


Abstract
Azelastine, an antiallergy and antiasthmatic drug, has been reported to be metabolized mainly to desmethylazelastine and 6-hydroxyazelastine in mammals. In the present study, the inhibitory effects of azelastine and its two metabolites on human cytochrome P-450 (CYP) isoform-dependent reactions were investigated to predict the drug interactions of azelastine using microsomes from human B-lymphoblast cells expressing CYP. The specific activities for human CYP isoforms included: 7-ethoxyresorufin O-deethylation (CYP1A1), phenacetin O-deethylation (CYP1A2), coumarin 7-hydroxylation (CYP2A6), 7-benzyloxyresorufinO-dealkylation (CYP2B6), S-warfarin 7-hydroxylation (CYP2C9), S-mephenytoin 4′-hydroxylation (CYP2C19), bufuralol 1′-hydroxylation (CYP2D6), chlorzoxazone 6-hydroxylation (CYP2E1), and testosterone 6β-hydroxylation (CYP3A4). In almost all the activities, desmethylazelastine exhibited stronger inhibition than azelastine and 6-hydroxyazelastine. Desmethylazelastine, but not azelastine and 6-hydroxyazelastine, uncompetitively inhibited CYP2B6 activity (Ki = 32.6 ± 4.8 μM). Azelastine, desmethylazelastine, and 6-hydroxyazelastine competitively inhibited CYP2C9 activity (Ki = 13.9 ± 1.8, 15.0 ± 3.1, and 17.0 ± 4.1 μM, respectively), CYP2C19 activity (Ki = 21.9 ± 2.2, 7.3 ± 1.6, and 9.3 ± 1.6 μM, respectively), and CYP2D6 activity (Ki= 1.2 ± 0.1, 1.5 ± 0.2, and 3.0 ± 0.5 μM, respectively). Azelastine and desmethylazelastine competitively inhibited CYP3A4 activity (Ki = 23.7 ± 4.6 and 13.2 ± 2.3 μM). 6-Hydroxyazelastine interfered with the determination of testosterone 6β-hydroxylation by HPLC. CYP1A2, CYP2A6, and CYP2E1 activities were not significantly inhibited by azelastine and the two metabolites. Among the human CYPs tested, the inhibitory effects of azelastine and its two metabolites were the most potent on human CYP2D6. In consideration of theKi values and the concentration of azelastine and desmethylazelastine in human livers after chronic oral administration of azelastine, the possibility of in vivo drug interaction of azelastine and other drugs that are mainly metabolized by CYP2D6 was suggested although it might not cause critical side effects. The inhibition of CYP2C9, CYP2C19, and CYP3A4 activity by azelastine and its two metabolites might be clinically insignificant.
Drug interactions can cause severe complications from medications. Clinically relevant drug interactions are often the result of the effects of cytochrome P-450 (CYP)1 enzymes during biotransformation (Muck, 1994). CYP comprises a superfamily of enzymes that have long been recognized as the primary enzymes responsible for human drug metabolism. Although the number of individual isoforms that have been identified and characterized is increasing (Nelson et al., 1996), the metabolism of xenobiotics in humans is handled mainly by enzymes from three families: CYP1, CYP2, and CYP3 (Spatzenegger and Jaeger, 1995).
