


Abstract
Expression and activities of cytochrome P450 enzymes are down-regulated in the liver during the host response to inflammation or infection, leading to alterations in drug clearance and toxin activation. This review focuses on recent studies on the mechanisms of this down-regulation, as well as the cytokines and cell types involved. Possible reasons for cytochrome P450 down-regulation are discussed.
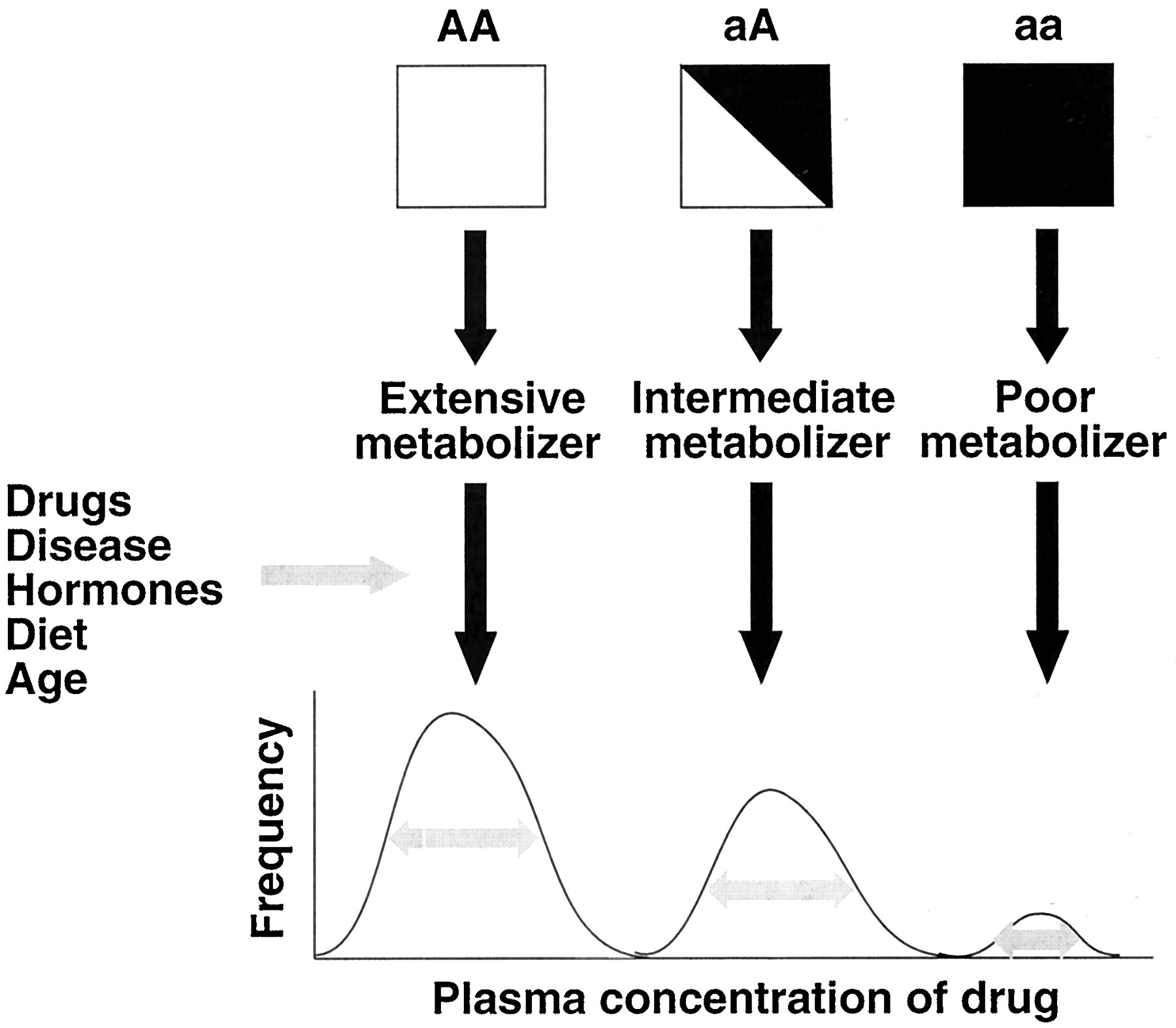
Causes of Interindividual Variation in Drug Metabolism
Cytochrome P450 (P4501) enzymes are responsible for the metabolic activation or inactivation of the majority of clinically used drugs and many toxins. There is substantial interindividual variation in the activities of P450 enzymes in humans, which can result in decreased or increased susceptibility to the beneficial or toxic effects of chemicals. A large component of interindividual variability is explained by polymorphisms in P450 genes that cause altered activity or expression of the encoded enzyme. However, the range of variability cannot be explained by genetic factors alone. For example, although the levels of the major human enzyme CYP3A4 in both liver and intestine vary greatly among individuals, there is no clear polymorphism in the distribution of activities of this enzyme. The expression of the drug-metabolizing P450 enzymes is also regulated by a variety of factors including drugs, hormones, development, and diet. Thus, it is the interaction of genetic and epigenetic factors that determines an individual's capacity to metabolize a given drug (Fig.1). In contrast to differences resulting from genetic polymorphisms, however, differences resulting from epigenetic factors may be short-lived. Traditionally, the most unpredictable component of drug responses has been attributable to genetic polymorphisms. With the advent of facile tests for genotypes at a given locus, we can consider that it is the variation caused by epigenetic factors that is now more unpredictable, whether between individuals or in the same person from one day to the next.

