


Abstract
Unexplained cases of CYP2D6 genotype/phenotype discordance continue to be discovered. In previous studies, several African Americans with a poor metabolizer phenotype carried the reduced function CYP2D6*10 allele in combination with a nonfunctional allele. We pursued the possibility that these alleles harbor either a known sequence variation (i.e., CYP2D6*36 carrying a gene conversion in exon 9 along the CYP2D6*10-defining 100C>T single-nucleotide polymorphism) or novel sequences variation(s). Discordant cases were evaluated by long-range polymerase chain reaction (PCR) to test for gene rearrangement events, and a 6.6-kilobase pair PCR product encompassing the CYP2D6 gene was cloned and entirely sequenced. Thereafter, allele frequencies were determined in different study populations comprising whites, African Americans, and Asians. Analyses covering the CYP2D7 to 2D6 gene region established that CYP2D6*36 did not only exist as a gene duplication (CYP2D6*36x2) or in tandem with *10 (CYP2D6*36+*10), as previously reported, but also by itself. This “single” CYP2D6*36 allele was found in nine African Americans and one Asian, but was absent in the whites tested. Ultimately, the presence of CYP2D6*36 resolved genotype/phenotype discordance in three cases. We also discovered an exon 9 conversion-positive CYP2D6*4 gene in a duplication arrangement (CYP2D6*4Nx2) and a CYP2D6*4 allele lacking 100C>T (CYP2D6*4M) in two white subjects. The discovery of an allele that carries only one CYP2D6*36 gene copy provides unequivocal evidence that both CYP2D6*36 and *36x2 are associated with a poor metabolizer phenotype. Given a combined frequency of between 0.5 and 3% in African Americans and Asians, genotyping for CYP2D6*36 should improve the accuracy of genotype-based phenotype prediction in these populations.
Cytochrome P450 2D6 (CYP2D6) is a major drug-metabolizing enzyme involved in the biotransformation of many clinically important medications, including antidepressant and antipsychotic drugs (Kirchheiner et al., 2004). CYP2D6 activity ranges considerably within a population and includes individuals with ultrarapid, extensive, intermediate (IM), and poor (PM) metabolizer status. Genetic variation within the CYP2D6 gene locus is the major contributing factor determining a subject's activity or capability to metabolize certain drugs (Zanger et al., 2004). The CYP2D gene locus on chromosome 22 contains three contiguous genes, CYP2D8, CYP2D7, and CYP2D6 (Kimura et al., 1989), with only the latter encoding a functional protein (Gaedigk et al., 2005b). Not only is the CYP2D6 gene highly polymorphic, with 58 allelic variants and numerous subvariants reported to date (http://www.imm.ki.se/CYPalleles; accessed Jan 5, 2006), but rearrangements within the gene locus have created alleles harboring two or multiple CYP2D6 genes, deleted the entire gene, or led to the creation of fused CYP2D7/2D6 genes. Despite vast efforts characterizing CYP2D6 allelic variants in many populations of different ethnicity, subjects exhibiting a phenotype that is discordant with their determined genotype persist. Most of these cases are poor metabolizers who carry at least one functional or reduced-function allele predicting an extensive or intermediate metabolizer phenotype. Follow-up studies of such cases have revealed novel alleles such as CYP2D6*40 and *42 in African Americans (Gaedigk et al., 2002, 2003a) and CYP2D6*21 and *44 in Japanese (Yamazaki et al., 2003).