Azelastine is a long-acting antiallergy and antiasthmatic drug that possesses properties beyond histamine H1receptor-blocking activity. These include antagonism of the chemical mediators adenosine, LTC4, LTD4, endothelin-1, and platelet activation factor, and inhibition of the generation and/or release of histamine, interleukin-1β, leukotrienes, and superoxide-free radicals (Perhach et al., 1989; Szelenyi, 1989). Azelastine has been reported to be metabolized to desmethylazelastine and 6-hydroxyazelastine (Fig.1) in mammals (Tatsumi et al., 1984). 6-Hydroxyazelastine is a major metabolite of azelastine in rats and guinea pigs (Tatsumi et al., 1984). In humans, desmethylazelastine is detected in plasma after the administration of azelastine (Pivonka et al., 1987). It has also been established that desmethylazelastine has pharmacologic activity equivalent to the parent drug (Perhach et al., 1989; Szelenyi, 1989). Recently, it has been reported that azelastine and desmethylazelastine inhibited the CYP2C19 and CYP2D6 activities in human liver microsomes (Morganroth et al., 1997). Furthermore, in our previous study (M.N., S.N., S. Tokudome, N.S., H.Y. and T.Y., submitted), we clarified that azelastineN-demethylation is catalyzed by CYP3A4, CYP2D6, and CYP1A2 in different human liver microsomes. To further characterize the inhibitory effects of azelastine and its metabolites on human CYP activities, we investigated the specific activities for human CYP isoforms using microsomes from human B-lymphoblast cells expressing human CYP in the presence of azelastine, desmethylazelastine, and 6-hydroxyazelastine.

Chemical structures of azelastine, desmethylazelastine, and 6-hydroxyazelastine.
Materials and Methods
Chemicals.
Azelastine hydrochloride [4-(p-chlorobenzyl)-2-(hexahydro-1-methyl-1H-azepin-4-yl)-1(2H)-phthalazinone hydrochloride] was provided by Eisai (Tokyo, Japan). Desmethylazelastine hydrobromide [4-(p-chlorobenzyl)-2-(hexahydro-1H-azepin-4-yl)-1(2H)-phthalazinone hydrobromide] and 6-hydroxyazelastine hydrochloride [6-hydroxy-4-(p-chlorobenzyl)-2-(hexahydro-1-methyl-1H-azepin-4-yl)-1(2H)-phthalazinone hydrochloride] were provided by Degussa Japan (Tokyo, Japan). 7-Ethoxyresorufin, 7-benzyloxyresorufin, resorufin, and chlorzoxazone were purchased from Sigma (St. Louis, MO). Phenacetin, acetaminophen, coumarin, and 7-hydroxycoumarin were purchased from Wako Pure Chemical Industries (Osaka, Japan). S-(−)-Warfarin, 7-hydroxywarfarin, S-(+)-mephenytoin, (±)-4′-hydroxymephenytoin, (±)-bufuralol hydrochloride, and 1′-hydroxybufuralol maleate were obtained from Ultrafine Chemicals (Manchester, UK). Testosterone, 6β-hydroxytestosterone, and 11β-hydroxytestosterone were purchased from Steraloids (Wilton, NH). NADP+, glucose 6-phosphate, and glucose 6-phosphate dehydrogenase were purchased from Oriental Yeast (Tokyo, Japan). Other chemicals were of the highest grade commercially available.
Enzyme Preparations.
Microsomes from human B-lymphoblast cells expressing CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2C9(Arg), CYP2C19, CYP2D6(Val), CYP2E1, and CYP3A4 were obtained from Gentest (Woburn, MA). Except for CYP1A2, CYP2B6, or CYP2C19, these were coexpressed with NADPH-CYP reductase. The CYP contents of these microsomes were provided in the data sheets by the manufacturer.
Enzyme Assays.