Interaction between genetic and epigenetic factors in the determination of drug metabolizing enzyme activities.
A, wild-type allele; a, mutant (deficient) allele. The gray arrows inside the curves denote variance influenced by epigenetic factors.
Regulation of P450 by Inflammation and Infection
In humans and animals, infections or inflammatory stimuli cause changes in the activities and expression levels of various forms of P450 in the liver, as well as in extrahepatic tissues such as kidney and brain (Morgan, 1997). In most cases, P450s and their activities are suppressed, but some are unaffected or induced under these conditions. P450 suppression can result in increased clinical toxicity of drugs with a low therapeutic index (Morgan, 1997). Conversely, some drugs must be converted to their pharmacologically or toxicologically active metabolites by P450 enzymes, and suppression of their metabolism during an inflammatory response can lead to a reduced therapeutic or toxic effect. On the other hand, the CYP4A family is induced by inflammatory stimuli (Morgan, 1997), and regulation of these fatty acid hydroxylases may be important in lipid homeostasis and termination of the action of inflammatory eicosanoids in these states (Devchand et al., 1996). The implications of these effects of inflammatory mediators are not limited to infections with live organisms or inflammatory diseases: interferons are used in the treatment of various cancers and viral infections, and various other cytokines are currently under investigation for treatment of cancers.
Much of the foregoing discussion is the topic of a recent extensive review (Morgan, 1997). The remainder of this article will discuss the author's view of some of the major issues confronting researchers in the field, with particular emphasis on the humoral, cellular and molecular mechanisms involved in hepatic P450 down-regulation and on possible reasons why this phenomenon occurs. In doing so, we will discuss contemporary work and evolving concepts that may shed light on some of these questions.
Extrahepatic P450.
Compared with the extensive study of hepatic P450, there is relatively little data on the effects of inflammation or infection on extrahepatic P450 expression, and therefore this is an area that requires much more research. Evidence suggests that, as in liver, P450s in extrahepatic tissues are likely to be regulated differentially by different inflammatory stimuli. For example CYP2E1 is induced in astrocytes during brain inflammation and CYP1A1 is down-regulated (Tindberg et al., 1996), whereas intracerebroventricular injection of bacterial lipopolysaccharide (LPS) results in suppression of CYP1A activity (Renton and Nicholson, 2000). Interestingly, two groups have found that injection of LPS in the brain has profound effects on CYP1A, -2B, -2E1, and -3A activities in the liver (Shimamoto et al., 1998; Renton and Nicholson, 2000). The effect on CYP1A activity occurs without a significant decrease in levels of CYP1A protein. Both groups have found that these effects are not dependent on sympathetic nerve activity (Shimamoto et al., 1999; Renton and Nicholson, 2000). These findings have important implications for the effects of infections of the central nervous system on systemic drug metabolism and suggest that other localized infections or inflammatory signals could also affect hepatic P450 activity.