CYP2D6*36, originally termed Ch2 or *10C (http://www.imm.ki.se/CYPallele), has been described in a tandem arrangement with CYP2D6*10B (CYP2D6*36 located upstream of CYP2D6*10B; Fig. 1) (Johansson et al., 1994). Regarding CYP2D6.36 function, Johansson et al. (1994) speculated that the 100C>T SNP (P34S) present in CYP2D6*10 and *36 is the major cause for protein destabilization and that the six-amino acid difference in exon 9 (i.e., the CYP2D7 exon 9 conversion) does not have a significant impact on CYP2D6.36 activity. Fukuda et al. (2000) later demonstrated that decreased clearance by CYP2D6.10 and CYP2D6.36 is caused not only by low protein expression, but also increased Km values. However, since the CYP2D6*36 gene did not appear to occur by itself, and activity was conferred by the downstream CYP2D6*10 gene on this allele, no further functional studies were pursued, and thus, it remained unclear whether CYP2D6*36 was an IM- or PM-associated allele. However, a discordant Japanese case was recently resolved by the discovery of a CYP2D6*36x2 gene duplication demonstrating that CYP2D6*36 is a loss-of-function allele as assessed by phenotyping with the probe drug debrisoquine (Chida et al., 2002).

Graphical display of CYP2D6 gene arrangements. Each line represents an allele as labeled, open boxes being genes and black boxes indicating the presence of a CYP2D7 exon 9 sequence. Primer binding sites are given by open (CYP2D6-specific) and black (CYP2D7 or exon 9 conversion-specific) circles. PCR products are labeled A to F with respective lengths as shown in kilobase pairs (kb). Fragments F-1 and F-2 are generated in nested PCRs using fragments B and C, respectively, as templates. Note that the primers for fragment D amplify products of different lengths from CYP2D6*36 and CYP2D6*5 alleles. The designation of PCR fragments (A to F) corresponds to that shown in Fig. 2.
Current genotyping protocols, including our own, define CYP2D6*10 and *36 by the presence and absence of two SNPs, 100C>T and 1846G>A, respectively. Consequently, the known *10A/B, *10x2, *36x2, and *36+*10 (tandem) alleles (Fig. 1) are not discriminated and are collectively genotyped as “CYP2D6*10”. Interestingly, three of our discordant cases had a “CYP2D6*10” in combination with a nonfunctional CYP2D6*4 allele, and we hypothesized that “CYP2D6*10” was in fact a masked CYP2D6*36x2 allele as described by Chida et al. (2002). To test this hypothesis, we conducted a detailed characterization of the CYP2D6/2D7 gene locus in phenotypic poor metabolizers with an assigned CYP2D6*10 allele.
Materials and Methods
Subjects. Study protocols, blood collection, and use of tissues were approved by the University of Missouri-Kansas City Adult and Pediatric Health Sciences Review Boards and the Morehouse School of Medicine Review Board (Atlanta, GA), respectively. All study participants gave written informed consent.
Subjects were phenotyped with dextromethorphan and genotyped previously. These studies comprised adult white Americans, n = 214 (Gaedigk et al., 1999) and African Americans, n = 281 (Gaedigk et al., 2002); African Americans with sickle cell disease (children >1 years of age), n = 126 (Alander et al., 2002); and an ethnically diverse pediatric population (subjects enrolled at 2 weeks of age), n = 155 (M. Blake, A. Gaedigk, R. E. Pearce, K. Adcock, S. Blaney, M. Christenson, L. James, G. L. Kearns, J. T. Wilson, and J. S. Leeder, manuscript in preparation).
An additional 81 genomic DNA samples were derived from an ethnically diverse tissue collection (ethnicity data were retrieved from data sheets accompanying the tissues) that was obtained from National Institute of Child Health and Human Development-supported tissue retrieval programs: the University of Maryland Brain and Tissue Bank for Developmental Disorders (Baltimore, MD) and the Central Laboratory for Human Embryology at the University of Washington (Seattle, WA). DNA samples of Asian Americans, n = 38, were collected for a previous study (Gaedigk et al., 1999) or from discarded, anticoagulated blood obtained for routine clinical management of hospitalized patients (self-reported ethnicity on hospital admission form).
DNA Isolation andCYP2D6Genotyping. DNA was isolated from whole blood or liver tissue using DNA Blood and DNeasy Tissue Kits (QIAGEN, Valencia, CA), respectively. A subset of DNAs were isolated from cheek scrapings (buccal brushes) using Gentra reagents as recommended (Gentra Systems, Minneapolis, MN). CYP2D6 genotyping was performed as described previously (Gaedigk et al., 1999, 2002, 2003a,b, 2005a,c) and included *2 through *12, *14, *15, *17, *28, *29, *35, *40, *41, *42, *45, *46, and *1x2, *2x2, and *4x2 gene duplications. CYP2D6 allele nomenclature throughout this report uses the recently revised allele definitions as established by the CYP2D6 nomenclature committee (http://www.imm.ki.se/CYPalleles).