7-Ethoxyresorufin and 7-benzyloxyresorufin O-dealkylase activity (EROD and BROD) in microsomes from B-lymphoblast cells expressing CYP1A1 and CYP2B6, respectively, were determined as described previously (Nakajima et al., 1998b). The concentrations of microsomal protein expressing CYP1A1 and CYP2B6 were 0.02 and 0.04 mg/ml, respectively. The incubation mixture including 0.1 μM 7-ethoxyresorufin or 2 μM 7-benzyloxyresorufin was incubated for 10 min at 37°C. The formed product was determined fluorometrically (excitation; 574 nm, emission; 596 nm) using an F-4500 fluorometer (Hitachi, Tokyo, Japan). Phenacetin O-deethylase activity (POD) in microsomes from B-lymphoblast cells expressing CYP1A2 was determined as described previously (Kobayashi et al., 1998). The concentrations of microsomal protein were 0.2 mg/ml. The incubation mixture including 10 μM phenacetin was incubated for 30 min at 37°C. Coumarin 7-hydroxylase activity (COH) in microsomes from B-lymphoblast cells expressing CYP2A6 was determined as described previously (Pearce et al., 1992). The concentrations of microsomal protein were 0.08 mg/ml. The incubation mixture including 1 μM coumarin was incubated for 10 min at 37°C. The formed product was determined fluorometrically (excitation: 371 nm; emission: 454 nm) using an F-4500 fluorometer (Hitachi). S-Warfarin 7-hydroxylase activity (S-WFOH) in microsomes from B-lymphoblast cells expressing CYP2C9 was determined as described previously (Lang and Döcker, 1995). The concentrations of microsomal protein were 0.05 mg/ml. The incubation mixture including 1 μM S-warfarin was incubated for 30 min at 37°C.S-Mephenytoin 4′-hydroxylase activity (S-MPOH) in microsomes from B-lymphoblast cells expressing CYP2C19 was determined as described previously (Nakajima et al., 1998a). The concentrations of microsomal protein were 0.5 mg/ml. The incubation mixture including 25 μM S-mephenytoin was incubated for 30 min at 37°C. Bufuralol 1′-hydroxylase activity (BFOH) in microsomes from B-lymphoblast cells expressing CYP2D6 was determined as described previously (Nakajima et al., 1998b). The concentrations of microsomal protein were 0.05 mg/ml. The incubation mixture including 2 μM bufuralol was incubated for 10 min at 37°C. Chlorzoxazone 6-hydroxylase activity (CZXOH) in microsomes from B-lymphoblast cells expressing CYP2E1 was determined as described previously (Court et al., 1997). The concentrations of microsomal protein were 0.5 mg/ml. The incubation mixture including 50 μM chlorzoxazone was incubated for 5 min at 37°C. Testosterone 6β-hydroxylase activity (TESOH) in microsomes from human B-lymphoblast cells expressing CYP3A4 was determined as described previously (Arlotto et al., 1991). The concentrations of microsomal protein were 0.5 mg/ml. The incubation mixture including 100 μM testosterone was incubated for 5 min at 37°C. For the measurement of POD, S-WFOH,S-MPOH, BFOH, CZXOH, and TESOH, the formed product was determined by HPLC.
With the exception of 7-ethoxyresorufin and 7-benzyloxyresorufin, which were dissolved in dimethyl sulfoxide, substrates and inhibitors were dissolved in methanol so that the final concentration of solvent in the incubation mixture was <1%.
HPLC Analysis.
HPLC analyses were performed using L-7100 pump (Hitachi), L-7400 UV detector (Hitachi), F-1080 fluorescence detector, L-7200 autosampler (Hitachi), L-7500 integrator (Hitachi), and 865-CO column oven (Jasco, Tokyo, Japan) equipped with a Capcell Pak C18UG120 (4.6 × 250 mm; 4 μm) column (Shiseido, Tokyo, Japan). The flow rate was 1.0 ml/min and the column temperature was 35°C. The mobile phase for POD was 10% CH3CN, 50 mM KH2PO4. The eluent was monitored at 245 nm. The mobile phase for S-WFOH was 40% CH3CN, 0.04% H3PO4. The eluent was monitored fluorometrically (excitation: 320 nm; emission: 415 nm). The mobile phase for S-MPOH was 20% CH3CN, 50 mM KH2PO4. The eluent was monitored at 204 nm. The mobile phase for BFOH was 18% CH3CN, 1 mM perchloric acid. The eluent was monitored fluorometrically (excitation: 252 nm; emission: 302 nm). The mobile phase for CZXOH was 20% CH3CN, 50 mM KH2PO4. The eluent was monitored at 295 nm. The mobile phases for TESOH were solvent A (CH3OH/H2O/CH3CN, 39:60:1) (v/v) and solvent B (CH3OH/H2O/CH3CN, 80:18:2) (v/v). Typical conditions for elution were as follows: 25% B (0–25 min); 25–80% B (25–28 min); 80–25% B (28–33 min). A linear gradient was used for all solvent changes. The eluent was monitored at 240 nm.
