


Abstract
Itraconazole (ITZ) has three chiral centers and is administered clinically as a mixture of four stereoisomers. This study evaluated stereoselectivity in ITZ metabolism. In vitro experiments were carried out using heterologously expressed CYP3A4. Only (2R,4S,2′R)-ITZ and (2R,4S,2′S)-ITZ were metabolized by CYP3A4 to hydroxy-ITZ, keto-ITZ, and N-desalkyl-ITZ. When (2S,4R,2′R)-ITZ or (2S,4R,2′S)-ITZ was incubated with CYP3A4, neither metabolites nor substrate depletion were detected. Despite these differences in metabolism, all four ITZ stereoisomers induced a type II binding spectrum with CYP3A4, characteristic of coordination of the triazole nitrogen to the heme iron (Ks 2.2–10.6 nM). All four stereoisomers of ITZ inhibited the CYP3A4-catalyzed hydroxylation of midazolam with high affinity (IC50 3.7–14.8 nM). Stereochemical aspects of ITZ pharmacokinetics were evaluated in six healthy volunteers after single and multiple oral doses. In vivo, after a single dose, ITZ disposition was stereoselective, with a 3-fold difference in Cmax and a 9-fold difference in Cmin between the (2R,4S)-ITZ and the (2S,4R)-ITZ pairs of diastereomers, with the latter reaching higher concentrations. Secondary and tertiary ITZ metabolites (keto-ITZ and N-desalkyl-ITZ) detected in plasma were of the (2R,4S) stereochemistry. After multiple doses of ITZ, the difference in Cmax and Cmin decreased to 1.5- and 3.8-fold, respectively. The initial difference between the stereoisomeric pairs was most likely due to stereoselective metabolism by CYP3A4, including stereoselective first-pass metabolism as well as stereoselective elimination. However, stereoselective elimination was diminished after multiple dosing, presumably as a result of CYP3A4 autoinhibition. In conclusion, the metabolism of ITZ is highly stereoselective in vitro and in vivo.
Itraconazole (ITZ) is a broad-spectrum triazole antifungal agent mainly used to treat infections caused by mixed dermatophyte and Candida (Haria et al., 1996). It is also used for prophylaxis in patients with immunodepression (Poirier and Cheymol, 1998). The antifungal activity of ITZ is caused by inhibition of the fungal cytochrome P450, 14α-demethylase, thus impairing the synthesis of ergosterol.
The ITZ molecule has three chiral centers (Fig. 1). The two chiral centers in the dioxolane ring are fixed in relation to one another, and the triazolomethylene and aryloxymethylene dioxolane-ring substituents are always cis to each other. The clinical formulation is a mixture of four stereoisomers (two enantiomeric pairs; Fig. 1). ITZ stereoisomers with (2S,4R) configuration in the dioxolane ring [(2S,4R)-ITZ] are 4-fold more potent in vitro against Candida albicans than those with the (2R,4S) configuration [(2R,4S)-ITZ] (Koch et al., 2000), but any studies assessing the antifungal activity of individual ITZ stereoisomers are unpublished.
Despite stereoselective differences in antifungal activity, the metabolism and disposition of ITZ stereoisomers are poorly understood. An electropherogram of a single plasma sample from one patient indicated differences in ITZ stereoisomer concentrations, and results from an in vitro incubation suggested that ITZ metabolism may be stereoselective (Breadmore and Thormann, 2003). However, only two component peaks of ITZ were resolved, their absolute stereochemistry was not identified, and no pharmacokinetic or enzyme kinetic analysis was conducted. The prospect that stereochemistry may be a major determinant of antifungal activity, pharmacokinetics, and the magnitude of drug-drug interactions suggest that a single stereoisomer of ITZ might be clinically superior to the mixture of ITZ stereoisomers that is currently administered.
The achiral pharmacokinetics of the unresolved mixture of ITZ stereoisomers has been well characterized. ITZ has a large volume of distribution (11 l/kg), an intermediate hepatic extraction ratio (0.55), and dose- and time-dependent pharmacokinetics that are best described using a three-compartment model (Haria et al., 1996; Poirier and Cheymol, 1998). After the rapid distribution phase, the slow distribution phase and the elimination phase might reflect the rapid and slow metabolism of stereoisomer pairs. Such stereoselective kinetics have been demonstrated by simulations (Tucker and Lennard, 1990) and have been reported previously for several drugs, including mephenytoin and warfarin (Wedlund et al., 1985; Levy and Boddy, 1991; Hutt and Tan, 1996).
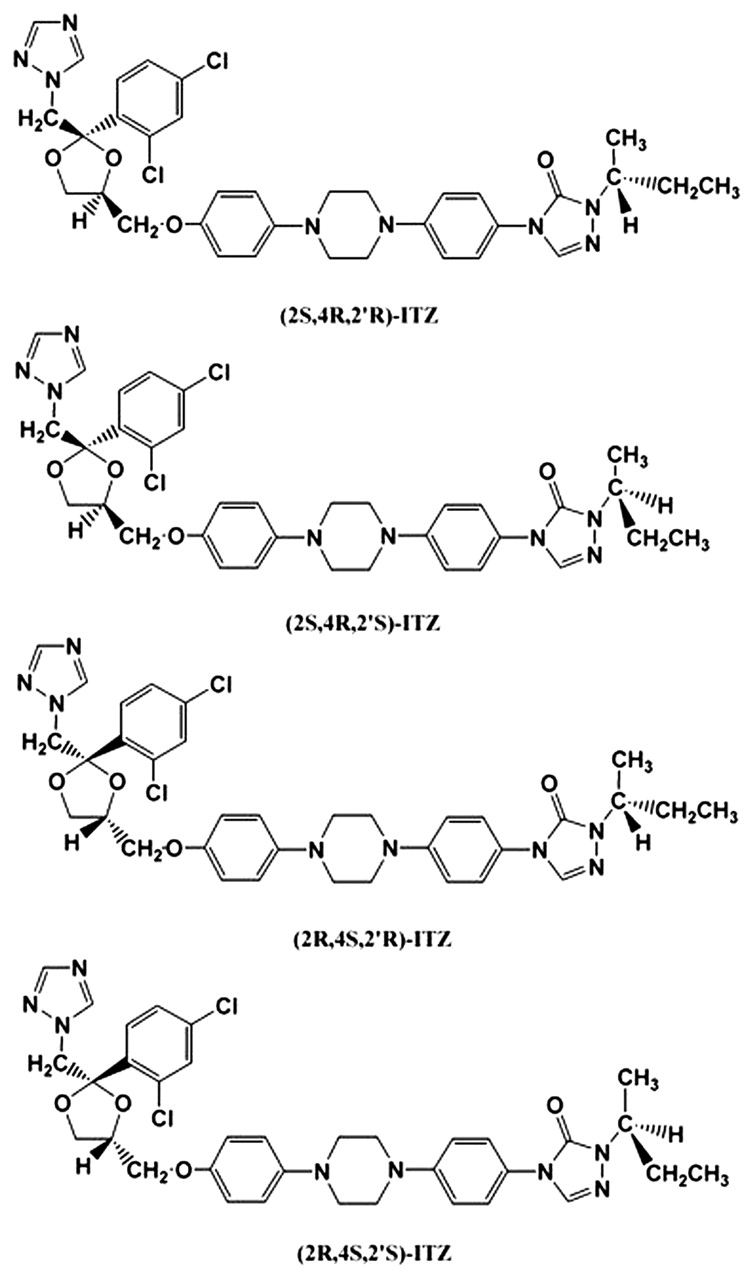
Structures of the four stereoisomers of itraconazole.