Cloning and Sequencing ofCYP2D6Alleles. A 6.6-kb CYP2D6-specific fragment that encompassed the entire gene (Fig. 1, fragment C) was amplified and cloned with the pCR-XL-TOPO cloning kit (Invitrogen, Carlsbad, CA). This fragment is only derived from the most downstream gene within the locus and was also used as genotyping template. Sequence analysis of clones was performed with DYEnamic ET dye terminator chemistry and a MegaBACE 500 capillary sequencer (Amersham Biosciences Corp., Piscataway, NJ). AY545216 served as CYP2D6*1 reference sequence (Gaedigk et al., 2005a).
PCRs and Conditions. Primer sequences and additional details pertaining to all PCRs performed for the study are presented in Table 1.
Primers and assay conditions for long and short range PCR reactions
Long-Range PCR. Reactions for PCR fragments A to D were carried out with JumpStart REDAccuTaq LA DNA Polymerase (Sigma, St. Louis, MO) in the presence of 5% DMSO. Reaction volumes were 8 μl, and composition was as recommended. Extension times were 11, 12, 7, and 6 min, respectively. Typically, 1 to 2 μl of the PCR were analyzed by agarose gel electrophoresis. PCR A, carried out with forward and reverse primers binding to CYP2D6 exon 9 and intron 2, respectively, yielded product only from duplicated CYP2D6 genes such as *1x2, *2x2, *4x2, and *10x2. Gene duplications such as CYP2D6*36x2 and the CYP2D6*36+*10 tandem did not amplify because of the presence of the exon 9 conversion. In contrast, a CYP2D6-specific forward primer binding to intron 6 (PCR B) allowed amplification of all duplication arrangements regardless of their exon 9 configuration (Fig. 1). Reaction D amplified a 3.6-kb fragment and was similar to that described by Chida et al. (2002) for the detection of CYP2D6*36x2. This primer set also amplified from CYP2D6*5, but produced a larger, ∼ 5-kb-long fragment.
CYP2D6*36 Genotyping. Reaction E, a novel CYP2D6*36 assay, contained two sets of primers. The set for CYP2D6*36 amplified a 597-bp product only if the gene carrying the exon 9 conversion was located at the downstream position in the locus. The assay does not, however, discriminate between CYP2D6*36 and *36x2. The second primer set amplified an 860-bp-long CYP3A7 product as internal control for PCR performance (present in all samples). This PCR was carried out in a volume of 8 μl with JumpStart REDTaq DNA Polymerase (Sigma). The entire reaction was separated on a 3% agarose gel containing Synergel (Diversified Biotech, Boston, MA). This assay is referred to as the “CYP2D6*36 duplex assay.”
Fragments F-1 and F-2 (Fig. 1) were generated from diluted PCR fragments B and C, respectively, with JumpStart REDTaq DNA Polymerase. Subsequent incubation with the restriction enzyme NcoI allowed the detection of the CYP2D7 exon 9 conversion. CYP2D6-derived fragments were cut once by NcoI (591 + 184 bp), whereas the fragment harboring the conversion remained uncut. Digestion patterns were resolved on 3% agarose gels containing Synergel. This assay is referred to as the “CYP2D6*36 NcoI assay.”
To detect the presence of the exon 9 conversion, the DNA of all study participants, regardless of their genotype, was assayed with the CYP2D6*36 NcoI assay. All positive DNAs were retested and confirmed with the CYP2D6*36 duplex assay on genomic DNA.