Rodent Models and Human Studies
The regulation of P450 by inflammatory cytokines has been studied extensively in cultured rat hepatocytes, and as detailed previously (Morgan, 1997), this closely reflects responses observed in vivo in most cases. A number of studies have been performed on the effects of cytokines on cultured human hepatocytes (Abdel-Razzak et al., 1993;Muntané-Relat et al., 1995), which have produced results quite similar to those in rat hepatocytes. Although this would suggest that the rat may be a good model for studying the effects of inflammation on drug metabolism in humans, this is difficult to confirm because there has been relatively little research on the effects of infectious or inflammatory disease on P450 expression in man. Those studies that have been performed to date (Morgan, 1997) have mostly studied the pharmacokinetics of drugs whose metabolism is catalyzed by a number of enzymes. Although most systemic inflammatory stimuli have been reported to cause a decrease in human drug metabolism, postmortem specimens of liver infected with hepatitis C or hepatitis B virus each had elevated levels of CYP2A6 in infected cells or cells adjacent to areas of fibrosis or inflammation (Kirby et al., 1996). In contrast, hepatitis A virus was associated with decreased clearance of a CYP2A6 substrate (Pasanen et al., 1997). These conflicting results illustrate the need for detailed studies using specific pharmacokinetic and gene expression probes to begin to determine the effects of different infectious or inflammatory diseases on the activities and expression of individual human P450 enzymes. There are several considerations that make such work in humans difficult, including the fact that the investigator is limited mostly to the study of patient populations with varying severity of disease who are already receiving drug therapy, factors that can greatly complicate interpretation. Shedlofsky et al. (1997)have demonstrated effects of low doses of LPS on drug clearance in human volunteers. The information provided by their important work suggests that P450s may be regulated by LPS in humans much as they are regulated in rodents. However, we do not know whether or not this approach can accurately predict qualitative or quantitative changes in drug metabolism during severe clinical infections.
P450 Down-Regulation: A Homeostatic Mechanism or a Pathophysiological Phenomenon?
The phenomenon of P450 suppression in liver following activation of host-defense response by infectious or inflammatory stimuli has been known for more than 30 years (reviewed in Morgan, 1997). Yet, the reason for this down-regulation continues to be a major question in the field. Our laboratory speculated earlier that the suppression of P450 expression might not be an adaptive or homeostatic response, but could be a consequence of the liver's need to devote its transcriptional machinery to the synthesis of acute-phase proteins that have important roles in controlling the systemic inflammatory response (Morgan, 1989). Arguing against this interpretation is the fact that different cytochrome P450s appear to be regulated by different cytokines (Morgan, 1997) and by different mechanisms as discussed hereunder, which would not be expected if a nonspecific mechanism was occurring.
Although the possibility remains that P450 down-regulation is a pathophysiological consequence of inflammatory processes, there are several known properties of P450 enzymes that provide a basis for rational speculation on why the organism might find it advantageous to suppress hepatic cytochrome P450 enzymes during inflammation. These are summarized in Fig. 2 and discussed below.

Putative functions of P450 enzymes during cellular responses to inflammation.
Reactive Oxygen Species.
P450 enzymes undergo uncoupled catalytic turnover, resulting in the formation of superoxide anion radicals and hydrogen peroxide (White and Coon, 1980). The degree of uncoupling differs among P450 isoforms, with CYP2E1 being particularly active in this respect (Dai et al., 1993). Elevated levels of these reactive oxygen species in the cell can cause oxidative stress, and in the presence of iron, can result in the generation of the highly reactive hydroxyl radical. Induced or ectopic expression of various P450 enzymes has been associated with markers of oxidative damage in cultured cells (Dai et al., 1993; Park et al., 1996). In addition, some models of inflammation (such as injection of LPS, a model of bacterial sepsis) cause the induction of the inducible form of nitric-oxide synthase, NOS2, and depression of cellular glutathione levels in the hepatocyte (Harbrecht et al., 1997). Reaction of nitric oxide with superoxide (from P450s) generates the highly reactive peroxynitrite, which can result in protein oxidation and nitration reactions, and depletion of reduced glutathione will render the cell more susceptible to oxidative stress. Thus, P450 down-regulation could be a mechanism to protect the cell from the deleterious effects of these oxidizing species.
Arachidonic Acid Metabolites.