Kinetic Analysis.
The concentrations of substrate ranged as follows: 7-ethoxyresorufin (50–500 nM), 7-benzyloxyresorufin (0.5–4 μM), phenacetin (2.5–500 μM), coumarin (1–10 μM), S-warfarin (1–10 μM),S-mephenytoin (10–100 μM), bufuralol (1–10 μM), chlorzoxazone (10–200 μM), and testosterone (25–100 μM), respectively. Kinetic parameters were estimated from the fitted curves using computer programs (KaleidaGraph, Synergy Software, Reading, PA; K · cat, BioMetallics, Princeton, NJ) designed for nonlinear regression analysis. All data were analyzed using the mean of duplicate determinations.
Results and Discussion
Azelastine has been reported to be metabolized to desmethylazelastine and 6-hydroxyazelastine in mammals (Tatsumi et al., 1984; Yang et al., 1992; Adusumalli et al., 1992). In the present study, the inhibitory effects of azelastine and these two metabolites on human CYP activities were determined using microsomes from human B-lymphoblast cells expressing each CYP isoform. To investigate the specific activity for each CYP isoform, substrate concentrations were set up to be close to the KM values in all activities.
EROD is catalyzed by CYP1A1 and CYP1A2 in humans (Eugster et al., 1993). However, it has been reported that the affinity of CYP1A1 for ethoxyresorufin is higher than that of CYP1A2 (Eugster et al., 1993). In our preliminary study, the KM value for EROD catalyzed by recombinant CYP1A1 of B-lymphoblast cells was determined to be 89 ± 21 nM. Therefore, EROD by CYP1A1 at a substrate concentration of 0.1 μM was examined for the specific activity of CYP1A1. Azelastine, desmethylazelastine, and 6-hydroxyazelastine inhibited EROD by CYP1A1 to a similar extent (Fig.2A). The IC50values were 29.2, 20.2, and 14.6 μM by azelastine, desmethylazelastine, and 6-hydroxyazelastine, respectively. POD is used as a marker activity of human CYP1A2 (Tassaneeyakul et al., 1993). However, it has been reported that the Eadie-Hofstee plot of the POD activity in human liver microsomes is biphasic (Boobis et al., 1981;Kahn et al., 1985), suggesting that at least two enzymes are involved. In these previous reports, the KM values for the high- and low-affinity components of the POD in human liver microsomes were 6 to 9 and 250 to 540 μM, respectively. In our preliminary study, the KM value for POD catalyzed by recombinant CYP1A2 of B-lymphoblast cells was determined to be 15.9 ± 1.5 μM. Therefore, POD catalyzed by CYP1A2 at substrate concentration of 10 μM was examined for the specific activity of CYP1A2. The inhibitory effects of azelastine and its two metabolites on POD by CYP1A2 were weak (IC50values were >100 μM; Fig. 2B).