Results
After extensive genotype analysis, several subjects presented with a genotype/phenotype discordance. Notably, all four cases were African Americans. As shown in Table 2, three individuals, one adult and two neonates, had a CYP2D6*4/*10 genotype. The neonates had been challenged with dextromethorphan on multiple occasions (coinciding with well baby visits), indicating that the phenotype assessments truly reflected their metabolizer status. A fourth case presented with a DM/DX ratio of 0.38, just above the antimode of 0.3 at 2 weeks of age, and thus was initially classified as a poor metabolizer. However, the neonate assumed intermediate metabolism when rechallenged at 1 and 2 months of age, suggesting that this subject's CYP2D6*10 allele may also be compromised.
Description of subjects presenting with genotype-phenotype discordance
CYP2D6 phenotype was determined from the urinary ratio of dextromethorphan to dextrorphan (DM/DX ratio) with PM status defined as a DM/DX ratio >0.3. One subject (case 4) presented as PM 2 weeks after birth, but acquired IM status (0.03 < DM/DX < 0.3 as defined by Gaedigk et al., 2003b) within 2 months of life. Each DM/DX ratio represents a phenotype assessment. Originally discordant and subsequently revised genotypes are presented.
To test whether any of the four subjects carried a CYP2D6*36x2 allele, we performed the PCR described by Chida et al. (2002). Indeed, all produced a 3.6-kb-long amplicon (fragment D) that suggested the presence of the exon 9 conversion in the most downstream CYP2D6 gene within the locus (Figs. 1 and 2D). However, fragment B, a duplication-specific, ∼10.5-kb product that covers the entire intergenic region as well as the “tail and head” parts of duplicated CYP2D6 genes, did not amplify in multiple attempts, whereas it was readily generated from any other high-quality DNA that had previously been identified to carry either a CYP2D6*1x1, *2x2, or *4x2 gene duplication. This observation implied that the four cases did not harbor a CYP2D6*36x2 gene duplication, but carried only one CYP2D6*36 gene copy (Figs. 1 and 2B). CYP2D6*36 and CYP2D6*36x2 designations are used within this report for alleles with one and two CYP2D6*36 gene copies, respectively. We also noted that fragment B, generated from the CYP2D6*36+*10 tandem allele, was approximately 0.5 kb longer than the amplicon generated from CYP2D6*1x1, *2x2, or *4x2 gene duplication events (Fig. 2B). This phenomenon is currently being further characterized.

Long-range PCR and genotyping assays on selected DNA samples. A to F correspond to the PCR fragments shown in Fig. 1. Respective marker bands are labeled to the left and PCR product(s) in each panel to the right in kilobase pairs (kb) or base pairs (bp). Genotypes of selected samples are as indicated on the top. A CYP2D6*5/*5 DNA in lane 8 served as control to demonstrate assay specificity (i.e., did not amplify any product with CYP2D6-specific primer pairs). Reactions A to E were carried out on genomic DNA; F-1 and F-2 were generated from fragments B and C, respectively. Fragment C is the 6.6-kb PCR product that is generated only from the most downstream CYP2D6 gene and serves as our routine genotyping template. This PCR was duplexed with primers generating a 3.5-kb-long fragment from *1x2, *2x2, and *4x2 gene duplications (note that the CYP2D6*36+*10 tandem did not yield the 3.5-kb “duplication” band). M, 100-bp and 1-kb DNA ladders (New England Biolabs, Beverly, MA).
The discovery of CYP2D6*36 in combination with another nonfunctional allele in phenotypic poor metabolizers demonstrated that this allele encodes a gene product that lacks appreciable activity toward dextromethorphan in vivo. To determine the frequency of CYP2D6*36 and to examine whether any alleles other than CYP2D6*36 harbor the exon 9 conversion, a total of 895 DNA samples (i.e., 1790 chromosomes) derived from either blood or tissue samples of white Americans, African Americans, Asian Americans and “other or unknown” ethnicity were genotyped using the CYP2D6*36 NcoI assay. In addition, all DNA samples positive for CYP2D6*10 were tested for CYP2D6*10x2 gene duplications and CYP2D6*36+*10 tandem arrangements by generating an ∼10.5-kb amplicon (fragment B; Figs. 1 and 2B) and genotyping this fragment using a nested PCR-restriction fragment length polymorphism assay (CYP2D6*36 NcoI assay, product F-1 in Figs. 1 and 2). As summarized in Table 3, CYP2D6*36 was only present in African Americans and in one Asian subject. The CYP2D6*36+*10 tandem allele was most abundant in the Asian cohort (half of the total CYP2D6*10 were tandem alleles; Table 3), but was absent in whites and detected in only one African-American subject and in two subjects of unknown ethnicity. In three individuals, genotyping results were compatible with either a CYP2D6*36/*36+*10 or *36x2/*10 assignment. No CYP2D6*10x2 or CYP2D6*36x2 duplications were found in any subject. It is interesting that CYP2D6*36, *36x2, and the *36+*10 tandem arrangement were absent in the white populations tested. The frequencies of CYP2D6*36 (including alleles that could be either CYP2D6*36 or CYP2D6*36x2) were 0.53 to 2.5% in African Americans, 2.63% in Asians, and 3.33% in the other or unknown group.
Allele frequencies in whites, African Americans, Asians, and two study populations of mixed ethnicity
*10 denotes a “collective” CYP2D6*10 assignment made by testing for 100T only. Combination of additional assays (see Fig 1 and Table 2) allowed further discrimination of collective CYP2D6*10 alleles as indicated.
We also designed and evaluated a novel assay to facilitate detection of CYP2D6*36 and *36x2 alleles directly from genomic DNA without the need to produce a long-range PCR product. A short 597-bp amplicon was generated alongside an internal amplification control product, only when the most downstream CYP2D6 gene contained the exon 9 conversion. This reaction was CYP2D6-specific and did not amplify from the CYP2D7 gene as demonstrated by using a CYP2D6*5/*5 DNA lacking the CYP2D6 gene on both alleles (Figs. 1 and 2E). All DNA samples that were CYP2D6*36-positive with the CYP2D6*36 NcoI assay were also positive with the novel assay, i.e., the CYP2D6*36 duplex assay.
The exon 9 conversion was also found in a single individual originally genotyped as CYP2D6*2x2/*4. As shown in Figs. 1 and 2 (lane 7), further characterization revealed that the CYP2D6*4 allele also harbored a gene duplication and that the conversion was located on both CYP2D6*4 gene copies (i.e., both *2x2 and *4x2 alleles must have generated fragment B, since F-1 amplified thereof was genotyped heterozygous for the exon 9 conversion; fragment A was derived from the CYP2D6*2x2 allele only, since F-1 was negative for the conversion; fragment C, generated from both alleles, yielded F-2 fragments with and without the conversion; and, finally, fragments D and E amplified from genomic DNA, indicating an exon 9 conversion-containing gene at the downstream position). Moreover, 1846G>A, the key CYP2D6*4 SNP, was confirmed to be present on both gene copies of the CYP2D6*4x2 allele. This was achieved by generating a PCR fragment encompassing exon 3 through 9 with primers that specifically amplified this gene structure and using it as genotyping template for 1846G>A (or *4) detection (not shown). The presence of this novel allele was further confirmed by pedigree analysis, which showed that both offspring of our case had inherited the exon 9 conversion-negative CYP2D6*2x2 allele. This allele was designated CYP2D6*4N by the P450 nomenclature committee.
During the course of this study, we also discovered a CYP2D6*4 allele that lacked the C>T SNP at position 100. The subject was a CYP2D6*4/*4 poor metabolizer who presented with a heterozygous 100C>T genotyping result (not shown). It is noteworthy that 4180G>C was also absent. This polymorphism is otherwise present on all defined CYP2D6*4 alleles except CYP2D6*4J, according to the nomenclature web site. In addition, four other SNPs that were found on five resequenced CYP2D6*4 alleles were also missing (Fig. 3). Absence of these SNPs on the novel CYP2D6*4 variant was again confirmed by genotyping and/or resequencing DNA from the parents of the subject. This allele has been designated CYP2D6*4M by the P450 nomenclature committee.
To complete the analysis of the novel alleles, the entire 6.6-kb PCR products (fragment C) from CYP2D6*4, *4M, *4N, *10, and *10 derived from a *36+*10 tandem and *36 were cloned and sequenced. The presence of SNPs and their locations in comparison to two reference sequences, AY545216 (CYP2D6*1 reference sequence previously published; Gaedigk et al., 2005a) and M33388, are shown in Fig. 3.
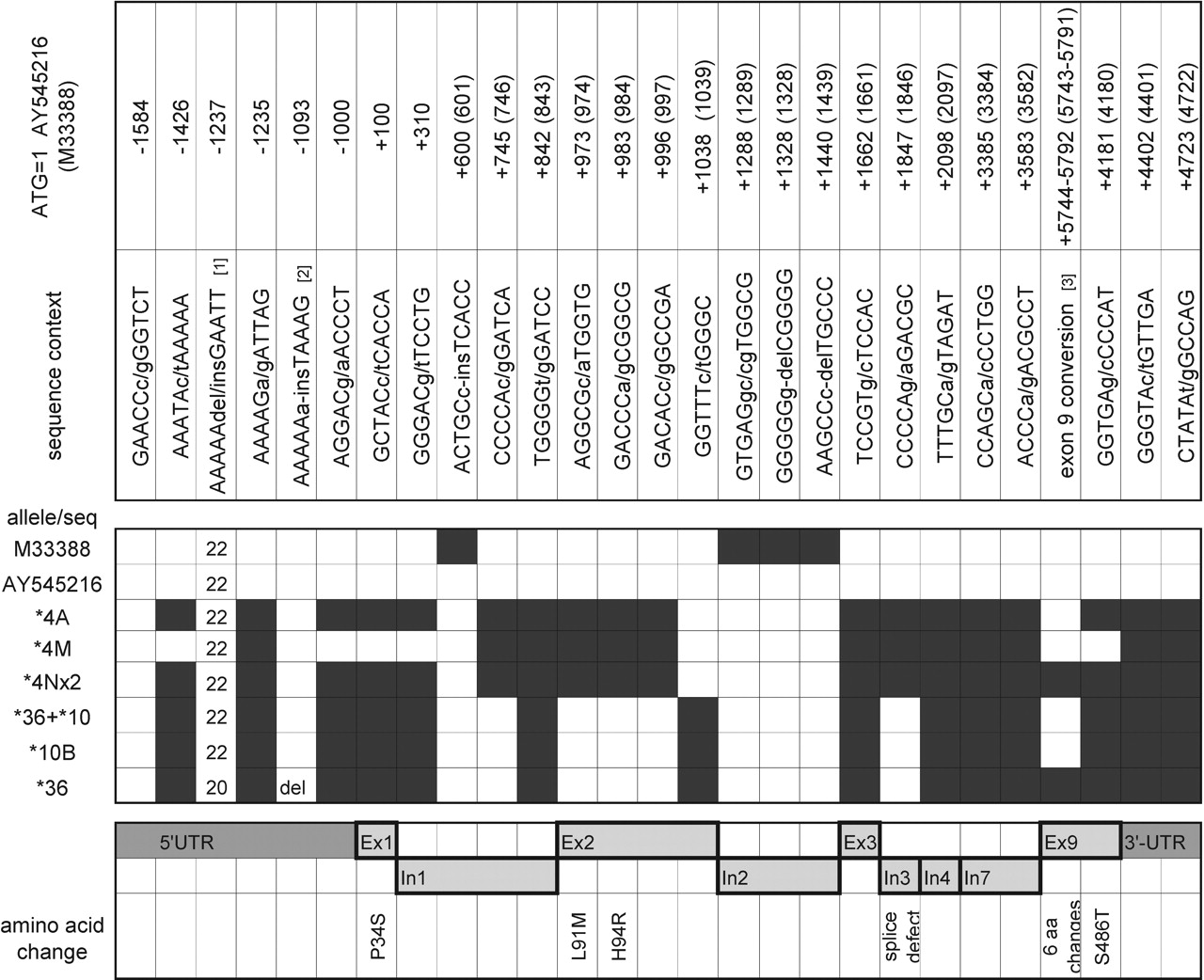
Display of SNPs in resequenced CYP2D6 allelic variants. The two panels on the top give the SNP sequence context and the SNP coordinates on two reference sequences, AY545216 and M33388. The nomenclature numbering is based on ATG = +1 of M33388. Sequence differences between AY545216 and M33388 lead to an off-set in numbering. In the checkerboard panel, open and black boxes indicate the absence and presence of respective SNPs. The line for CYP2D6*4A comprises sequence data for two white and three African-American alleles that were identical; each other line represents a single allele. For CYP2D6*4Nx2 and the CYP2D6*36+*10 tandem arrangement, the sequence is derived from the most downstream gene. The bottom panel shows the location of the 5′-UTR, exons (Ex), introns (In), and the 3′-UTR. Amino acid changes due to nonsynonymous SNPs are shown. 1 Due to PCR errors, the absolute number of A may vary from the number shown. 2 The CYP2D6*36 allele has an A-del at this position. 3 P469A, T470A, H478S, G479R, F481V, A482S.
Discussion
Three cases with a poor metabolizer phenotype and an initial discordant CYP2D6*4/*10 genotype were characterized by long-range PCR, genotyping, cloning and sequencing. Results are consistent with the presence of a “single” CYP2D6*36 gene in the most downstream position within the CYP2D6 locus. The CYP2D6*36x2 gene duplication has been characterized only in Japanese, to date, and this is the first report describing an allele carrying a single CYP2D6*36 gene copy. In addition, allele frequencies of this novel allele were determined in white, African American, and Asian population samples. This discovery not only resolved the genotype to phenotype discordance in our cases, but also provided ample evidence that CYP2D6*36 was indeed responsible for poor metabolism in vivo, as previously suggested by Fukuda et al. (2000) and Chida et al. (2002). Kinetic data on bufuralol 1′-hydroxylation and venlafaxine O-demethylation showed significantly higher Km values for CYP2D6*36-derived protein compared with that of CYP2D6*1 and *10 gene products (Fukuda et al., 2000), and a CYP2D6*21/*36x2 subject presented as a poor metabolizer toward the probe drug debrisoquine (Chida et al., 2002). The data presented here extend these findings to dextromethorphan and suggest that CYP2D6.36 protein has limited or no appreciable activity toward other drugs known to be metabolized or bioactivated through the CYP2D6 pathway.
The fourth case presented here exhibited a metabolic ratio of DM/DX of 0.38 at 2 weeks of age and was, by strict application of the antimode of 0.3, a poor metabolizer. This initial discordance, however, subsided with age (Table 1), suggesting that this infant had a particularly slow onset of CYP2D6 expression compared with other subjects studied at this age. The subject's revised genotype (CYP2D6*2/*36), with one nonfunctional allele, may have contributed to the delayed expression in this case; however, factors other than genotype may also have played a role. It should be noted that the onset of CYP2D6 expression occurs within days after birth and generally is consistent with genotype at 2 weeks of age. The acquisition of CYP2D6 activity during the first year of life is further described and discussed in detail elsewhere (M. Blake, A. Gaedigk, R. E. Pearce, K. Adcock, S. Blaney, M. Christenson, L. James, G. L. Kearns, J. T. Wilson, and J. S. Leeder, manuscript in preparation).
Considering the frequency of CYP2D6*36, between 0.5% and 2.6% in African Americans and Asians, CYP2D6*36 is one of the more common PM alleles in these populations, next only to CYP2D6*4 and CYP2D6*5 (Nishida et al., 2000; Gaedigk et al., 2002; Ji et al., 2002a,b). In light of our limited sample number of Asians and the absence of detailed information concerning the ethnicity of those samples (i.e., country of origin, degree of admixture), the frequency of CYP2D6*36 will require further analysis in defined Asian populations. Because the CYP2D7 exon 9 conversion has been found almost exclusively on a CYP2D6*10 background, and Asians exhibit a high frequency of the CYP2D6*10 allele (30–50%), it is tempting to speculate that revealing CYP2D6*36 may, at least in part, explain the wide range of enzymatic activity observed between subjects of various CYP2D6*10 genotypes (Kim et al., 2004).
Moreover, CYP2D6*36 may have originated in Africa and spread to Asia, but not Europe, since it has not been observed in any of our white samples, either as a single gene copy or in tandem with CYP2D6*10 (i.e., CYP2D6*36+*10). This observation clearly suggests follow-up studies in African populations. To facilitate such studies and, also, to make diagnostic genotyping easy and reliable, we have developed an assay that can be performed directly on genomic DNA, is specific (i.e., amplifies only from the most downstream CYP2D6 gene within the locus), and has the potential for adaptation to high-throughput genotyping platforms.
The discovery of CYP2D6*4M and CYP2D6*4Nx2 along with CYP2D6*36 underscores the highly polymorphic nature of the CYP2D6 locus. Although the presence of the exon 9 conversion on CYP2D6*4N is of no functional consequence (i.e., 1846G>A is the detrimental mutation causing aberrant splicing), knowledge of such gene structures is important for the accurate interpretation of genotyping results (i.e., correct allocation of SNPs or sequence variations to an allele). Likewise, absence of the 100C>T SNP on CYP2D6*4M does not have a functional consequence. However, the majority of CYP2D6*4 alleles (i.e., *4A-L) are characterized by the linkage of 100T and 1846A, and many genotyping approaches are interpreting results based on 100C/1846A linkage to assign CYP2D6*4 and 100C/1846G (lack of the “*4” SNP) to assign CYP2D6*10. Lack of 100T on a fraction of CYP2D6*4 alleles may therefore interfere with proper allele assignment, e.g., a subject heterozygous for 100C/T and 1846G/A likely is a CYP2D6*1/*4 with both SNPs located on the CYP2D6*4 allele, but a CYP2D6*4/*10 genotype is possible with 100T on CYP2D6*10 and 1846A on CYP2D6*4. The presented case is our first to reveal the absence of 100T on CYP2D6*4M as it was paired with a second “normal” CYP2D6*4; in other cases, however, it may have remained “masked.” It is also the first to be entirely sequenced. There are only two reports in the literature describing lack of 100T on CYP2D6*4. In one study, a single Basque individual lacked 100T on a CYP2D6*4 allele (Fuselli et al., 2004), whereas 17 of 40 (40%) and 16 of 62 (26%) CYP2D6*4 in Nicaraguans and Spanish subjects, respectively, lacked this SNP (Agundez et al., 1997). The family of our case, however, reported German and English origins and is unaware of any Spanish or Hispanic ancestors.
In conclusion, CYP2D6*36 has been overlooked in the past, and its contribution to the polymorphic expression of CYP2D6 was probably underestimated. The discovery of three poor metabolizers carrying this allele demonstrates loss of function, whereas allele frequencies between 0.5 and 3% emphasize its important contribution to intermediate and poor metabolism in African Americans and Asians. It is highly recommended to include CYP2D6*36 testing for reliable phenotype prediction, especially when genotyping is performed in African Americans, subjects of African descent, and Asians.
Acknowledgments
We gratefully acknowledge the technical assistance of Liliane Ndjountché and Darren Baker. We also thank the participating families for their contribution.
Footnotes
-
Supported by R01 ES10855-05 from the National Institute of Environmental Health Sciences and in part by an intramural grant from the Katherine B. Richardson Associates Endowment Fund.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.008292.
-
ABBREVIATIONS: IM, intermediate metabolizer; PM, poor metabolizer; kb, kilobase pair(s); PCR, polymerase chain reaction; SNP, single-nucleotide polymorphism; bp, base pair(s); DM/DX ratio, dextromethorphan to dextrorphan ratio; UTR, untranslated region.
- Received November 9, 2005.
- Accepted January 12, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}