ITZ is metabolized by CYP3A4 sequentially to three metabolites (Fig. 2), OH-ITZ, keto-ITZ, and N-desalkyl-ITZ (ND-ITZ) (Isoherranen et al., 2004), which are all present in plasma after administration of ITZ (Isoherranen et al. unpublished observations). Because keto-ITZ and ND-ITZ are formed sequentially from ITZ via OH-ITZ, it was suggested that the keto-ITZ and ND-ITZ observed in plasma would have a stereochemical composition identical to that of the ITZ stereoisomers that are metabolized to OH-ITZ. Only CYP3A4 forms OH-ITZ from ITZ in vitro (Isoherranen et al., 2004), and clinically, CYP3A4-catalyzed processes appear to make up a major elimination pathway for ITZ (Ducharme et al., 1995). Of the circulating metabolites, the pharmacokinetics of OH-ITZ has been best characterized. The half-life of OH-ITZ has been consistently described to be shorter than that of ITZ (Heykants et al., 1989; Barone et al., 1993; Ducharme et al., 1995), which is inconsistent with conventional pharmacokinetic theory. To explain this unusual behavior, it was hypothesized that formation of OH-ITZ is stereoselective. The ITZ stereoisomers that are converted to OH-ITZ would have a short half-life (the half-life of the α-phase of ITZ elimination) equivalent to or shorter than the half-life of OH-ITZ. The clinically observed terminal half-life of ITZ would correspond to the elimination of stereoisomer(s) of ITZ that is (are) not metabolized by CYP3A4 to OH-ITZ or is (are) metabolized at a much slower rate.
ITZ is a potent inhibitor of CYP3A4 and causes significant drug-drug interactions when coadministered with other CYP3A4 substrates (Olkkola et al., 1996; Backman et al., 1998; Neuvonen et al., 1998; Florea et al., 2003; Mahnke et al., 2003). The mixtures of stereoisomers of OH-ITZ, keto-ITZ, and ND-ITZ are also potent inhibitors of CYP3A4 in vitro, and thus these stereoisomers may contribute to the inhibition of CYP3A4 observed in clinical situations (Isoherranen et al., 2004). The magnitude of the in vivo interactions observed with ITZ is greater than that predicted from in vitro data when reversible inhibition is assumed. Several reasons have been offered to explain the in vitro-in vivo discrepancy, including nonspecific protein binding, selective uptake of ITZ into hepatocytes, and inhibitory metabolites of ITZ (Yamano et al., 1999, 2001; Isoherranen et al., 2004). Stereoselective elimination of ITZ and its metabolites, together with CYP3A4 inhibition by stereoisomers of ITZ or the metabolites, may contribute to the poor prediction of CYP3A4 inhibition in vivo.

Structures of the metabolites of itraconazole: OH-ITZ, keto-ITZ, and ND-ITZ. Only one of the possible stereoisomers formed in vivo is depicted for each metabolite.
The goals of this study were to determine whether the metabolism of ITZ by CYP3A4 and the inhibition of CYP3A4 by ITZ are stereoselective, and investigate the stereoselective disposition of ITZ in vivo. To address these aims, the four ITZ stereoisomers were incubated with heterologously expressed CYP3A4, and ITZ depletion as well as formation of metabolites was measured. The IC50 values of the stereoisomers of ITZ were determined, and spectral titrations of ITZ stereoisomers with CYP3A4 were performed to obtain Ks values. In addition, the stereoselective pharmacokinetic behavior of ITZ was studied in six healthy volunteers during administration of ITZ for 7 days.
Materials and Methods
Chemicals, Recombinant P450s, and Human Liver Microsomes. The four ITZ stereoisomers and four of the eight possible OH-ITZ stereoisomers (2R,4S,2′S,3′R)-OH-ITZ, (2R,4S,2′R,3′S)-OH-ITZ, (2R,4S,2′S,3′S)-OH-ITZ, and (2R,4S,2′R,3′R)-OH-ITZ were obtained from Sepracor Inc. (Marlborough, MA). The mixtures of ITZ and OH-ITZ stereoisomers were purchased from Research Diagnostics Inc., (Flanders, NJ). keto-ITZ was generously provided by Dr. Jan Heeres, Janssen Pharmaceutica N. V. (Beerse, Belgium), and ND-ITZ was prepared as previously reported (Heeres et al., 1984; Isoherranen et al., 2004). Midazolam, 1′-hydroxymidazolam (OH-MDZ), and 1′-[2H2]-hydroxymidazolam were gifts from Roche Laboratories (Nutley, NJ). Acetonitrile was purchased from Fischer Scientific Co. (Fairlawn, NJ), ammonium acetate from J.T. Baker (Phillipsburg, NJ), and NADPH from Sigma-Aldrich (St. Louis, MO). Ultrapure water, filtered through a Barnstead Nanopure filter system, was used throughout the study.
Supersomes containing cDNA-expressed CYP3A4 coexpressed with P450 reductase and cytochrome b5 were purchased from BD Gentest (Woburn, MA). Purified CYP3A4, expressed in Escherichia coli, was a gift from Josh Pearson, Department of Medicinal Chemistry, University of Washington.