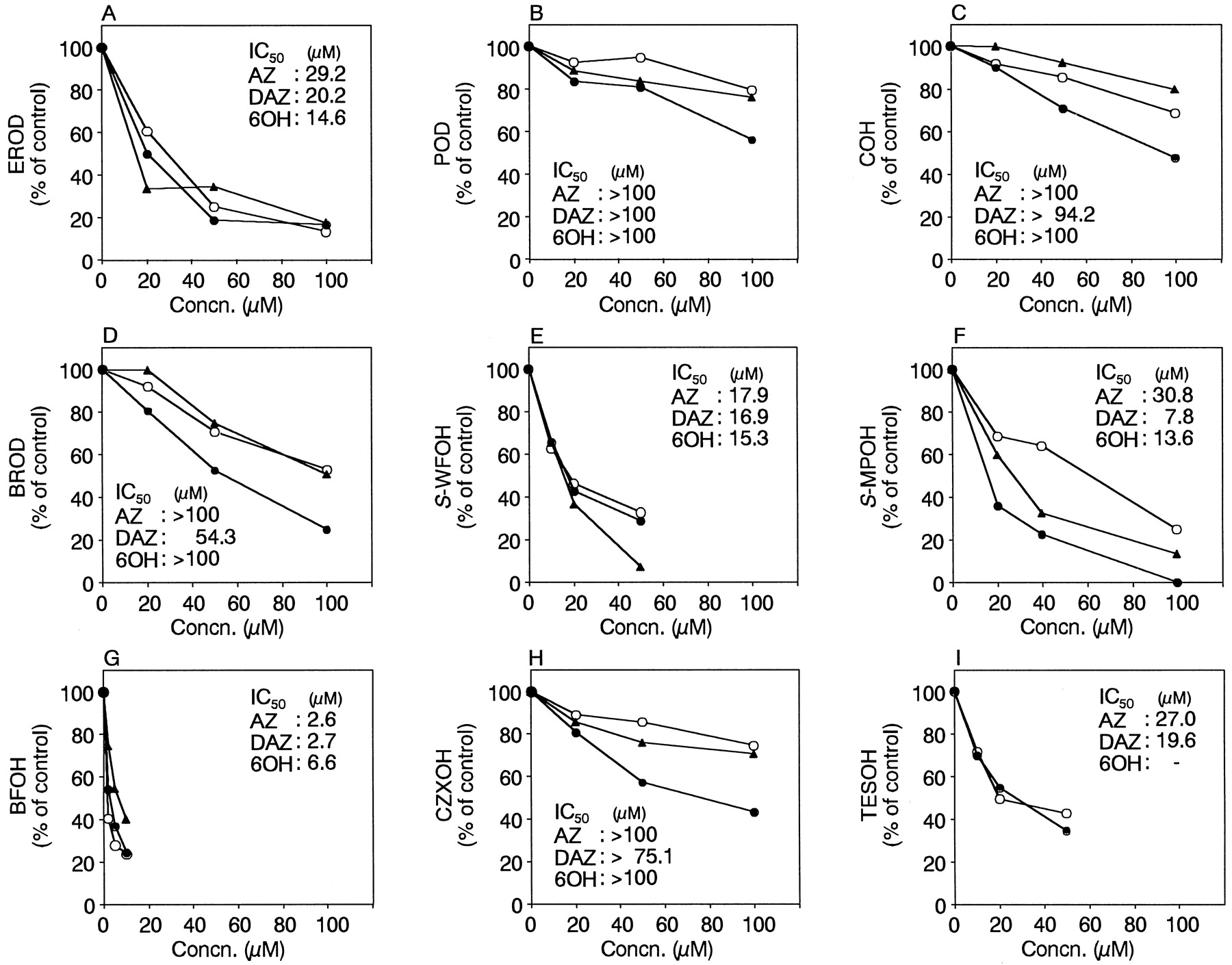
Inhibitory effects of azelastine (○), desmethylazelastine (●), and 6-hydroxyazelastine (▴) on human CYP activities.
A, EROD by recombinant CYP1A1 was determined at a 7-ethoxyresorufin concentration of 0.1 μM. The control activity was 6.2 pmol/min/pmol CYP. B, POD by recombinant CYP1A2 was determined at a phenacetin concentration of 10 μM. The control activity was 0.9 pmol/min/pmol CYP. C, COH by recombinant CYP2A6 was determined at a coumarin concentration of 1 μM. The control activity was 1.4 pmol/min/pmol CYP. D, BROD by recombinant CYP2B6 was determined at a 7-benzyloxyresorufin concentration of 2 μM. The control activity was 0.3 pmol/min/pmol CYP. E, S-WFOH by recombinant CYP2C9 was determined at a S-warfarin concentration of 1 μM. The control activity was 7.9 fmol/min/pmol CYP. F,S-MPOH by recombinant CYP2C19 was determined at aS-mephenytoin concentration of 25 μM. The control activity was 0.3 pmol/min/pmol CYP. G, BFOH by recombinant CYP2D6 was determined at a bufuralol concentration of 2 μM. The control activity was 1.5 pmol/min/pmol CYP. H, CXZOH by recombinant CYP2E1 was determined at a chlorzoxazone concentration of 50 μM. The control activity was 1.5 pmol/min/pmol CYP. I, TESOH by recombinant CYP3A4 was determined at a testosterone concentration of 100 μM. The control activity was 12.6 pmol/min/pmol CYP. 6-Hydroxyazelastine interfered with the quantification of 6β-hydroxytestosterone. Each data point represents the mean of duplicate determinations. The IC50values of azelastine (AZ), desmethylazelastine (DAZ), and 6-hydroxyazelastine (6OH) are shown as μM.
COH is solely catalyzed by CYP2A6 in humans (Yun et al., 1991). In our preparatory investigation, the KM value for COH catalyzed by recombinant CYP2A6 of B-lymphoblast cells was determined to be 0.8 ± 0.1 μM. The value was similar to that of a previous report (Shimada et al., 1996). Accordingly, COH by CYP2A6 at a substrate concentration of 1 μM was investigated for the specific activity of CYP2A6. The inhibitory effects of azelastine, desmethylazelastine, and 6-hydroxyazelastine on COH by CYP2A6 were weak (IC50 values were >100, 94.2, and >100 μM, respectively; Fig. 2C). BROD is catalyzed by the CYP2B6 in humans (Ekins et al., 1998). In our preliminary experiment, theKM value for BROD catalyzed by recombinant CYP2B6 of B-lymphoblast cells was determined to be 1.1 ± 0.3 μM (mean of three different determinations). The value was similar to that of a previous report (Ekins et al., 1998). Consequently, BROD by CYP2B6 at a substrate concentration of 2 μM was investigated for the specific activity of CYP2B6. Azelastine and 6-hydroxyazelastine had few inhibitory effects on BROD by CYP2B6 (Fig. 2D). Desmethylazelastine exhibited weak inhibition on BROD by CYP2B6 (IC50value was 54.3 μM).
S-WFOH is catalyzed by CYP2C9 in humans (Kaminsky and Zhang, 1997). In our preliminary study, the KMvalue for S-WFOH catalyzed by recombinant CYP2C9 of B-lymphoblast cells was determined to be 1.5 ± 0.2 μM. The value was similar to a previous report (4 μM; Rettie et al., 1992). Accordingly, S-WFOH by CYP2C9 at a substrate concentration of 1 μM was investigated for the specific activity of CYP2C9. Azelastine, desmethylazelastine, and 6-hydroxyazelastine inhibitedS-WFOH by CYP2C9 to a similar extent (Fig. 2E). The IC50 values were 17.9, 16.9, and 15.3 μM by azelastine, desmethylazelastine, and 6-hydroxyazelastine, respectively.S-MPOH is catalyzed by CYP2C19 in humans (Chiba et al., 1993). In our preparatory investigation, theKM value for S-MPOH catalyzed by recombinant CYP2C19 of B-lymphoblast cells was determined to be 25.2 ± 8.2 μM. Therefore, S-MPOH by CYP2C19 at substrate concentration of 25 μM was examined for the specific activity of CYP2C19. Azelastine, desmethylazelastine, and 6-hydroxyazelastine inhibited the S-MPOH by CYP2C19 (IC50 values were 30.8, 7.8, and 13.6 μM, respectively; Fig. 2F).
BFOH is catalyzed by CYP2D6 in humans (Kronbach et al., 1987). In our preparatory experiment, the KM value for BFOH catalyzed by recombinant CYP2D6 of B-lymphoblast cells was determined to be 2.6 ± 0.1 μM. Therefore, BFOH by CYP2D6 at a substrate concentration of 2 μM was investigated for the specific activity of CYP2D6. Azelastine, desmethylazelastine, and 6-hydroxyazelastine exhibited potent inhibition of BFOH by CYP2D6 (IC50 values were 2.6, 2.7, and 6.6 μM, respectively; Fig. 2G).