P450 down-regulation during an inflammatory response could be related to their function in the formation of biologically active metabolites of arachidonic acid. Epoxygenation of arachidonic acid catalyzed by P450s results in formation of four epoxyeicosatrienoic acids (EETs) (Campbell, 2000), each of which has anti-inflammatory properties as evidenced by their ability to inhibit tumor necrosis factor-α (TNFα)-induced expression of vascular cell adhesion molecule-1 and activation of the transcription factor NF-κB (Node et al., 1999). Support of an in vivo anti-inflammatory role for P450s is provided by the fact that inducers of P450 attenuate, whereas P450 inhibitors potentiate, the febrile response to low doses of LPS (Kozak et al., 2000, and references therein). 11,12-EET produced by endothelial cells also hyperpolarizes vascular smooth muscle and thereby functions as a paracrine vasodilator in many vascular beds (Campbell, 2000). P450s of both the CYP2J and -2C subfamilies have been detected in endothelial cells, and each can catalyze EET formation (Node et al., 1999;Fisslthaler et al., 2000). Moreover, inflammatory cytokines cause a down-regulation of CYP2C expression in cultured porcine aortic endothelial cells, resulting in reduced EET-dependent relaxation of the arteries (Kessler et al., 1999). Thus, P450 down-regulation in endothelial cells during inflammation may serve to prevent EET inhibition of the host inflammatory response, and at the same time it may help to attenuate the life-threatening hypotension associated with septic shock. Whether or not EETs generated in the liver could be endocrine factors involved in the regulation of other tissues and cell types (such as vascular smooth muscle) is not known. Alternatively, the down-regulation of hepatic P450 expression might have no physiological purpose per se, but instead could be a consequence of mechanisms that evolved for the purpose of suppressing these enzymes in extrahepatic cell types.
P450-Derived Nitric Oxide.
Lastly, the reason for down-regulation of P450 enzymes in the liver could be related to their ability to form nitric oxide (Fig. 2). Rat CYP3A enzymes can form NO from N-hydroxyarginine (Renaud et al., 1993), and inhibition of CYP3A activity in hepatocytes inhibits LPS and cytokine-stimulated production of NO and citrulline by more than 90% without affecting N-hydroxyarginine formation (Kuo et al., 1995). It has been suggested that under some conditions, P450 may have a physiological role in metabolizing excessN-hydroxyarginine generated by NOS2 (Kuo et al., 1995). Indeed, LPS-induced NO formation in mice is potentiated by dexamethasone induction of CYP3A, and the potentiation is inhibited by troleandomycin, an inhibitor of CYP3A enzymes (Fantuzzi et al., 1995). NO stimulates TNFα production, and inhibition of NO production blocks TNFα release. In accordance with a physiological role for P450s in NO generation, nonspecific P450 inhibitors block LPS-induced TNFα production in mice (Fantuzzi et al., 1993). These studies should be interpreted with caution because of possible nonspecific effects of the drugs used. With that caveat, they suggest the interesting possibility that down-regulation of P450 during endotoxemia, by reducing NO formation in the hepatocyte, could protect the cell from NO toxicity and at the same time regulate the inflammatory response by inhibiting TNFα production. However, the induction of hepatocyte NOS2 does not occur in all models of inflammation (e.g., sterile inflammation) that result in down-regulation of hepatic P450 (Morgan, 1997), and thus cannot be the only reason for P450 suppression.
It may be noted that some of the proposed roles of P450 down-regulation in the response to inflammation are apparently contradictory. Thus, it appears that mediators derived from different P450s could either augment (NO, CYP3A) or inhibit (EETs, CYP2C, CYP2J) inflammatory responses. However, the proposed physiological effects would require only down-regulation of these specific forms and would occur in different tissues. Therefore, to explain the suppression of other P450s, specific functions for them in the inflammatory process will have to be identified as well. On the other hand, it is possible that catalytic uncoupling or some other property common to most P450 enzymes is the primary reason for the evolution of these suppressive mechanisms.
Humoral Factors and Cell Types Involved in P450 Down-Regulation
Cytokines.
It is recognized that different models of inflammation and infection have the potential to suppress different subsets of hepatic P450s in vivo (Morgan, 1997; Sewer et al., 1997). Likewise, cytokines administered in vivo or in vitro have enzyme-selective effects on P450 expression (Muntané-Relat et al., 1995; Morgan, 1997). Although many studies have found that individual P450 enzymes can be down-regulated by multiple cytokines in the context of hepatocyte cultures, it has not always been clear that the effects are physiologically relevant in terms of the cytokine concentrations and exposure times studied. Therefore, a crucial question is: what cytokines are important for the down-regulation of which P450s caused by a given stimulus in vivo? Warren et al. (1999) tested the role of TNFα in P450 down-regulation by LPS in mice, using animals deficient in both the p55 and p75 receptors for this cytokine. LPS caused similar decreases in hepatic microsomal CYP1A, -2B, -3A, and -4A proteins and/or activities in both wild-type and TNFα receptor-deficient animals. Responses of CYP2E1- and CYP2D9-dependent activities were attenuated in the knockout mice, but this may be partly due to the decrease in basal activities of these enzymes in the receptor-deficient animals. Siewert et al. (2000) observed a similar lack of effect of interleukin (IL)-6 gene deletion on suppression of CYP1A2, -2A5, -2E1, and -3A11 mRNAs following LPS administration to mice. The authors argued convincingly that their observations and those of Warren et al. (1999) may be explained by functional redundancy of the various cytokines released during LPS-induced inflammation (Siewert et al., 2000). Another point is that the LPS receptor, CD14, has recently been shown to be expressed in hepatocytes (Liu et al., 1998), and LPS injected in vivo could be acting directly on the hepatocyte to modulate P450 expression. In support of this contention, we and others have observed direct effects of LPS on CYP2C11, NOS2 and acute phase protein expression in hepatocyte cultures (Sewer and Morgan, 1997;Panesar et al., 1999). It should also be considered that different doses of LPS might affect P450 expression by different mechanisms.