Human liver microsomes, devoid of significant amounts of CYP3A5 protein, were selected from the University of Washington Human Liver Bank, and equal amounts of microsomal protein from the five different preparations were pooled for subsequent experimentation (Isoherranen et al., 2004).
In Vitro Incubations. Stereoselective metabolism of ITZ was studied in vitro using heterologously expressed CYP3A4 (Supersomes). ITZ stereoisomers were dissolved in acetonitrile and added to the incubations at a concentration of 10 or 50 μM to give a final acetonitrile concentration in all incubations of 1% and concentrations of the ITZ stereoisomers of 100 nM and 500 nM. The incubations were performed in 100 mM potassium phosphate (KPi) buffer (pH 7.4) with 1 mM EDTA, using a concentration of 10 pmol/ml CYP3A4 (0.084 mg microsomal protein/ml). All incubations were performed in duplicate. A starting incubation volume of 1.8 ml was used. After preincubation for 4 min at 37°C, the reaction was initiated with NADPH (final concentration 1 mM). At 0, 1, 2, 5, and 10 min, 200-μl samples were transferred to tubes containing 200 μl of ice-cold acetonitrile to quench the reaction. The samples were vortexed, centrifuged at 10,000g for 10 min, and the supernatant was transferred to an high-performance liquid chromatography vial for high performance liquid chromatography-mass spectrometry (LC-MS) analysis. The concentrations of the parent ITZ stereoisomers and the metabolites (OH-ITZ, keto-ITZ, and ND-ITZ) were determined using a previously described LC-MS method (Isoherranen et al., 2004).
Inhibition of CYP3A4 by Itraconazole Stereoisomers. IC50 values for (2R,4S,2′R)-ITZ, (2R,4S,2′S)-ITZ, (2S,4R,2′R)-ITZ, and (2S,4R,2′S)-ITZ inhibition of the CYP3A4-catalyzed 1′-hydroxylation of midazolam were determined. The incubations were carried out in duplicate in 100 mM KPi buffer (pH 7.4) with 1 mM EDTA and 5 pmol/ml CYP3A4. The NADPH concentration in all incubations was 1 mM. A sub-Km concentration of 1 μM midazolam was used in all incubations. ITZ stereoisomers were added to the 500-μl incubations at nominal concentrations of 0, 5, 25, 50, 100, 250, 500, and 1000 nM. After preincubation for 4 min at 37°C, the reaction was initiated with NADPH (final concentration 1 mM). At 0 min and 2 min, a 200-μl sample was collected into a vial containing 200 μl of ice-cold acetonitrile. Internal standard solution (D2-labeled OH-MDZ, 100 ng/ml, 10 μl) was added, and the mixture was vortexed and centrifuged. The inhibitor concentration was measured at time 0 min and at time 2 min, and the mean inhibitor concentration was calculated for the incubation period (i.e., the inhibitor concentration was corrected for depletion). The concentration of OH-MDZ was measured in the 2-min sample.
Concentrations of OH-MDZ were measured using a Hewlett Packard (Palo Alto, CA) series 1100 MSD system operating in the positive ion electrospray mode with selected ion monitoring, according to a previously reported method (Isoherranen et al., 2004). Separation was achieved using a Zorbax Eclipse XDB-C8 5-μm column (2.1 mm i.d. × 50 mm; Agilent Technologies, Palo Alto, CA) equipped with a Phenomenex (Torrance, CA) SecurityGuard C8 guard column.
Determination of Ligand-Induced Binding Spectra. The optical titrations of the ligand-induced binding spectra were performed with an Aminco DW2 dual beam spectrophotometer as upgraded by Olis Instruments, Inc. (Bogart, GA). Matched cuvettes containing CYP3A4 (370 nM) in KPi buffer (pH 7.4) with 20% glycerol (to stabilize the purified CYP3A4 and prevent denaturation and aggregation) at 37°C were used. Ligand was added in 1-μl increments of 25 μM solution to the sample cuvette. Matching volumes of solvent (acetonitrile) were added into the reference cuvette. Ligand-induced difference spectra were recorded using Supersomes and purified CYP3A4 with the mixture of itraconazole stereoisomers, and identical (same extinction coefficient, and absorbance maximum and minimum) behavior of the two enzyme systems was confirmed. However, only purified CYP3A4 was used for the quantitative spectral titration experiments to improve spectral quality and decrease noise in the spectra. Use of purified CYP3A4 also allowed recording of absolute binding spectra of each ligand.
Clinical Study Design and Blood Sampling. Six healthy adult volunteers, two male and four female (age range 18–33 years), participated in the study after each had given written consent. The study protocol was approved by the institutional review board of the University of Washington and by the Scientific Advisory Committee of the General Clinical Research Center of the Medical Center of the University of Washington. The study consisted of a 7-day administration of ITZ oral solution (10 ml of 10 mg/ml once daily on the morning). The subjects fasted overnight before each ITZ dose. Blood samples (4 ml) were collected into heparinized collection tubes on day 1 and day 7 of the study at time points 0, 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h after drug administration via an indwelling venous catheter.
Determination of Itraconazole and Metabolite Concentrations. Concentrations of ITZ, OH-ITZ, keto-ITZ, and ND-ITZ in the inhibition experiments were measured by LC-MS using a Hewlett Packard series 1100 MSD system operating in the positive ion electrospray mode with selected ion monitoring and equipped with HP Chemstation data analysis software using a previously published method (Isoherranen et al., 2004). Analyte separation was achieved using a Zorbax Eclipse XDB-C8 5-μm column (2.1 mm i.d. × 50 mm; Agilent) equipped with a Phenomenex C8 guard column (2.1 mm i.d × 4 mm). Calibration curves for ITZ, OH-ITZ, and keto-ITZ were prepared between concentrations 1 nM and 1000 nM. For ND-ITZ, the concentration range was 1 nM to 100 nM. The limit of detection for ITZ and its metabolites was at 1 nM and the limit of quantification was at 5 nM.