CZXOH is catalyzed by CYP2E1 and CYP1A1 in humans (Carriere et al., 1993). In our preparatory experiment, theKM value for CZXOH catalyzed by recombinant CYP2E1 of B-lymphoblast cells was determined to be 19.2 ± 1.8 μM. The KM value for CZXOH in human liver microsomes has been reported to be 40 μM (Peter et al., 1990). Although CYP1A1 and CYP2E1 had similar Vmaxvalues, the KM value of CYP2E1 was ∼23 times lower that that of CYP1A1 (Yamazaki et al., 1995). Furthermore, CYP1A1 is not expressed at an appreciable level in human liver in almost all cases (Guengerich, 1992). Therefore, the role of CYP1A1 in CZXOH at a physiologic chlorzoxazone concentration of 30 to 60 μM seems to be minor. Accordingly, CZXOH by CYP2E1 at a substrate concentration of 50 μM was investigated for the specific activity of CYP2E1. The inhibitory effects of azelastine and 6-hydroxyazelastine on CZXOH by CYP2E1 were slight (Fig. 2H). Desmethylazelastine exhibited weak inhibition of CZXOH by CYP2E1 (IC50 value was 54.3 μM). TESOH is catalyzed by CYP3A4 in humans (Waxman et al., 1988). In our preliminary study, the KMvalue for TESOH catalyzed by recombinant CYP3A4 of B-lymphoblast cells was determined to be 69.0 ± 9.3 μM. Consequently, TESOH by CYP3A4 at a substrate concentration of 100 μM was investigated for the specific activity of CYP3A4. Azelastine and desmethylazelastine inhibited the activity (IC50 values were 27.0 and 19.6 μM, respectively; Fig. 2I). 6-Hydroxyazelastine interfered with the quantification of 6β-hydroxytestosterone.
In an inhibition study, the Ki value is a better parameter to define the interaction of an inhibitor with a particular enzyme, as the IC50 value varies with the substrate concentration. Thus, the Kivalues and inhibitory patterns (competitive, noncompetitive, or uncompetitive) were investigated for the activities that were inhibited by azelastine, desmethylazelastine, and 6-hydroxyazelastine (Table1). BROD catalyzed by CYP2B6 was uncompetitively inhibited by desmethylazelastine (Ki = 32.6 ± 4.8 μM). TheKi values of azelastine, desmethylazelastine, and 6-hydroxyazelastine for S-WFOH catalyzed by CYP2C9 were 13.9 ± 1.8, 15.0 ± 3.1, and 17.0 ± 4.1 μM, respectively. The inhibitory pattern was competitive for azelastine, and mixed-type of competitive and noncompetitive for desmethylazelastine and 6-hydroxyazelastine. S-MPOH catalyzed by CYP2C19 was competitively inhibited by azelastine, desmethylazelastine, and 6-hydroxyazelastine (Ki = 21.9 ± 2.2, 7.3 ± 1.6, and 9.3 ± 1.6 μM, respectively). TESOH catalyzed by CYP3A4 was competitively inhibited by azelastine and desmethylazelastine (Ki = 23.7 ± 4.6 and 13.2 ± 2.3 μM). Finally, BFOH catalyzed by CYP2D6 was competitively inhibited by azelastine, desmethylazelastine, and 6-hydroxyazelastine (Ki = 1.2 ± 0.1, 1.5 ± 0.2, and 3.0 ± 0.5 μM, respectively). In our previous study (M.N., S.N., S. Tokudome, N.S., H.Y. and T.Y., submitted), we confirmed that the transformation of azelastine to desmethylazelastine in human liver microsomes is catalyzed mainly by CYP3A4 and CYP2D6 and to a smaller extent, CYP1A2. The KM value of CYP2D6 for azelastine N-demethylation was 1.4 μM. It is reasonable that the Ki value of azelastine for BFOH would be close to the KM value of CYP2D6 for azelastine N-demethylation.
Ki values and inhibitory patterns of azelastine and its metabolites for human CYP activities1-a
Recently, Morganroth et al. (1997) reported the inhibitory effects of azelastine and desmethylazelastine on CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 activities using human liver microsomes as follows: 1) S-MPOH (CYP2C19) in human liver microsomes was inhibited by azelastine and desmethylazelastine (theKi values were 7.0–9.1 and 3.4–8.2 μM, respectively), 2) the metabolism of S-metoprolol (CYP2D6) in human liver microsomes was inhibited by azelastine (theKi value was 1.7–12.1 μM) but not by desmethylazelastine, and 3) no inhibitory effects of azelastine and desmethylazelastine on CYP1A2, CYP2A6, CYP2C9, CYP2E1, and CYP3A4 activities were reported. The reason for the contradiction between the previous report (Morganroth et al., 1997) and the present study in which the inhibition of CYP2C9 and CYP3A4 by azelastine and desmethylazelastine was indicated is unknown. However, it might due to the difference of substrate, because carbamazepine andR-warfarin were utilized as marker probes for CYP3A4 in the previous report. Concerning CYP2C9, a marker probe used is same, i.e.,S-warfarin. Therefore, the contradiction might be due to the differences of substrate concentrations and/or in vitro techniques. Furthermore, the difference in the source of enzymes, i.e., human liver microsomes or recombinant CYPs might possibly cause the contradiction.
If an enzyme reaction proceeds with a single enzyme and is inhibited competitively by other drugs, velocity (V) is expressed by eq. 1.
 Equation 1where I and S indicate the concentrations of inhibitor and substrate, respectively. In addition, the inhibition type is noncompetitive, V is expressed by eq. 2.
Equation 1where I and S indicate the concentrations of inhibitor and substrate, respectively. In addition, the inhibition type is noncompetitive, V is expressed by eq. 2.
When it is assumed that KM ≫ S, eq. 3 can be adequately approximated to eq. 4 by combining eqs. 1 or 2.
The results obtained in the present study that the inhibitory effects of azelastine on CYP2D6 activity were the most potent among the CYP isoforms tested were consistent with those of our previous study (M.N., S.N., S. Tokudome, N.S., H.Y. and T.Y., submitted) in which theKM value of CYP2D6 for azelastineN-demethylation was smaller than those of CYP3A4 and CYP1A2. Although the percent contribution of CYP2D6 for azelastineN-demethylation was estimated to be 1.4 to 45.1% (M.N., S.N., S. Tokudome, N.S., H.Y., and T.Y., submitted), the affinity would be high enough to cause drug interaction. Thus, even if the contribution of a CYP isoform to certain types of drug metabolism is relatively low, it is important to define the affinity of the CYP for the drug metabolism to predict the possibility of drug interaction.
In conclusion, the present study suggested that azelastine and its metabolites have the strong competitive inhibitory effects on CYP2D6. The possibility of an interaction of azelastine and other drugs that are mainly metabolized by CYP2D6 in humans should be considered, although it might not cause critical side effects in clinical use.
Acknowledgments
We thank Mikie Suzuki for the skillful technical assistance and Brent Bell for reviewing the manuscript.
Footnotes
-
Send reprint requests to: Dr. Miki Nakajima, Division of Drug Metabolism, Faculty of Pharmaceutical Sciences, Kanazawa University, Takara-machi 13–1, Kanazawa 920-0934, Japan. E-mail:nmiki{at}kenroku.kanazawa-u.ac.jp
- Abbreviations used are::
- CYP
- cytochrome P-450
- BFOH
- bufuralol 1′-hydroxylase activity
- BROD
- 7-benzyloxyresorufin O-dealkylase activity
- COH
- coumarin 7-hydroxylase activity
- CZXOH
- chlorzoxazone 6-hydroxylase activity
- EROD
- 7-ethoxyresorufin O-deethylase activity
- POD
- phenacetin O-deethylase activity
- S-MPOH
- S-mephenytoin 4′-hydroxylase activity
- S-WFOH
- S-warfarin 7-hydroxylase activity
- TESOH
- testosterone 6β-hydroxylase activity
- Received January 21, 1999.
- Accepted March 30, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}