In contrast to the above findings in the LPS model, it was clearly demonstrated that IL-6 gene deficiency blocks the suppression of CYP1A2, -2A5, and -3A11 (with only a partial effect on CYP2E1) in mice treated with turpentine (Siewert et al., 2000). Unlike LPS, which causes release of many different cytokines including TNFα, IL-1, IL-6, and interferon-γ (Morgan, 1997), the effects of turpentine-induced sterile inflammation on hepatic acute-phase proteins are achieved mainly via IL-6 (Siewert et al., 2000). Thus, as noted by the authors, these results suggest that P450s should be considered as intracellular negative acute-phase proteins.
An important caveat for the use of knockout mice to deduce the roles of specific mediators or signaling pathways caused by an in vivo stimulus is that elimination (or inhibition) of a cytokine, receptor or signaling protein has the potential to affect P450 expression indirectly, e.g., by modifying the profile of other cytokines regulated by the deleted gene. The use of mice with hepatocyte-specific deletions of cytokine receptors would thus be highly desirable. This caveat notwithstanding, these approaches hold considerable promise for testing the roles of cytokines such as IL-1, TNFα, IL-6, transforming growth factor-β, and interferons in different models of inflammation or infection.
Non-Cytokine Components of the Inflammatory Response.
Because the above cytokines are the major factors involved in hepatic acute-phase protein induction during inflammation, their ability to suppress P450 expression has been studied intensively. However, it should be borne in mind that inflammatory stimuli also cause a stress response involving changes in circulating levels of many hormones, including glucocorticoids and epinephrine from the adrenal cortex and glucagon from the pancreas, and these have the potential to be involved in P450 regulation. For example, the expression of CYP2C11 and -3A2 is suppressed after injection of low doses of dexamethasone in rats. Glucocorticoids exert bimodal effects on CYP2C11 mRNA expression in hepatocytes: induction at low (resting) concentrations, and suppression at high (stress) concentrations (Morgan, 1997). Glucagon and epinephrine receptors in the liver are both coupled to adenylate cyclase activation, and cAMP inhibits phenobarbital-induced CYP2B1 and CYP3A expression (Sidhu and Omiecinski, 1995). More work is needed to determine the contribution of these hormones to P450 down-regulation in models of infectious and inflammatory disease.
Cytochrome P450 expression in rodents, and especially the rat, is highly sensitive to the temporal pattern of growth hormone in the plasma (Shapiro et al., 1995). We showed that LPS produced the same suppression of CYP2C6, -2C7, -2C12, and -2E1 mRNAs in intact or hypophysectomized female rats with or without growth hormone supplementation (Morgan, 1993), implying that the suppression was not caused by altering growth hormone secretion. However, the same experiments have not been performed in male rats, and it is possible that different inflammatory stimuli could have effects on P450 expression by perturbing growth hormone regulation.
One important aspect of inflammation that has been largely overlooked is the reduction in food intake (hypophagia) that occurs in some of these models. We discovered that, in addition to the suppression of multiple hepatic P450s in liver, LPS treatment produces induction of renal CYP4A mRNAs in mice and rats (Sewer et al., 1997;Barclay et al., 1999) and hepatic CYP4A in rats only (Sewer et al., 1997). Starvation is well known to cause induction of CYP4A expression, and to suppress the hepatic levels of other P450s to a lesser extent (Imaoka et al., 1990). Although our recent studies with rats or mice pair-fed to LPS-treated animals indicate that hypophagia is not a major factor in P450 suppression or induction by LPS (Barclay et al., 1999;Mitchell et al., 2000), we found that the more profound hypophagia caused by particulate irritant injection in rats does contribute to CYP4A induction by these agents (Mitchell et al., 2000). CYP2E1 is another P450 whose expression in both rat liver and kidney is induced by fasting, the latter by a post-translational mechanism (Ronis et al., 1998). We found that, although LPS suppresses CYP2E1 expression in liver, it induces CYP2E1 in rat kidney (Sewer et al., 1997). The possible role of hypophagia in the renal induction of CYP2E1 by LPS has not been determined. However, fasting for 24 h has no significant effect on CYP2C29, -3A11, or -2A5 mRNA expression in mouse liver (Barclay et al., 1999), or on CYP2C11 or -2E1 mRNAs in rat liver (E. Morgan and M. B. Sewer, unpublished observations). Therefore, hypophagia should not be a concern to investigators studying the acute effects of an inflammatory stimulus on these P450 mRNAs. However, this parameter should be controlled when more chronic models of inflammation, or different P450s, are being studied.