Stereoselective Analysis. A stereoselective high-performance liquid chromatography-mass spectrometry assay was developed to analyze the concentrations of itraconazole stereoisomers in plasma and to determine the absolute configuration of formed metabolites. The stereoisomers were separated using a Chiralpak AS-RH 5-μm column (150 mm × 2.1 mm i.d.; Chiral Technologies Inc., Exton, PA) with a mobile phase flow rate of 0.2 ml/min. The initial mobile phase was 30% acetonitrile/70% aqueous 5 mM ammonium acetate. The acetonitrile concentration increased linearly to 70% between 4 and 50 min, then to 80% over an additional 2 min. Ions monitored were m/z 705 for ITZ, m/z 721 for OH-ITZ, m/z 719 for keto-ITZ, and m/z 649 for ND-ITZ, all [MH+] ions. The drying gas flow was set at 10 l/min, nebulizer pressure at 25 psig, gas temperature at 300°C, and the capillary voltage at 5500 V. The fragmentor was set at 160 V for all ions. The incubation samples were prepared identically to the nonchiral assay. For plasma samples, 300 μl of acetonitrile was added to 100 μl of plasma, and the samples were centrifuged at 10,000g for 10 min and then transferred to autosampler vials. A 20-μl sample was injected on the column. Only two of the four stereoisomers of ITZ were clearly separated using this method; the (2R,4S,2′R)-ITZ and (2R,4S,2′S)-ITZ eluted together (Fig. 3a). Therefore, pharmacokinetic analysis was conducted separately for the two pairs of stereoisomers based on their dioxolane ring stereochemistry, with (2R,4S,2′R)-ITZ and (2R,4S,2′S)-ITZ as one pair and (2S,4R,2′R)-ITZ and (2S,4R,2′S)-ITZ as a second pair. The keto-ITZ stereoisomers eluted as three peaks, and the elution order of the keto-ITZ stereoisomers was assumed to follow the elution order of the corresponding stereoisomers of ITZ (Fig. 3a). This allowed tentative assignments of the peaks corresponding to (2R,4S)-keto-ITZs and (2S,4R)-keto-ITZs. Similarly, it was assumed that the ND-ITZ stereoisomers eluted in an order similar to that of the ITZ stereoisomers, based on the stereochemistry on the dioxolane ring; i.e., the (2S,4R)-stereoisomers eluted first. It was not possible to separate all eight of the OH-ITZ stereoisomers in the commercial mixture, and of these, only the four (2R,4S)-stereoisomers were available. Therefore, only partial characterization of the formation of OH-ITZ stereoisomers was possible.
Data Analysis. All nonlinear fitting was performed using WinNonlin (Pharsight, Mountain View, CA) data analysis software. All pharmacokinetic parameters were obtained using the noncompartmental approach and standard pharmacokinetic methods. The peak concentrations (Cmax) and times at which peak concentrations were reached (tmax) were obtained directly from the plasma concentration versus time data. IC50 values were calculated by standard nonlinear fit to the data. The Ks values for ITZ stereoisomers were obtained by fitting the “Morrison” equation (Segel, 1993) to the spectral titration data:  in which [E] is the total CYP3A4 concentration in solution, [S] is the total substrate concentration added, and [ES] is the concentration of the CYP3A4-ITZ complex as calculated from the titration data using the extinction coefficient for [ES] measured from the ratio of absorbance (difference of absorbance between 424 nm and 390 nm) at saturation and the total enzyme concentration.
in which [E] is the total CYP3A4 concentration in solution, [S] is the total substrate concentration added, and [ES] is the concentration of the CYP3A4-ITZ complex as calculated from the titration data using the extinction coefficient for [ES] measured from the ratio of absorbance (difference of absorbance between 424 nm and 390 nm) at saturation and the total enzyme concentration.

Ion chromatograms demonstrating the stereoselective separation of itraconazole and its metabolites. a, separation of reference mixtures of ITZ (4 stereoisomers), OH-ITZ (8 stereoisomers), keto-ITZ (4 stereoisomers), and ND-ITZ (2 stereoisomers). The peak annotation is as follows: A, (2R,4S)-ND-ITZ; B, (2S,4R)-ND-ITZ; C, (2R,4S)-keto-ITZ stereoisomer pair; D, (2S,4R,2′S)-keto-ITZ; E, (2S,4R,2′R)-keto-ITZ; F, G, and H, unknown combinations of OH-ITZ stereoisomers: peak H has only (2S,4R)-stereochemistry; I, (2R,4S)-ITZ; J, (2S,4R,2′S)-ITZ; K, (2S,4R,2′R)-ITZ. The elution order of ITZ stereoisomers was determined to be (2R,4S,2′S)-ITZ, (2R,4S,2′R)-ITZ, (2S,4R,2′S)-ITZ, and (2S,4R,2′R)-ITZ after injections of individual stereoisomers. For the OH-ITZ stereoisomers, (2R,4S,2′S,3′R)-OH-ITZ, (2R,4S,2′R,3′R)-OH-ITZ, and (2R,4S,2′R,3′S)-OH-ITZ had identical retention times when injected separately. Only the four OH-ITZ stereoisomers with the (2R,4S) configuration were available, and therefore, it was not possible to identify the combination of stereoisomers eluting in each of the F, G, and H peaks. The assignment of the elution order of the keto-ITZ stereoisomers (2R,4S,2′S)-keto-ITZ and (2R,4S,2′R)-keto-ITZ first, together, then (2S,4R,2′S)-keto-ITZ and (2S,4R,2′R)-keto-ITZ, and ND-ITZ stereoisomers was based on the elution order of the ITZ stereoisomers. b, separation of the stereoisomers of ITZ metabolites in an incubation with (2R,4S,2′S)-ITZ. Peaks A and C are as described for a. Peak L was assigned as (2R,4S,2′S,3′R)-OH-ITZ based on comparison to reference material shown in d. c, stereoselective separation of ITZ and its metabolites in the plasma of volunteer E. Peaks were assigned as described in a. d, stereoselective separation of (2R,4S,2′S,3′S)-OH-ITZ (peak M) and (2R,4S,2′S,3′R)-OH-ITZ (peak L).
Statistical Analysis. Data are expressed as mean values ± S.D. Statistical analysis was performed with the SYSTAT statistical analysis program (SYSTAT Inc., Evanston, IL). The Wilcoxon matched pair test was used to compare the peak and trough concentrations between the two pairs of stereoisomers.
