determined in a liquid scintillation counter (model LS 6000LL; Beckman, Galway, Ireland) after separation by HPLC described as below.
Uptake Study.
2 × 105 cells were seeded in 24-well plates 24 h before the experiments. Uptake of [3H]VCR (0.05 μM) and [3H]DGX (0.05 μM) by the cultured cells was examined at 37°C in uptake medium consisting of Hanks’ balanced salt solution (pH 7.4) supplemented with 0.3% BSA. A warm plate was used to adjust the temperature to 37°C. At 2 h after initiation of the uptake experiments, the cells were washed four times with ice-cold phosphate-buffered saline and then solubilized with 250 μl 1 N NaOH. The radioactivity was counted in a liquid scintillation counter (model LS 6000LL; Beckman).
HPLC Analysis of [3H]VCR.
The analysis of [3H]VCR was accomplished by HPLC as described previously (Belle et al., 1992) after extraction with diethyl ether (Tellingen et al., 1993) with a modification. Briefly, tissues were homogenized with 2 ml ice-cold saline using a homogenizer (model Ultra-Turrax T25, IKA Labortechnik, Staufen, Germany). Ten micrograms of unlabeled VCR was added to the 100 μl of plasma and prepared specimens of bile, urine, and tissue as an internal standard. The mixtures were subjected to vortex mixing for 10 s. Samples were extracted twice with 2 ml of diethyl ether. The diethyl ether layer was collected and evaporated under a gentle nitrogen stream at 37°C. The residue was reconstituted in 200 μl HPLC solvent and 100 μl reconstituted solution was injected onto the HPLC column. Eluent corresponding to the [3H]VCR peak was collected and the radioactivity was counted. The radioactivity was corrected for the recovery calculated from the unlabeled internal standard data.
The HPLC system consisted of a pump (model L-6200; Hitachi, Ltd., Tokyo, Japan), an analytical column (5 μm CN-bonded phase, 150- × 4.6-mm, YMC-PACK CN A502; Yamamura Chemical Laboratories Co., Ltd., Kyoto, Japan), a guard column (5 μm CN-bonded phase, C-KGC-524C-3; Yamamura Chemical Laboratories Co., Ltd.), an auto-sampler (model 851-AS; Jusco Co., Tokyo, Japan), a spectrophotometer detector (model L-4200; Hitachi, Ltd.), and a fraction collector (model L-5200; Hitachi, Ltd.). The wavelength for the analysis was 210 nm. The mobile phase consisted of acetonitrile, H2O, and phosphoric acid (20:80:0.16, v/v/v). The flow rate was 0.5 ml/min.
HPLC Analysis of [3H]DGX.
The analysis of [3H]DGX was accomplished by HPLC as described previously (Stone and Soldin, 1988) after extraction with ethanol with a modification. Briefly, tissues were homogenized in 2 ml of ice-cold saline using a homogenizer (model Ultra-Turrax T25; IKA Labortechnik). One microgram of unlabeled DGX was added to the 100 μl of plasma and prepared specimens of bile, urine, and tissue as an internal standard. The mixture was subjected to vortex mixing for 10 s. Four volumes of ethanol was added, mixed for 10 min, and centrifuged for 10 min at 3000 rpm. Supernatant was collected and evaporated in a centrifugal evaporator. The residue was reconstituted in 200 μl eluent and 100 μl reconstituted solution was injected onto the HPLC column. Eluent corresponding to the [3H]DGX peak was collected and the radioactivity was counted. The radioactivity was corrected for the recovery calculated from the unlabeled internal standard data.
The HPLC system was the same as that described previously except that an analytical column (5 μm C18-bonded phase, 150- × 4.6-mm, Shim-pack CLC ODS (M); Shimadzu Co., Kyoto, Japan) with a guard column (5 μm C18-bonded phase, TSKgel ODS-Prep; Tosoh Co., Tokyo, Japan) was used. The wavelength for the analysis was 220 nm. The mobile phase consisted of tetrahydrofuran:H2O (23:77, v/v). The flow rate was 0.6 ml/min.
Determination of Kinetic Parameters.
From the time profiles of the plasma concentration of [3H]VCR and [3H]DGX, the area under the plasma concentration-time curve (AUC) to the last sampling point (fraction of dose/ml × min) was performed by the log-trapezoidal rule.
The cumulative amount of ligands excreted into the bile (Xbile) and urine (Xurine) up to time T, 120 min and 360 min for VCR and DGX, respectively, [Xbile, T (fraction of dose) and Xurine, T (fraction of dose)] was calculated from:
Biliary clearance (CLbile in ml/min/kg body weight) and urinary clearance (CLR in ml/min/kg body weight) were obtained from the equations:
Statistical Method.
The results are shown as the mean ± S.E. of the number of determinations. ANOVA followed by Fisher’s t test was used to determine the significance of differences between the means of two groups, with p < .05 as the minimum level of significance.
Results
Effect of PSC 833 on Elimination of [3H]VCR and [3H]DGX.
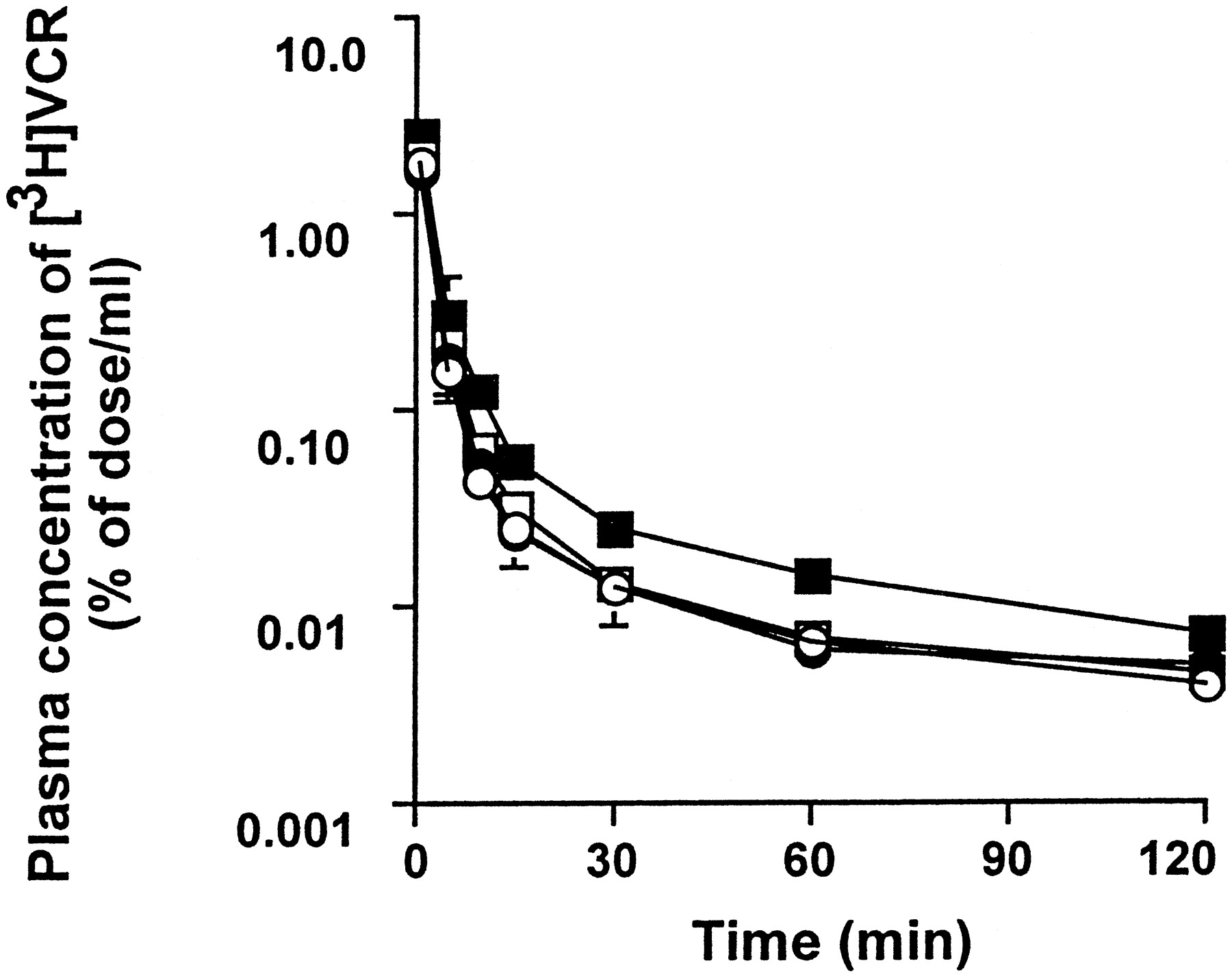
The dose-dependent effect of PSC 833 on the plasma concentration profiles of [3H]VCR and [3H]DGX is shown in Figs.1 and 2, respectively. PSC 833 significantly increased the AUC0–120 min of [3H]VCR and AUC0–360 min of [3H]DGX in a dose-dependent manner (Tables 1 and2). In the present study, the total body clearance was not determined due to the difficulty in determining the long half-life in the terminal phase.

Plasma concentration-time profiles of VCR in rats.
[3H]VCR (0.5 mg/kg) was given i.v. to bile duct-cannulated SD rats which had received the i.v. injection of PSC 833 [0 (○), 0.1 (●), 0.3 (■), and 3 (▪) mg/kg] 30 min before the experiments. All values are given as the mean ± S.E. of three independent experiments. Where the vertical bars are not indicated, the S.E. is contained within the limits of the symbol.

Plasma concentration-time profiles of DGX in rats.
[3H]DGX (0.25 mg/kg) was given i.v. to bile duct-cannulated SD rats that had received the i.v. injection of PSC 833 [0 (○), 0.1 (●), 0.3 (■), and 3 (▪) mg/kg] 30 min before the experiments. All values are given as the mean ± S.E. of four independent experiments. Where the vertical bars are not indicated, the S.E. is contained within the limits of the symbol.
Pharmacokinetic parameters of [3H]VCR after i.v. administration to rats
Pharmacokinetic parameters of [3H]DGX after i.v. administration to rats
CLbile and CLR of [3H]VCR and [3H]DGX are also listed in Tables 1 and 2. The CLbile of [3H]VCR and [3H]DGX was significantly reduced by PSC 833 (Tables 1 and 2). Moreover, 0.1 mg/kg PSC 833 was sufficient to significantly reduce CLbile of DGX, whereas 3 mg/kg PSC 833 was needed to significantly reduce the CLbile of VCR (Tables1 and 2). In addition, a significant reduction in the CLR of [3H]VCR was observed when rats were treated with 3 mg/kg PSC 833 (Table 1). For [3H]DGX, the CLR was not significantly affected by PSC 833 (Table 2).
Effect of PSC 833 on Tissue Disposition of [3H]VCR and [3H]DGX.
Tables 3 and4 demonstrate the dose-dependent effect of PSC 833 on the tissue disposition of [3H]VCR and [3H]DGX, respectively. In most tissues, the tissue-to-plasma concentration ratio (Kp) of [3H]VCR and [3H]DGX at 2 and 6 h after i.v. administration was not significantly affected by PSC 833 treatment (Tables 3 and 4). However, theKp of [3H]DGX in the brain was significantly increased by administration of PSC 833 (3 mg/kg; Table 4). In contrast, the Kp of [3H]VCR in the liver was significantly reduced by the same dose of PSC 833 (Table 3).
Effect of PSC 833 on the tissue-to-plasma concentration ratio of [3H]VCR
Effect of PSC 833 on the tissue-to-plasma concentration ratio of [3H]DGX
Effect of PSC 833 on Uptake of [3H]VCR and [3H]DGX by AH66 Cells.
Figure 3 shows the concentration-dependent effect of PSC 833 on the uptake of [3H]VCR and [3H]DGX by AH66. PSC 833 increased the cellular uptake of [3H]VCR and [3H]DGX at 2 h in a concentration-dependent manner.

Effect of PSC 833 on the uptake of VCR and DGX by AH66 cells.
In the presence of indicated concentrations of PSC 833, the uptake of [3H]VCR (0.05 μM; A) and [3H]DGX (0.01 μM; B) was examined at 2 h after initiation of the experiments. All values are given as the mean ± S.E. of four and eight independent experiments for VCR and DGX, respectively.
Discussion
In the present study, we studied the dose-dependent effect of PSC 833 on the disposition of P-gp substrates, particularly focusing on its effect on biliary and urinary excretion. Because biliary excretion is the predominant pathway for the elimination of VCR and P-gp may play a role in the urinary and biliary excretion of DGX, these two P-gp substrates were used as model compounds. The concentration-dependent effect of PSC 833 on the accumulation of VCR and DGX by AH66 cells suggests that VCR and DGX can be substrates for and PSC 833 can be the inhibitor of rat P-gp, respectively (Fig. 3). To determine the potency of inhibition by PSC 833, it is important to examine its effect on the disposition of P-gp substrates in tumor cells at their tracer concentrations (i.e., much less than theirKm values). However, in our previous study (Song et al., 1998b), we found a difficulty in determining theKm values for P-gp-mediated transport of VCR in tumor cells because the cell-to-medium concentration ratio of [3H]VCR was decreased by adding unlabeled VCR to the medium, presumably due to the saturation of intracellular binding. In the present study, we set the concentration of [3H]VCR and [3H]DGX as low as possible in in vitro experiments (0.05 μM and 0.01 μM, respectively). The concentration of [3H]VCR is much lower than its Km values (0.140 and 24.8 μM for high- and low-affinity sites, respectively), which were determined in membrane vesicles from P-gp-expressing cell line, DC-3F/VCRd-5L (Tamai and Safa, 1990). In addition, the concentration of VCR and DGX in in vivo experiments are lower than theirKm values; indeed, the plasma concentration of VCR and DGX were less than 0.003 μM and 0.002 μM, respectively (Figs. 1 and 2).
In the present in vivo studies, we examined the effect of PSC 833 (0.1, 0.3, and 3 mg/kg) on the disposition of VCR and DGX because its effect has been demonstrated to be concentration-dependent (Desrayaud et al., 1998). The plasma concentration of PSC 833 after i.v. administration of 0.1, 0.3, and 3 mg/kg of PSC 833 is 0.0333, 0.101, and 1.62 μM at 30 min and 0.0150, 0.0432, and 0.577 μM at 150 min, respectively (Song et al., 1998a). Because in in vitro experiments we and others have reported that IC50 of PSC 833 for P-gp is 0.05 to 0.1 μM and that the maximum inhibitory effect of PSC 833 is obtained at 1 to 2 μM (Lum et al., 1993; Song et al., 1998b), the different degrees of inhibition of P-gp may be obtained after administration of these three doses of PSC 833. However, we must be cautious in such in vitro-in vivo comparison because high plasma protein binding is reported (Simon et al., 1998) to date. Nevertheless, one of the ex vivo clinical studies in healthy subjects demonstrated the IC50 of PSC833 ranging from 0.5 to 1 μM, measuring rhodamine 123 uptake into a human myeloma cell line (M. Lehnert, C. Pfister and D. Russell, unpublished observations).
PSC 833 reduced the CLbile of VCR to approximately 20% of the control, presumably by inhibiting P-gp-mediated excretion across the bile canalicular membrane (Table 1). This hypothesis is also supported by our previous finding that 50 μM verapamil reduced the biliary excretion of VCR, but not that of taurocholic acid, in perfused rat liver (Watanabe et al., 1995). In addition, by using isolated bile canalicular membrane vesicles,Böhme et al. (1994) showed that PSC 833 inhibits the P-gp-mediated uptake of daunorubicin with aKi of 0.3 μM.
The inhibitory effect of PSC 833 on the urinary excretion of VCR (Table1) may be accounted for by its inhibitory effect on the P-gp located on the brush-border membrane in the renal tubular epithelial cells. This hypothesis is further supported by the previous study by de Lannoy et al. (1994) who found that the urinary excretion of [3H]VCR was reduced by 26 ± 5% in dogs when their plasma CsA concentration was maintained at >1.3 μM by i.v. infusion. The Kp value of VCR in the liver was significantly reduced by 3 mg/kg PSC 833 (Table 3), which may be accounted for by inhibition of uptake and/or intracellular binding.
The effect of PSC 833 on the disposition of DGX can be discussed in relation to the pharmacokinetic data on this compound inmdr1 knockout mouse strains (Mayer et al., 1996; Schinkel et al., 1997). Up to 72 h after administration of DGX to wild-type and mdr1a (−/−) mice, 33 and 73%, respectively, of the i.v. DGX (0.2 mg/kg) was excreted into urine (Mayer et al., 1996). The increase in the DGX excreted into urine in the knockout mice may be accounted for by the increased plasma AUC and, consequently, these data suggest that P-gp may not necessarily be the predominant mechanism for the urinary excretion of DGX (Mayer et al., 1996; Schinkel et al., 1997). This hypothesis is consistent with our experimental data showing that PSC 833 had no significant effect on the urinary excretion of DGX (Table 2). However, controversial results have been reported by others.de Lannoy et al. (1992) examined the urinary excretion of DGX in the canine kidney in vivo using the single-pass multiple indicator dilution method and found that the urinary recovery of DGX was reduced to 54% of the control by a blood concentration >1 μM CsA. In the same manner, Hori et al. (1993) found that 8 μM quinidine or verapamil in the perfusate reduced the urinary excretion of DGX to approximately 50% of the control in the isolated perfused rat kidney. At the present time, we do not have any convincing explanation to account for the discrepancy. An interspecies difference in the contribution of P-gp to the urinary excretion of DGX, and/or a differential inhibitory effect of MDR modifiers on the other transporter(s) involved in the urinary excretion of DGX are possible reasons.