Role of Kupffer Cells.
LPS administration is a classical model of bacterial sepsis, and it is probably the best characterized model for in vivo P450 down-regulation by inflammation. In response to LPS, Kupffer cells, the resident macrophages of the liver, produce TNFα, IL-1, IL-6, and transforming growth factor-β, and are thought to be a major source of these substances acting on the hepatocyte. In accordance with this view, an elegant study by Milosevic et al. (1999), comparing the responses of hepatocytes cocultured with Kupffer cells with those of hepatocytes alone, showed that suppression of phenobarbital-induced CYP2B1 mRNA by LPS was mediated by TNFα release from the Kupffer cells. However, as noted above, LPS receptors are known to be present on hepatocytes, and we have detected potent effects of LPS on expression of CYP2C11 in cultured hepatocytes. Although a role of low-level contamination of Kupffer cells cannot be ruled out, this suggests that LPS can act directly on the hepatocyte to affect CYP expression. Clearly, additional work is required to determine the role of Kupffer cells in LPS regulation of P450 expression both in vitro and in vivo.
Molecular Mechanisms of Down-Regulation
Many of the effects of in vivo inflammation, interferons, and inflammatory cytokines on P450 levels in liver and cultured hepatocytes can be attributed to decreases in the levels of specific P450 mRNAs (Morgan, 1997). Different cytokines down-regulate different P450s both in vivo and in vitro, implying the existence of distinct regulatory mechanisms for different cytokines. The decreases in P450 mRNAs caused by cytokines and other inflammatory stimuli have generally been assumed to be due to inhibition of P450 gene transcription. Similarly, because the decreases in the cognate proteins usually display slower kinetics of suppression, it may be thought that the mRNA suppression is the primary reason for the decrease in the proteins. However, very little is known about the effects of inflammation on specific P450 translation, or mRNA or protein degradation, and these are areas that deserve closer attention.
Transcriptional Regulation.
Two different mechanisms have been proposed to be involved in down-regulation of aryl hydrocarbon-induced CYP1A1 by cytokines. The aryl hydrocarbon receptor, required for dioxin-dependent CYP1A1 induction, was shown to exhibit physical interaction and mutual functional repression with the cytokine-activated transcription factor NF-κB (Tian et al., 1999). Secondly, the CYP1A1 gene is repressed by oxidative stimuli via modulation of the binding of nuclear factor-1 to the CYP1A1 promoter (Morel and Barouki, 1998). Suppression of CYP1A1 promoter activity by TNFα treatment was dependent on an intact NF-1 binding site, and treatment with the antioxidant pyrrolidine dithiocarbamate also inhibited the effect of the cytokine (Morel and Barouki, 1998), suggesting that TNFα regulates CYP1A1 via redox regulation of NF-1. However, this interpretation in tempered by the fact that pyrrolidine dithiocarbamate is also an inhibitor of NF-κB activation.
IL-2 down-regulates the expression of CYP2C11 and CYP3A in rat hepatocytes, and Tinel et al. (1999) have proposed that this is related to IL-2 induction of the proto-oncogene transcription factor c-myc, based on the fact that an antisense oligonucleotide to c-myc, and several drugs that block c-myctranscription also blocked CYP2C11 and -3A down-regulation by IL-2. We have recently reported that the CYP2C11 promoter contains a low-affinity binding site for NF-κB, and that mutation of the promoter to inhibit NF-κB binding also abolishes suppression of a CYP2C11 promoter-reporter gene construct by either IL-1 or LPS (Iber et al., 2000). Although these findings argue strongly for a role of NF-κB in this model system, it remains to be determined if this mechanism plays a significant role in the in vivo suppression.