Results
Stereoselective Metabolism of Itraconazole in Vitro. Metabolism of ITZ was dependent on the stereochemistry of the dioxolane ring. Incubations of the individual ITZ stereoisomers with heterologously expressed CYP3A4 showed that OH-ITZ is formed only from the two (2R,4S)-ITZ-stereoisomers, (2R,4S,2′R)-ITZ and (2R,4S,2′S)-ITZ. No OH-ITZ formation could be detected for (2S,4R,2′S)-ITZ and (2S,4R,2′R)-ITZ, even at high substrate concentrations (1000 nM) and an incubation time of 30 min. The absence of (2S,4R,2′S)-ITZ and (2S,4R,2′R)-ITZ metabolism by CYP3A4 was further confirmed by showing that no significant depletion of these stereoisomers occurred in the incubations. Results from experiments with pooled human liver microsomes confirmed the results obtained using the expressed enzyme system: OH-ITZ was formed only from the (2R,4S)-stereoisomers in human liver microsomes.
CYP3A4-catalyzed 3′-hydroxylation of the ITZ side chain can produce as many as four possible OH-ITZ stereoisomers from the two (2R,4S)-ITZ stereoisomers: (2R,4S,2′S,3′R)-OH-ITZ, (2R,4S, 2′R,3′S)-OH-ITZ, (2R,4S,2′S,3′S)-OH-ITZ, and (2R,4S,2′R,3′R)-OH-ITZ. Separation of (2R,4S,2′S,3′S)-OH-ITZ from the three other (2R,4S)-OH-ITZ stereoisomers was attained (Fig. 3d), but separation of all four of these stereoisomers of OH-ITZ was not achieved. Analysis of the hydroxylated products from incubations of (2R,4S,2′S)-ITZ showed that (2R,4S,2′S,3′R)-OH-ITZ was formed, whereas only trace amounts of (2R,4S,2′S,3′S)-OH-ITZ were detected (Fig. 3b), demonstrating stereoselective formation of the hydroxylated metabolite. Separation of the two possible OH-ITZ stereoisomers formed from (2R,4S,2′R)-ITZ was not achieved and, therefore, similar analysis could not be conducted for this stereoisomer. A greater amount of OH-ITZ was formed from (2R,4S,2′S)-ITZ. After a 10-min incubation, the amount of OH-ITZ produced from (2R,4S,2′S)-ITZ was approximately 5 times that produced from (2R,4S,2′R)-ITZ (21 pmol versus 3.8 pmol in 200 μl, respectively).

Scheme showing the stereoselective sequential metabolism pathways of itraconazole. The formation of both (2R,4S,2′R,3′R)-OH-ITZ and (2R,4S,2′R,3′S)-OH-ITZ from (2R,4S,2′R)-ITZ and the formation of ND-ITZ from both (2R,4S)-keto-ITZ stereoisomers has not been confirmed experimentally.
Sequential metabolism was observed in incubations with both (2R,4S)-ITZ stereoisomers. keto-ITZ and ND-ITZ, both of the (2R,4S)-configuration based on a retention pattern analogous with that of ITZ (Fig. 3, a and b), were detected in all incubations with these stereoisomers of ITZ. The formation of keto-ITZ and ND-ITZ from the four different OH-ITZ stereoisomers with the (2R,4S)-stereochemistry of the dioxolane ring was confirmed in incubations of the individual stereoisomers with heterologously expressed CYP3A4 (data not shown). Based on the experiments described above, a scheme for the sequential metabolism of ITZ stereoisomers was constructed (Fig. 4).
Inhibition and Binding of CYP3A4 by Itraconazole Stereoisomers. The inhibition of CYP3A4-mediated midazolam hydroxylation by the stereoisomers of ITZ was studied using CYP3A4 Supersomes. Despite the short incubation time (2 min), significant depletion of (2R,4S,2′S)-ITZ and (2R,4S,2′R)-ITZ was observed in incubations with the nominal inhibitor concentrations below 100 nM. Formation of OH-ITZ and keto-ITZ was observed in all of these incubations as well. Concentrations of metabolites produced in these incubations were 2 to 44 nM for OH-ITZ and 1 to 12 nM for keto-ITZ. In some incubations with the (2R,4S)-ITZ isomers, the concentration of OH-ITZ exceeded that of ITZ. Neither depletion of the inhibitor nor formation of any metabolites was observed in incubations using either (2S,4R,2′S)-ITZ or (2S,4R,2′R)-ITZ as the inhibitor. Their apparent IC50 values are summarized in Table 1, and a determination of a representative IC50 value is shown in Fig. 5. Interestingly, the two ITZ stereoisomers that underwent metabolism by CYP3A4 also had 2- to 4-fold lower IC50 values than the stereoisomers that were not metabolized. It was not possible to separate the inhibitory contribution of the metabolites from the contribution of the parent drug isomers, and it should be noted that the lower IC50 values might be a result of inability to account for the effects of additional inhibitory species. Therefore, to compare the intrinsic binding affinity of the ITZ stereoisomers to CYP3A4, spectral titrations with the stereoisomers were undertaken and their Ks values were determined.
Binding characteristics of the four stereoisomers of ITZ
All four ITZ stereoisomers, regardless of their ability to undergo metabolism by CYP3A4, induced type II binding spectra with purified CYP3A4, suggesting that the triazole nitrogen of these compounds coordinates the heme iron of CYP3A4. The spectral characteristics obtained from the stereoisomers that were metabolized were indistinguishable from the stereoisomers that were not metabolized by CYP3A4; the difference spectrum of each compound was characterized by a maximum at 434 nm and a minimum at 390 nm. From these spectral titrations, it appeared that the (2R,4S)-ITZ pair might have a slightly higher apparent extinction coefficient with CYP3A4 than the (2S,4R)-ITZ pair, but the difference was not significant. All four stereoisomers exhibited high affinity for CYP3A4. The Ks values for the four ITZ stereoisomers are summarized in Table 1. Due to the depletion of free ligand by binding to CYP3A4, the quadratic equation (Morrison equation) was fitted to the spectral titration data. The spectral titration and the fit of the Morrison equation to the data from (2S,4R,2′S)-ITZ are shown in Fig. 5.