In the present study, we found that the CLbile of DGX was significantly reduced by i.v. administration of PSC 833 (Table3), which is consistent with the previously reported results inmdr1 knockout mice; the amount of DGX excreted into the bile up to 90 min after i.v. administration was 21 to 24%, 16%, and 14% in the wild-type, mdr1a(−/−), andmdr1a/1b(−/−) mouse strains, respectively (Mayer et al., 1996; Schinkel et al., 1997). The significant increase in plasma AUC suggests a reduction in CLbile in the knockout mouse (Mayer et al., 1996; Schinkel et al., 1997). The reduction in the intestinal excretion of DGX in the mutant mice, however, was much more marked than that observed in the biliary excretion; indeed, the amount of DGX excreted into the intestine up to 90 min after i.v. administration was 16%, 2.2%, and 1.6% in the wild-type,mdr1a(−/−), and mdr1a/1b(−/−) mouse strains, respectively (Mayer et al., 1996; Schinkel et al., 1997). Collectively, multiple excretion mechanisms including that mediated by P-gp may be involved in the hepatobiliary excretion of DGX, whereas the intestinal excretion of this ligand is predominantly mediated by P-gp.
The Kp value of DGX in normal tissues, except brain, was not significantly affected by PSC 833 (Table 4), which is consistent with a previous observation in themdr1a/1b(−/−) mouse strain (Schinkel et al., 1997). An i.v. dose of 3 mg/kg PSC 833 increased theKp of DGX in the brain, at 6 h after administration, to a level approximately 5-fold that in control rats (Table 4). This result is also consistent with a previous finding in the mdr1a/1b knockout mouse. Although the total radioactivity after i.v. administration of [3H]DGX (1 mg/kg) was reported, theKp of [3H]DGX in the brain was approximately 9-fold higher than in the wild-type mice at both 90 min and 4 h after i.v. administration (Schinkel et al., 1997).
How can we account for the difference in the dose of PSC 833 required to alter the CLbile and the brainKp of DGX? One-tenth milligram per kilogram PSC 833 was sufficient to reduce the CLbile of DGX, whereas 3 mg/kg PSC 833 was needed to increase the Kp of DGX in the brain. The difference in the mechanism responsible for the transport of DGX across the bile canalicular membrane and across the luminal membrane of cerebral endothelial cells may account for the discrepancy. Alternatively, it is also possible that the intracellular unbound concentration of PSC 833 may differ between hepatocytes and cerebral endothelial cells.
In marked contrast to the effect of PSC 833 on theKp of DGX in the brain, the same dose of PSC 833 (3 mg/kg) did not affect the Kp of VCR in the brain at 2 h after i.v. administration (Table 3). Our result is consistent with the previous finding by Lemaire et al. (1996), who showed that 10 mg/kg PSC 833 increased theKp of CsA in the brain, but not that of VCR, at 2 h after i.v. administration, although they demonstrated that the penetration of VCR across the blood-brain barrier was enhanced by simultaneously administered PSC 833 by determining the isotope content associated with the brain parenchyma after washing out the blood remaining in the cerebral vascular system.
In conclusion, we could demonstrate the differential effect of PSC 833 on the disposition of VCR and DGX in several tissues. Althoughmdr1 knockout mouse strains can be excellent animal models for predicting possible adverse effects of the P-gp substrates that are concomitantly administered with the MDR modifiers (Mayer et al., 1996;Schinkel et al., 1994, 1995), pharmacokinetic studies to directly determine the effect of the modifiers are still important because the transport of antitumor drugs in normal tissues may be mediated not only by P-gp but also by other transporter(s) inhibited by the MDR modifiers.
Footnotes
-
Send reprint requests to: Dr. Yuichi Sugiyama, Graduate School of Pharmaceutical Sciences, The University of Tokyo, 7–3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail:sugiyama{at}seizai.f.u-tokyo.ac.jp
-
This work was supported in part by a grant-in-aid from the Ministry of Education, Science, Sports, and Culture of Japan and Core Research for Evolutional Sciences and Technology of Japan Science and Technology Corporation.
- Abbreviations used are::
- MDR
- multidrug resistance
- P-gp
- P-glycoprotein
- VCR
- vincristine
- DGX
- digoxin
- CsA
- cyclosporin A
- AUC
- area under the plasma concentration-time curve
- Xbile
- amount of the ligand excreted into the bile
- Xurine
- amount of the ligand excreted into the urine
- CLbile
- biliary clearance
- CLR
- urinary clearance
- SD
- Sprague-Dawley
- Kp
- tissue-to-plasma concentration ratio
- Received June 23, 1998.
- Accepted February 10, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}