Considering the specificity of suppression of different P450 genes by different cytokines, it is likely that there will be multiple mechanisms for their transcriptional suppression. For example, expression of C/EBPα in HepG2 cells increases the expression of several P450 mRNAs (Jover et al., 1998), indicating the importance of this factor in expression of some liver-specific P450 genes. It is likely that C/EBPα and related proteins will be involved in regulation of some P450 genes during an inflammatory response, because during an acute-phase response to inflammation or infection, IL-6 causes a decrease in C/EBPα and an induction of C/EBPβ in the hepatocyte. C/EBPβ and/or C/EBPδ induction by IL-6 and other cytokines is one mechanism of induction of acute-phase genes (Alam et al., 1992). Because of the clear role of IL-6 in the down-regulation of some P450 mRNAs in vivo following turpentine administration (Siewert et al., 2000), other IL-6-regulated transcription factors such as Stat3 (Kaptein et al., 1996) should be considered for their role in P450 regulation. In addition to C/EBPα, other transcription factors required for basal transcription of liver-specific genes could be involved in P450 down-regulation. For example, the rat hepatic sodium-dependent bile acid transporter ntcp is down-regulated by LPS treatment in vivo, and this is due to decreased levels of hepatocyte nuclear factor-1 and an unidentified factor both of which are critical for ntcp transcription (Trauner et al., 1998).
Clearly, some progress has been made toward understanding transcriptional mechanisms of P450 regulation by inflammatory mediators. However, it should be noted that transcriptional regulation has been directly demonstrated in only a few cases: e.g., for cytokine suppression of dioxin-induced CYP1A1 and -1A2 transcription in hepatocytes and for LPS suppression of CYP2C11 in rats in vivo (reviewed in Morgan, 1997)
Role of Peroxisome Proliferator-Activated Receptor-α (PPARα) in P450 Down-Regulation.
PPARα is a nuclear receptor that functions as a central regulator of fatty acid catabolism, in response to fasting and stress situations. During a study on the role of PPARα in CYP4A induction, we made the surprising discovery that down-regulation of three hepatic murine P450 mRNAs by LPS treatment was blocked or attenuated in PPARα-null mice (Barclay et al., 1999). Moreover, the same P450s are down-regulated after treatment with the PPARα ligand clofibrate, and this effect was also blocked or attenuated in the PPARα-null animals (Barclay et al., 1999). Other studies have shown that peroxisome proliferators can suppress the expression of negative acute-phase genes and CYP2C11 in rat liver (Corton et al., 1998). The simplest explanation for the requirement for PPARα in the inflammatory down-regulation of murine P450s by LPS is that during inflammation, a PPARα ligand is generated that activates PPARα in the hepatocyte, somehow resulting in P450 suppression. However, there are a number of other potential explanations, and more work is needed to elucidate the role of this receptor in P450 down-regulation.
Oxidative Stress.
The possible role of oxidative signaling in suppression of CYP1A1 transcription by TNFα has already been discussed. Another role of reactive oxygen species in P450 regulation by inflammatory stimuli is indicated by the work of El-Kadi et al. (2000). Incubation of human hepatocytes with sera from humans with a viral infection, or sera from rabbits injected with turpentine, causes a decrease in CYP1A2-associated theophylline metabolism without affecting the levels of CYP1A2 or CYP1A1 proteins (El-Kadi et al., 2000). This decrease in P450 catalytic activity can be partially prevented with antioxidants and potentiated by inhibitors of antioxidant enzymes (El-Kadi et al., 2000). As discussed previously, therefore, this inactivation could be designed to prevent further generation of P450-derived reactive oxygen species. Oxidative signals may play a role in regulation of transcription and activities of other P450s as well.
Nitric Oxide.
The putative role of nitric oxide, produced by NOS2 during an inflammatory response, on the down-regulation of P450 catalytic activities as well as protein and mRNA expression, is a controversial subject that cannot be dealt with comprehensively here. Much of the data and arguments for and against this theory are presented in a previous review (Morgan, 1997). The use of NOS inhibitors in vitro and in vivo has been reported to attenuate declines in P450 activities, protein and mRNA levels produced by inflammatory stimuli in some laboratories. The ability of NO inhibition to attenuate declines in some P450-dependent activities is a relatively uncontested finding, as is the fact that gaseous NO, and generators of NO-derived reactive nitrogen species, can inhibit P450 catalysis in vitro. In fact, NO inhibitors can attenuate decreases in vivo drug clearance caused by liver inflammation (Blobner et al., 1999). However, some laboratories including our own have found little or no effect of NOS2 inhibition (or gene deletion) on suppression of various P450 mRNAs and proteins. One possible explanation is that NO-dependent mechanisms are dose-dependent and occur only at high doses of LPS; our studies that failed to find NO-dependent effects on P450 regulation in vivo employed a moderate dose of LPS (1 mg/kg) (Sewer et al., 1998; Sewer and Morgan, 1998). Most of the studies that found NO-dependent effects have used doses of 2 mg/kg or more (Muller et al., 1996; Khatsenko et al., 1997; Takemura et al., 1999). This is an easily testable hypothesis.