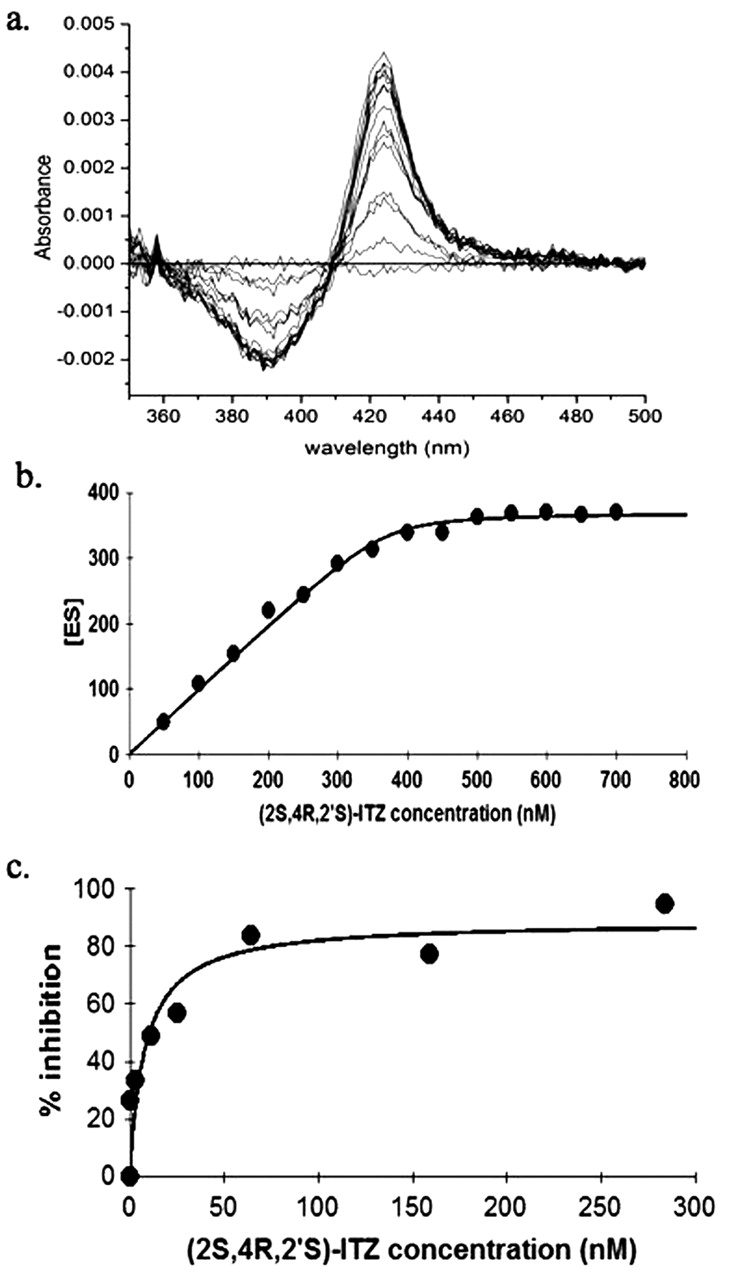
a, spectral titration of (2S,4R,2′S)-ITZ with CYP3A4. The lines represent the substrate-induced binding spectra at increasing ITZ concentrations (50 nM-700 nM). b, determination of the binding constant for (2S,4R,2′S)-ITZ. The line represents the fit of the Morrison equation to the spectral titration data. c, inhibition of midazolam hydroxylation by (2S,4R,2′S)-ITZ. The line represents the best fit to determine the IC50.
Stereoselective Pharmacokinetics of Itraconazole in Vivo. The disposition of ITZ was studied in six healthy subjects after single and multiple doses of ITZ. Because incomplete chromatographic separation of (2R,4S,2′R)-ITZ and (2R,4S,2′S)-ITZ was obtained, the pharmacokinetic parameter estimates for this pair of stereoisomers were calculated as the sum of the total concentration of (2R,4S,2′R)-ITZ and (2R,4S,2′S)-ITZ. For comparison, the plasma concentrations of (2S,4R,2′R)-ITZ and (2S,4R,2′S)-ITZ were added and pharmacokinetic analysis was performed on the sum of these two stereoisomers. The plasma concentration versus time curves of the (2S,4R,2′R)-ITZ and (2S,4R,2′S)-ITZ were superimposable, and these two stereoisomers had identical pharmacokinetic characteristics (data not shown). The analysis of the two pairs, instead of each stereoisomer separately, was justified based on the stereoselective metabolism of the stereoisomers by CYP3A4 in vitro. The pharmacokinetic analysis was essentially performed for the stereoisomers that undergo metabolism by CYP3A4 in vitro versus those that do not.

Plasma concentration versus time curves for the pairs of ITZ stereoisomers in subject E on day 1 (a) and day 7 (b) of the study.
Full stereoselective pharmacokinetic analysis was carried out in one of the six subjects (subject E). The plasma concentration versus time curves of the ITZ stereoisomeric pairs on study day 1 and day 7 in this subject are shown in Fig. 6. The pharmacokinetic parameter estimates obtained are presented in Table 2. The disposition of ITZ was stereo-selective in this subject, and the stereoselective indexes (the parameter ratio between the high and low stereoisomer pair value) ranged from 2.0 to 7.2. The largest difference between the stereoisomeric pairs was observed in oral clearance. On day 1, the (2R,4S)-ITZ pair had over 7-fold greater oral clearance than the (2S,4R)-ITZ pair. The apparent terminal half-life of the (2R,4S)-ITZ pair was half that of the (2S,4R)-ITZ pair. The pharmacokinetic parameters obtained on day 1 differed from those of day 7 for both stereoisomeric pairs in this subject. For the (2R,4S)-ITZ pair, the AUC increased 6.9-fold (AUCinf versus AUC0-24) with a corresponding decrease in oral clearance. The Cmax of the (2R,4S)-ITZ pair increased 5.5-fold between the two study occasions. For the (2S,4R)-ITZ pair, the increase in AUC and Cmax was more modest, 2-fold, between day 1 and day 7 of the study. Stereoselective pharmacokinetics was observed on day 7 of the study, at steady state, but to a lesser extent than on day 1. At steady state, the AUC of the (2S,4R)-ITZ pair was 3 times the AUC for the (2R,4S)-ITZ pair. It is interesting that in this subject, the difference in the Cmax value between the two pairs of stereoisomers disappeared at steady state, suggesting loss of a stereoselective first-pass effect.
Pharmacokinetic parameters for ITZ stereoisomer pairs in subject E after a single dose administration (day 1) of a 100-mg oral solution of itraconazole and after multiple dose administration (100 mg/day) for 7 days
The peak and trough concentrations were analyzed for all six volunteers, and the results are shown in Table 3. On day 1 of the study, the peak concentration of the (2S,4R)-ITZ pair was 3 times and the trough concentration, 10 times that of the (2R,4S)-ITZ pair (p < 0.05). The difference decreased at steady state, and on day 7 of the study, the peak concentration of the (2S,4R)-ITZ pair was only 1.5-fold greater, and the trough concentration 4-fold greater than that of the (2R,4S)-ITZ pair, but the difference was still significant (p < 0.05). Accumulation, demonstrated by a significant increase in the trough concentrations between day 1 and day 7 of the study, was observed for both stereoisomeric pairs, although it was greater for the (2R,4S) pair; 20-fold for (2R,4S)-ITZ and 8-fold for (2S,4R)-ITZ.
The peak and trough concentrations of the two enantiomeric pairs of ITZ in the six healthy volunteers after a single dose administration (day 1) of a 100-mg oral solution of itraconazole and after multiple dose administration (100 mg/day) for 7 days
The peak and trough plasma samples of the volunteers as well as the plasma samples of subject E were analyzed for stereochemistry of OH-ITZ, keto-ITZ, and ND-ITZ metabolites. In plasma, ND-ITZ and keto-ITZ were of the (2R,4S)-configuration (Fig. 3c). Because OH-ITZ potentially consists of eight stereoisomers, the absolute configuration of the formed OH-ITZ could not be confirmed, but the lack of formation of (2R,4S,2′S,3′S)-OH-ITZ in vivo was established, since this stereoisomer was separated chromatographically from the others.
To estimate the relative contribution of the individual pairs of itraconazole stereoisomers to the in vivo inhibition of CYP3A4, the ratios between the peak and trough plasma concentrations and the IC50 values were calculated (Table 4). All values except the trough concentration/IC50 on day 1 for (2R,4S)-ITZ were greater than 1, suggesting that both stereoisomer pairs would cause an in vivo drug-drug interaction, but the relative contribution of the different stereoisomers varied over the dosing interval as suggested by the change in the ratio between parameter values for the stereoisomer pairs.
The inhibitor concentration to IC50 ratios for the two pairs of ITZ stereoisomers, calculated using peak and trough concentrations of the two enantiomeric pairs after a single dose administration (day 1) of a 100-mg oral solution of itraconazole and after multiple dose administration (100 mg/day) for 7 days
Discussion
The goals of this study were to determine whether the binding of ITZ to CYP3A4, the metabolism of ITZ by CYP3A4, and the pharmacokinetic behavior of ITZ in vivo are stereoselective. The major finding is that the metabolism of ITZ by CYP3A4 is highly stereo-selective. Only two of the four ITZ stereoisomers underwent metabolism by CYP3A4.
The major stereochemical determinant in the metabolism of ITZ is the absolute stereochemistry of the dioxolane ring portion of the molecule. It was notable that the dioxolane ring remote from the site of metabolism had such a critical effect on oxidation of ITZ. One possibility is that some active site residue(s) steers the molecule into the metabolically productive orientation; perhaps via coordination of the triazole to an acidic amino acid at the active site. Further experiments are needed to more thoroughly assess the influence of the dioxolane ring on the orientation of ITZ stereoisomers at the CYP3A4 active site.
Stereochemistry of the 2′-position of ITZ pyrazolone-butyl side chain also affected oxidation. For (2R,4S,2′S)-ITZ, 5 times the amount of OH-ITZ was produced when compared with (2R,4S,2′R)-ITZ under these experimental conditions. Based on the similar IC50 and Ks values, the affinity of both stereoisomers for CYP3A4 was similar and, therefore, the larger amount of OH-ITZ formed from (2R,4S,2′S)-ITZ suggests a faster catalytic rate from this stereoisomer. The Km and Vmax of (2R,4S,2′S)-ITZ and (2R,4S,2′R)-ITZ were not determined because the metabolism of ITZ stereoisomers is complicated by sequential metabolism, formation of inhibitory metabolites, and potential multiple binding orientations in the active site. Future experiments will be needed to characterize and model the kinetics of the sequential oxidations of these stereoisomers.
The mixture of ITZ stereoisomers induces a characteristic type II binding spectrum with CYP3A4, suggesting that a triazole-ring nitrogen coordinates with the heme iron, producing a characteristic low spin complex. However, ITZ also undergoes metabolism by CYP3A4 at the aliphatic side chain distal to the triazole nitrogens. The apparent existence of at least two distinct enzyme-substrate complexes leads to the hypothesis that the two stereoisomers of ITZ that undergo metabolism by CYP3A4 would yield a type I binding spectrum with CYP3A4 and the two that are not turned over would demonstrate type II binding spectra. However, it was not possible to differentiate spectrally between the binding of the two stereoisomers that undergo metabolism and the two that do not, since all four ITZ stereoisomers produced a type II binding spectrum of similar intensity. This observation demonstrates that the triazole nitrogen accesses and coordinates the heme iron, regardless of the absolute stereochemistry in the dioxolane ring of ITZ, but stereochemistry is a crucial determinant for a second, metabolically productive orientation. At present, it is not known whether the two stereoisomers of ITZ that are not turned over by CYP3A4 have two different orientations at the active site of CYP3A4. It is likely, however, that the inhibition of CYP3A4 by these two stereoisomers, (2S,4R), is due to the coordination of CYP3A4 heme by the triazole nitrogen.
The stereoselective disposition of ITZ in vivo reflects the stereoselective metabolism of ITZ by CYP3A4 and supports the conclusion that CYP3A4 is important for elimination of ITZ. The stereoisomer pair that is metabolized by CYP3A4 ((2R,4S,2′R)-ITZ and (2R,4S,2′S)-ITZ) had higher oral clearance and smaller AUC than the other pair, (2S,4R,2′S)-ITZ and (2S,4R,2′R)-ITZ, in vivo. The 7-fold difference in oral clearance between the stereoisomeric pairs on day 1 is most likely due to stereoselectivity in the first-pass metabolism, as well as hepatic metabolic clearance of ITZ. The contribution of gut metabolism to the stereoselective pharmacokinetics of ITZ is also supported by the more modest stereoselectivity in the half-life when compared with oral clearance and AUC.
Presumably, as a consequence of the stereoselective metabolism of the (2R,4S)-ITZs during first pass, the peak concentrations of this pair were significantly lower than those of the (2S,4R) pair. Interestingly, stereoselectivity was diminished at day 7 of the pharmacokinetic study. The decrease in stereoselectivity might be attributed to autoinhibition of intestinal and, possibly, hepatic CYP3A4 by the ITZ stereoisomers, leading to reduced first-pass metabolism. Autoinhibition of CYP3A4 is also in agreement with the greater than predicted accumulation of (2R,4S)-ITZ. It is noteworthy that despite the shorter half-life after single dose, based on the increased trough levels, the (2R,4S)-ITZ stereoisomers accumulated significantly more than the (2S,4R)-ITZ stereoisomers. This observation supports the view that the autoinhibition observed after multiple doses of the four ITZ stereoisomers is due to saturation of CYP3A4-mediated metabolism of the (2R,4S)-ITZ pair. Also, based on the in vitro stereoselective metabolism data (including substrate depletion experiments), a maximum of 50% of the systemic clearance of ITZ is CYP3A4-mediated.
Interpretation of the pharmacokinetic data of ITZ stereoisomers is complicated by possible stereoisomer-stereoisomer interactions. All four ITZ stereoisomers are high affinity ligands of CYP3A4, and it is likely that the (2S,4R)-ITZ stereoisomers inhibit the elimination of the (2R,4S)-ITZs at steady state. The higher plasma concentrations of (2S,4R)-ITZ at steady state and after single dose suggest that a significant portion of the hepatic CYP3A4 is bound by these stereoisomers, despite the fact that they are not substrates of CYP3A4. It is also possible that the kinetic nonlinearity after ITZ dosing is a result of stereoisomer-stereoisomer interaction instead of saturation of the enzyme by the species undergoing metabolism. To obtain additional information about the mechanisms of kinetic nonlinearity, each of the ITZ stereoisomers should be administered separately to humans.
In previous studies, in which stereochemical factors were not explored, the half-life of OH-ITZ has been found to be consistently shorter than that of the parent compound, and it has been suggested that this phenomenon reflects predominant formation of OH-ITZ during the first pass (Ducharme et al., 1995). In this study, it was hypothesized that the half-life of OH-ITZ is dependent on the half-life of the stereoisomers of ITZ that undergo metabolism by CYP3A4 and therefore contribute to the formation of OH-ITZ in vivo, following classical formation rate-limited kinetics. Indeed, the half-life of (2R,4S)-ITZs, 10 h, was shorter than or equal to that calculated for OH-ITZ in this subject (10–14 h), in agreement with formation rate-limited metabolite kinetics. However, the plasma concentration versus time curves of ITZ stereoisomers still showed biphasic elimination, despite the stereoselective analysis, consistent with significant tissue distribution.
A modest stereoselective ratio was observed in the IC50 values for ITZ stereoisomers, although it is not clear whether this is due to contribution of the formed inhibitory metabolites. A previous study has shown that the inhibition of CYP3A4 by ketoconazole, a related azole antifungal, is stereoselective (Dilmaghanian et al., 2004). Whether the metabolism of ketoconazole by CYP3A4 is stereoselective as well has not been reported. It is not immediately evident which stereoisomers of ITZ are most important in terms of in vivo inhibition of CYP3A4 and drug-drug interactions. On day 1, the ratio between Cmax and IC50 was 23.4 for both stereoisomer pairs (Table 4), but at the trough concentration, a 5-fold greater ratio of Cmin/IC50 was observed for the (2S,4R)-ITZ pair. In contrast, on day 7 (steady state) of the study, the Cmax to IC50 ratio was twice as high for the (2R,4S)-ITZ pair as for the (2S,4R)-ITZ pair and the Cmin/IC50 value was approximately equal for the two pairs. At present, it is not possible to conclude which stereoisomer contributes the most to clinical inhibition of CYP3A4. In addition, it is likely that the circulating metabolites do contribute to CYP3A4 inhibition, but the inhibitory potency of the stereoisomers of the metabolites has not yet been determined.
The results presented here suggest that a single ITZ stereoisomer, i.e., (2S,4R,2′R)-ITZ or (2S,4R,2′S)-ITZ, might be clinically superior to the mixture of ITZ stereoisomers. This would have advantages over the currently used stereoisomer mixture. It would not undergo metabolism by CYP3A4, eliminating the formation of inhibitory metabolites and having potentially more predictable multiple dose kinetics than the mixture of stereoisomers, its elimination would not be subject to stereoisomer-stereoisomer interactions via CYP3A4, it would have equivalent plasma concentrations after administration of lower doses of drug due to lower oral clearance, and it may be a less potent inhibitor of CYP3A4 than the other stereoisomers, based on its IC50 and lack of inhibitory metabolites. In addition, based on the patent data (Koch et al., 2000), this stereoisomer should be a more potent antifungal than the mixture.
In conclusion, this study showed that the pharmacokinetic behavior of ITZ is stereoselective, most likely due to stereoselective metabolism of ITZ by CYP3A4. All stereoisomers bind to the CYP3A4 heme by the triazole nitrogen, but at least two of the stereoisomers [the (2R,4S) pair] also adapt to a binding orientation in which the alkyl side chain is presented to the heme iron. Thus, predicting in vivo inhibition of CYP3A4 by the itraconazole stereoisomer mixture currently administered is exceedingly complicated, due to different plasma concentrations and inhibitory potencies of the stereoisomers, formation of inhibitory metabolites, and potential enatiomer-enantiomer interactions between the stereoisomers.
Footnotes
-
This study was supported in part by National Institutes of Health Grants PO1 GM32165, K24 DA00417, and P30 ES07033. A portion of this work was conducted through the Clinical Research Center Facility at the University of Washington and supported by the National Institutes of Health Grant M01-RR-00037.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.008508.
-
ABBREVIATIONS: ITZ, itraconazole; OH-ITZ, hydroxy-itraconazole; keto-ITZ, keto-itraconazole; ND-ITZ, N-desalkyl-itraconazole; OH-MDZ, 1′-hydroxymidazolam; P450, cytochrome P450; KPi, potassium phosphate; LC-MS, high-performance liquid chromatography-mass spectrometry; AUC, area under the curve.
- Received November 22, 2005.
- Accepted January 12, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}