Some of the most convincing evidence for participation of NO in down-regulation of P450 mRNAs and proteins has been obtained with phenobarbital-inducible CYP2B enzymes. Administration of L-NAME to rats treated with phenobarbital and LPS blocked the down-regulation of CYP2B1/2 activity, mRNA and protein (Khatsenko et al., 1997). Inhibitors of NOS also blocked the suppression of CYP2B1/2 proteins by a cytokine cocktail in short-term cultures of rat hepatocytes (Carlson and Billings, 1996), although the regulation of CYP2B1/2 mRNAs in this system was not reported. Incubation of CYP2B1 with peroxynitrite in vitro results in the formation of 2 mol of nitrotyrosine per mol of CYP2B1, and the formation of nitrotyrosine correlates with loss of enzyme activity (Roberts et al., 1998). It remains to be determined whether P450 nitration is causative for inhibition of catalytic activity and/or reduction in CYP2B1 protein content.
Conclusion
From the above discussion, it will be obvious that, to predict the effects of infection or inflammation on drug metabolism, it is important to understand which stimuli regulate which forms of P450. It is also important to understand the mechanisms of these processes, because this would allow us to predict the effects of other diseases, drugs, or toxins that activate the same pathways on drug clearance or toxicity. It would also provide potential mechanisms to modulate the metabolism of a given drug, if that should become therapeutically necessary. Also, given the putative roles of P450s in physiological processes associated with inflammation, control of P450 down-regulation could be a potential target for drug or gene therapy.

Edward T. (Eddie) Morganwas born in Millport, Scotland, and received the B.Sc. (Honours) degree in pharmacology from the University of Glasgow in 1976. He received the Ph.D. degree in pharmacology from the same institution in 1979, working with Garth Powis and Paul Skett on the effects of ethanol on hepatic microsomal drug metabolism.
He worked as a postdoctoral fellow in Minor Coon's laboratory at the University of Michigan before moving to Jan-Åke Gustafsson's laboratory at the Karolinska Institute as a Visiting Scientist. In 1986, he joined the faculty at Emory University, where he is now Professor of Pharmacology. Throughout his career, Dr. Morgan has held an interest in cytochrome P450 regulation. This began with his work in Glasgow and Ann Arbor on ethanol-inducible CYP2E1 and continued in his work in Sweden on sexually differentiated and growth hormone-regulated P450 expression. His laboratory at Emory University was the first to discover that the decreases in P450-associated metabolism in the liver evoked by inflammatory stimuli are associated with the decreased expression of multiple cytochrome P450 mRNAs, and he has continued to make significant contributions to our understanding of the mechanisms behind these phenomena.
Dr. Morgan is Past-Chair of the Drug Metabolism Division of ASPET and a member of the Editorial Board of Drug Metabolism and Disposition. He recently completed a five-year term of service as Associate Editor of Molecular Pharmacology.
Footnotes
-
Send reprint requests to: Edward T. Morgan, Ph.D., Department of Pharmacology, Emory University, 1510 Clifton Rd., Atlanta, GA 30322. E-mail: etmorga{at}bimcore.emory.edu
-
This work was supported by Grants GM46897 and GM53093 from the National Institute of General Medical Sciences.
- Abbreviations used are::
- P450
- cytochrome P450
- LPS
- bacterial lipopolysaccharide
- NO
- nitric oxide
- NOS2
- inducible NO synthase
- EET
- epoxyeicosatrienoic acid
- TNFα
- tumor necrosis factor-α
- IL
- interleukin
- NF
- nuclear factor
- PPARα
- peroxisome proliferator-activated receptor-α
- Received October 11, 2000.
- Accepted December 12, 2000.
- The American Society for Pharmacology and Experimental Therapeutics
References





{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Causes of Interindividual Variation in Drug Metabolism
- Regulation of P450 by Inflammation and Infection
- Rodent Models and Human Studies
- P450 Down-Regulation: A Homeostatic Mechanism or a Pathophysiological Phenomenon?
- Humoral Factors and Cell Types Involved in P450 Down-Regulation
- Molecular Mechanisms of Down-Regulation
- Conclusion
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters