


Abstract
Breast cancer resistance protein (BCRP/ABCG2) is an ATP-binding cassette efflux transporter, important in drug disposition and in the development of multidrug resistance in cancer. Flavonoids, a large class of natural compounds widely present in the diet and herbal products, have been shown in vitro to be BCRP inhibitors. The flavonoid chrysin is a potent inhibitor of BCRP, inhibiting the efflux of mitoxantrone with an IC50 of 0.39 μM in BCRP-overexpressing human MCF-7 breast cancer cells. The purpose of this study was to investigate the potential pharmacokinetic interactions between chrysin and nitrofurantoin (a specific BCRP substrate) in rats. In Madin-Darby canine kidney cells expressing human BCRP or murine Bcrp1, the polarized transport of nitrofurantoin was effectively inhibited by chrysin at concentrations of 20 and 100 μM. Compared with the vehicle-treated group, p.o. coadministration of chrysin (200 mg/kg) significantly increased the area under the curve (AUC) and Cmax of nitrofurantoin (10 mg/kg) by 1.76- (p < 0.01) and 1.72-fold (p < 0.05), respectively. When nitrofurantoin (2 mg/kg) was given i.v., administration of chrysin (50 mg/kg i.p.) significantly increased the AUC of nitrofurantoin (123 ± 34.0 versus 91.5 ± 18.0 μg/ml · min in controls, p < 0.05). Moreover, the cumulative hepatobiliary excretion of nitrofurantoin (1.5 mg/kg i.v.) was significantly decreased by approximately 75% at the end of 120 min after the coadministration of chrysin (50 mg/kg i.p.). Taken together, these results indicate that the flavonoid chrysin significantly inhibits nitrofurantoin transport mediated by human BCRP and murine Bcrp1. Bcrp1 inhibition by chrysin is likely one potential mechanism for the observed chrysin-nitrofurantoin pharmacokinetic interactions in rats.
Flavonoids, a large class of polyphenolic compounds present in the diet and many herbal products, have long been associated with a variety of biochemical and pharmacological activities important in cancer prevention and health promotion (Middleton et al., 2000; Havsteen, 2002). The flavonoid chrysin (5,7-dihydroxyflavone) is present at high levels in honey and propolis (Siess et al., 1996) and in many plant extracts (Williams et al., 1997). In addition to its reported anticarcinogenic (Cardenas et al., 2006), antiviral (Critchfield et al., 1996), antioxidant (Lapidot et al., 2002), and anti-inflammatory (Cho et al., 2004) activities, chrysin has been shown as a potent inhibitor of aromatase (Jeong et al., 1999), an enzyme responsible for the conversion of androstenedione and testosterone into estrone and estradiol, respectively (Meinhardt and Mullis, 2002). As such, chrysin acts as a testosterone-boosting agent and is currently available as dietary supplements used for increasing lean body mass. The products on the market typically contain 500 mg of chrysin per capsule (iHerb Inc., Monrovia, CA; VitaDigest, Walnut, CA), and the suggested dosage can be as high as 6 capsules per day. On consumption of these dietary supplements, high concentrations of chrysin in the intestine would be expected, increasing the potential for flavonoid-drug interactions.
Recently, because of an increased public interest in alternative medicine and disease prevention, the use of dietary supplements and herbal preparations containing high doses of flavonoids for health maintenance has become very popular (Sparreboom et al., 2004). As a result, flavonoid-drug interactions have been increasingly evidenced in animal and clinical studies (Dresser et al., 2002; Choi et al., 2004). In some cases, these flavonoid-drug interactions can be serious or even life-threatening. This is true especially when flavonoids or dietary supplements are used concomitantly with narrow therapeutic index drugs, such as digoxin (Wang et al., 2004). On the other hand, coadministration of drug candidates with flavonoids, which are inhibitors of known drug transporters and metabolizing enzymes, may represent a strategy to improve the bioavailability of the coadministered drug. For example, flavonoid flavone has been shown to increase the bioavailability of paclitaxel (a dual substrate of P-glycoprotein and CYP3A) in rats in a concentration-dependent manner (Choi et al., 2004).
The mechanisms underlying most of the reported flavonoid-drug interactions have been ascribed to the inhibition of P-glycoprotein and/or drug-metabolizing enzymes. Very little information is currently available on flavonoid-drug interactions mediated by other major efflux drug transporters. Breast cancer resistance protein (BCRP) is a recently identified ATP-binding cassette transporter, important in conferring cellular resistance to many chemotherapeutic agents, such as anthracyclines, camptothecin derivatives, mitoxantrone, and nucleoside analogs (Mao and Unadkat, 2005). In addition, BCRP has been shown to play a critical role in the disposition of dietary carcinogens (van Herwaarden et al., 2003) and several clinically important drugs, such as imatinib mesylate (Breedveld et al., 2005), topotecan, and cimetidine (Jonker et al., 2005). In recent preclinical and clinical studies, inhibition of BCRP by GF120918 has been shown to significantly increase the bioavailability and decrease the biliary excretion of topotecan (a BCRP substrate) (Jonker et al., 2000; Kruijtzer et al., 2002). Therefore, inhibition of BCRP might represent an additional mechanism underlying drug-drug interactions.
Flavonoids represent a class of BCRP inhibitors, with chrysin being one of the most potent ones in inhibiting the efflux of mitoxantrone with an IC50 of 0.39 ± 0.13 μM (Zhang et al., 2004a,b). Therefore, we hypothesized that inhibition of BCRP by the flavonoid chrysin might alter the pharmacokinetics of drugs that are BCRP substrates. In a recent study, nitrofurantoin, an antibiotic used in the treatment of urinary tract infections, was shown to be specifically transported by human BCRP and murine Bcrp1 but not by P-glycoprotein and multidrug resistance-associated protein 2 (MRP2) (Merino et al., 2005). Moreover, chrysin was shown to exhibit very weak, if any, inhibitory effects on MRP1 and MRP2 (van Zanden et al., 2005). Additionally, studies in MRP1-expressing H69-AR cells have found no increased accumulation of nitrofurantoin in the presence of cyclophosphamide, further suggesting that nitrofurantoin may not be a substrate for MRP1 (Wang and Morris, unpublished results). Given the transporter specificity of nitrofurantoin and inhibition spectrum of chrysin, BCRP inhibition by chrysin is likely an important, if not the only, mechanism underlying the interaction between chrysin and nitrofurantoin.
The objective of this study was to examine the effects of chrysin on human BCRP- and murine Bcrp1-mediated transport of nitrofurantoin using polarized cells lines, and to investigate the potential pharmacokinetic interaction between chrysin and nitrofurantoin in rats.
Materials and Methods
Animals. Female Sprague-Dawley (SD) rats (body weight ∼220 g) were purchased from Harlan (Indianapolis, IN). Animals were housed in a temperature-controlled environment with a 12-h light/dark cycle and received a standard diet with free access to tap water. Animals were acclimatized to this environment for at least 1 week. All the animal studies were reviewed and approved by the University at Buffalo Institutional Animal Care and Use Committee.
Materials. Chrysin, glycofurol, polyethylene glycol 400, nitrofurantoin, and furazolidone were purchased from Sigma-Aldrich (St. Louis, MO). Dulbecco's modified Eagle's medium, Hanks' buffered salt solution (HBSS), fetal bovine serum, penicillin, and streptomycin were purchased from Invitrogen (Carlsbad, CA). Fumitremorgin C (FTC) was a gift from Dr. Susan E. Bates (National Cancer Institute, Bethesda, MD). All the other reagents or solvents used were either analytical or high-performance liquid chromatography (HPLC) grade.
Cell Culture. The polarized Madin-Darby canine kidney cell line (MDCK-II) was used in the transport studies. MDCK-mock (MDCK-II cells transfected with empty vector), MDCK-Bcrp1 (MDCK-II cells transfected with murine Bcrp1), and MDCK-BCRP (MDCK-II cells transfected with human BCRP) cells were gifts from Dr. Alfred Schinkel (Netherlands Cancer Institute, Amsterdam, The Netherlands). The expression of human BCRP and murine Bcrp1 in transfected MDCK cells has been characterized previously (Jonker et al., 2000; Pavek et al., 2005) and was confirmed in our laboratory (data not shown). The MDCK cells were cultured in 75-cm2 flasks with Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum at 37°C in a humidified 5% CO2/95% air atmosphere. The culture medium also contained 100 units/ml penicillin and 100 μg/ml streptomycin. The cells were trypsinized every 3 to 4 days for subculturing.
Nitrofurantoin Bidirectional Transport Studies. Transport studies using MDCK cell monolayers were performed as described previously (Zhang et al., 2005) with minor modifications. In brief, MDCK-Bcrp1, MDCK-BCRP, and MDCK-mock cells were seeded on Transwell polycarbonate inserts (six-well, 0.4-μm pore size; Transwell 3412, Corning Glassworks, Corning, NY) at a density of approximately 106 cells/well. Cells were grown for 5 to 7 days, and medium was replaced every day. On the day of the transport study, cell monolayers were first washed with HBSS (pH 7.2) twice and incubated at 37°C for a total of 30 min. Transport buffer (HBSS, pH 7.2) containing specified concentrations of chrysin or 10 μM FTC or 0.1% dimethyl sulfoxide (control) was then loaded into both apical (1.5 ml) and basolateral (2.6 ml) chambers. After a 15-min incubation, nitrofurantoin (10 μM) was added to either the apical or basolateral chamber (donor chamber). The aliquots (100 μl) were taken from the opposite chamber (receiver chamber) at 30, 60, and 90 min after the addition of nitrofurantoin and replaced with the same volume of fresh transport buffer. The samples were stored at –80°C until HPLC analysis. The integrity of the monolayer was measured by monitoring the flux of [3H]mannitol, a paracellular marker, across cell monolayers. The apparent permeability coefficients (Papp) of mannitol in both apical to basolateral (Papp, AP-BL) and basolateral to apical (Papp, BL-AP) directions were measured in triplicate, and these values were all lower than 1.0 × 10–6 cm/s. The apparent permeability coefficient was calculated as follows: Papp = (ΔQ/Δt) × [1/(A × C0)], where ΔQ/Δt is the rate of nitrofurantoin appearing in the receiver chamber, which was obtained from the slope of the regression line on the transport-time profile of nitrofurantoin across MDCK cell monolayers, C0 is the initial concentration of nitrofurantoin loaded in the donor chamber, and A is the cell monolayer surface area (4.71 cm2).
Pharmacokinetic Studies. Pharmacokinetic studies were performed in a manner similar to that described previously (Merino et al., 2005; Zhang et al., 2005) with some modifications. In brief, jugular vein cannulation was performed in rats following an i.m. injection of 90 mg/kg ketamine and 10 mg/kg xylazine (Henry Schein, Melville, NY) at least 3 days before pharmacokinetic studies. For oral drug interaction studies, rats were kept fasting overnight with free access to tap water. A specified dose of the test compounds (chrysin dissolved in glycofurol) or the vehicle glycofurol (as control) was administered to rats by gavage. Chrysin was dissolved in glycofurol at such a concentration that the specified dose was delivered as 10 μl/g body weight drug solution. Five minutes later, the animals were administered 10 mg/kg nitrofurantoin p.o. [3 mg/ml dissolved in 50% (v/v) ethanol and 50% (v/v) polyethylene glycol 400]. The blood samples (150 μl) were taken from the jugular vein cannula at 0 (predose), 5, 15, 30, 60, 120, 240, 360, 480, and 720 min after nitrofurantoin administration. For i.v. administration of nitrofurantoin, a dose of 50 mg/kg chrysin (dissolved in dimethyl sulfoxide at a concentration of 20 mg/ml) or vehicle (dimethyl sulfoxide) was first given to rats via i.p. injection. Five minutes later, a dose of 2 mg/kg nitrofurantoin [1 mg/ml dissolved in 10% (v/v) ethanol, 40% (v/v) polyethylene glycol 400, and 50% phosphate-buffered saline] was given to the rats via the jugular vein cannula. The blood samples (150 μl) were taken at 0 (predose), 2, 7, 15, 30, 60, and 120 min after nitrofurantoin administration. Urine samples were collected over the time periods of 0 to 2, 2 to 4, and 4 to 8 h after nitrofurantoin administration. All the samples were stored at –80°C until the time of HPLC analysis. Four to seven rats were used for each group.
Bile Duct Cannulation Experiment. Bile duct cannulation was carried out as described previously (Jackson et al., 2000) under anesthesia (a combination of 90 mg/kg ketamine and 10 mg/kg xylazine). A dose of 50 mg/kg chrysin (dissolved in dimethyl sulfoxide at a concentration of 20 mg/ml) or vehicle (dimethyl sulfoxide) was first given to rats via i.p. injection. Five minutes later, a dose of 1.5 mg/kg nitrofurantoin [1 mg/ml dissolved in 10% (v/v) ethanol, 40% (v/v) polyethylene glycol 400, and 50% phosphate-buffered saline] was administered via tail vein injection. Bile was collected in 15-min fractions for 120 min after the administration of nitrofurantoin. Bile samples were stored at –80°C until HPLC analysis. Four to five animals were used for each group.
HPLC Analysis. The concentration of nitrofurantoin in rat plasma, urine, bile, and buffer samples from transport studies was analyzed by a previously published method (Merino et al., 2005) with minor modifications. In brief, an aliquot of 50 μl of sample was spiked with 5 μl of 12.5 μg/ml furazolidone solution (internal standard), and then 50 μl of cold methanol (–20°C) was added. The mixture was vortexed vigorously for 60 s and incubated at –20°C for 15 min. The organic and water phases were separated by centrifugation at 16,000g for 10 min at 4°C, and 30 μl of the supernatant was injected into HPLC system. The HPLC analysis was performed using an Alltech Alltima C18 column (250 × 4.6 mm, 5-μm particle size). The system consisted of a Waters 1525 pump, 717 plus autosampler, a 2847 UV detector, and a Waters Breeze workstation. The composition of the mobile phase was 25 mM potassium phosphate buffer (pH 3)/acetonitrile (75:25). The mobile phase was delivered isocratically with a flow rate of 1.0 ml/min. UV absorbance was measured at 366 nm. Nitrofurantoin appeared on the chromatograph at approximately 8 min with no interference peaks. The standard curve was linear over the concentration range of 0.01 to 20 μg/ml with a regression coefficient greater than 0.99. The lower limit of quantitation of nitrofurantoin was 10 ng/ml.
Pharmacokinetic Analysis. The pharmacokinetic parameters of nitrofurantoin were obtained by noncompartmental analysis using WinNonlin version 2.1 (Pharsight, Mountain View, CA). The area under the plasma concentrationtime curve (AUC) was calculated using the trapezoidal method. The terminal half-life (t1/2) was calculated as ln2/k, and k was determined from the slope of the terminal regression line. The systemic clearance (CL) and oral clearance (CL/F) were calculated as the i.v. and p.o. dose divided by AUC, respectively. The bioavailability (F) was determined by (AUCp.o./Dosep.o.)/(AUCi.v./Dosei.v.).
Statistical Analysis. Data were analyzed for statistically significant differences using analysis of variance followed by a Bonferroni or Dunnett's post hoc test or by the two-sided unpaired Student's t test. All the p values <0.05 were considered statistically significant.
Results
Effects of Chrysin on in Vitro Transport of Nitrofurantoin. Flavonoid chrysin has been shown to inhibit BCRP-mediated efflux of mitoxantrone (Zhang et al., 2004b); however, its effects on BCRP-mediated efflux of nitrofurantoin have not been evaluated. Therefore, we first looked at the effects of chrysin on nitrofurantoin transport in both MDCK parental cells (MDCK-mock) and MDCK cells expressing either murine Bcrp1 (MDCK-Bcrp1) or human BCRP (MDCK-BCRP). As shown in Fig. 1, the polarized transport of nitrofurantoin (10 μM) was observed in both MDCK-Bcrp1 and MDCK-BCRP cells in the absence of inhibitors, with Papp, BL-AP values significantly higher than the corresponding Papp, AP-BL values (9.69 ± 0.41 versus 0.70 ± 0.23 × 10–6 cm/s, p < 0.001 and 11.7 ± 0.80 versus 2.91 ± 0.21 × 10–6 cm/s, p < 0.001 for MDCK-Bcrp1 and MDCK-BCRP cells, respectively). When the specific Bcrp1/BCRP inhibitor FTC (10 μM) was used, the Bcrp1/BCRP-mediated transport was completely inhibited, resulting in vectorial transport patterns similar to that of the MDCK parental cells. In the presence of chrysin (20 and 100 μM), apically directed transport of nitrofurantoin was significantly decreased, and basolaterally directed transport was significantly increased in both MDCK-Bcrp1 and MDCK-BCRP cells, resulting in efflux ratios (Papp, BL-AP/Papp, AP-BL) close to unity. For example, in MDCK-Bcrp1 cells, the efflux ratios were decreased from 13.9 (without inhibitors) to 1.75 (with 20 μM chrysin) and 1.29 (with 100 μM chrysin), and in MDCK-BCRP cells, the efflux ratios were decreased from 4.02 (without inhibitors) to 1.08 (with 20 μM chrysin) and 0.77 (with 100 μM chrysin). These results suggest that chrysin indeed inhibits both murine Bcrp1- and human BCRP-mediated transport of nitrofurantoin in MDCK cells. In MDCK parental cells, nitrofurantoin was consistently transported somewhat more efficiently in the basolateral direction than in the apical direction, indicating low endogenous basolaterally directed transport.
Effects of Chrysin on Nitrofurantoin Pharmacokinetics in SD Rats. The expression of Bcrp1 has been previously reported at high levels in the intestinal, renal, and hepatic tissues in female SD rats (Tanaka et al., 2005). To assess whether the in vitro chrysin-nitrofurantoin interactions via Bcrp1/BCRP also occurred in vivo, the pharmacokinetics of nitrofurantoin, following its p.o. and i.v. administration, were determined in SD female rats with and without the coadministration of chrysin. As shown in Fig. 2, p.o. coadministration of chrysin (200 mg/kg) significantly increased the AUC and Cmax of nitrofurantoin (10 mg/kg) by 1.76-fold [605 ± 163 in the chrysin-treated group versus 344 ± 101 μg/ml · min in the vehicle-treated (control) group, p < 0.01] and 1.72-fold (1.73 ± 0.52 in the chrysin-treated group versus 1.01 ± 0.47 μg/ml in the vehicle-treated group, p < 0.05), respectively. In contrast, the apparent oral clearance of nitrofurantoin (CL/F) was significantly decreased from 31.0 ± 8.65 ml/min/kg in the control group to 17.9 ± 6.58 ml/min/kg in the 200 mg/kg chrysin-treated group (p < 0.05). The terminal t1/2, however, was not significantly changed following the coadministration of chrysin. At a lower p.o. dose of 50 mg/kg, chrysin had no significant effects on nitrofurantoin pharmacokinetics (Table 1). When nitrofurantoin (2 mg/kg) was given i.v., administration of chrysin (50 mg/kg) i.p. significantly increased the AUC of nitrofurantoin (123 ± 34.0 versus 91.5 ± 18.0 μg/ml · min in controls, p < 0.05) (Fig. 3). Taking the AUC of i.v. administered nitrofurantoin in vehicle-treated rats as 100%, the bioavailability of nitrofurantoin administered p.o. was significantly increased from 60.2 ± 15.3% in the control group to 114 ± 17.9% in the 200 mg/kg chrysin-treated group (p < 0.01). These results indicate that chrysin indeed interacts with nitrofurantoin in vivo, altering both the bioavailability and elimination of nitrofurantoin.
Pharmacokinetic parameters of nitrofurantoin in SD female rats after p.o. and i.v. administration of nitrofurantoin with or without the coadministration of chrysin
The plasma concentration-time profile of nitrofurantoin in SD rats after p.o. and i.v. administration with control vehicle or the specified doses of chrysin was determined as described under Materials and Methods. The pharmacokinetic parameters were obtained by noncompartmental analysis using WinNonlin. Data are expressed as mean ± S.D. The number of animals for each treatment group is specified in parentheses after the corresponding group names.
Effects of Chrysin on the Hepatobiliary and Urinary Excretion of Nitrofurantoin in SD Rats. To investigate the effects of chrysin on nitrofurantoin hepatobiliary excretion, 1.5 mg/kg nitrofurantoin was administered to bile duct cannulated rats via a tail vein injection 5 min after the i.p. injection of vehicle or 50 mg/kg chrysin. Biliary excretion of nitrofurantoin was measured in 15-min bile collections, obtained over 120 min. As shown in Fig. 4, the hepatobiliary excretion rate of nitrofurantoin was substantially decreased in chrysin-treated rats for the first 60 min. After that, the excretion rate in both groups gradually became similar. The cumulative hepatobiliary excretion of nitrofurantoin was significantly decreased from 20.1 ± 4.54 μg (or 5.16 ± 0.96% of dose) in control rats to 5.39 ± 2.24 μg (or 1.21 ± 0.50% of dose) in chrysin-treated rats (p < 0.001) at the end of 120 min, indicating that approximately 75% of the biliary excretion of nitrofurantoin over 2 h was abolished on the administration of chrysin.
To assess the effects of chrysin on the urinary excretion of nitrofurantoin, a 2-mg/kg dose of nitrofurantoin was administered to SD rats via i.v. injection 5 min after the i.p. administration of control vehicle or 50 mg/kg chrysin. Levels of nitrofurantoin in urine samples were measured by HPLC. Most of the nitrofurantoin in urine was excreted during the first 0 to 8 h. In contrast to the biliary excretion of nitrofurantoin, chrysin had no significant effects on the renal excretion of nitrofurantoin. The percentage of the dose excreted in urine at the end of 8 h was 39.0 ± 10.2% in control rats and 35.8 ± 8.79% in chrysin-treated rats (p > 0.05).
Discussion
The recent resurgence of scientific interest in flavonoids has resulted in a dramatic increase in the consumption of flavonoid-containing products in the general population for health maintenance and disease prevention (Havsteen, 2002; Sparreboom et al., 2004). Increasing evidence from recent animal and human studies has indicated that flavonoids may interact with many clinically important drugs, causing favorable or adverse pharmacokinetic interactions (Kruijtzer et al., 2002; Choi et al., 2004; Wang et al., 2004). However, the mechanisms underlying most of these reported interactions are largely related to the modulation of drug-metabolizing enzymes and/or P-glycoprotein (Harris et al., 2003; Sparreboom et al., 2004). To date, far less information is available on flavonoid-drug interactions mediated by other major efflux drug transporters. Therefore, the objective of this study was to determine the potential in vitro and in vivo flavonoid-drug interactions mediated by BCRP, a recently identified efflux transporter important in conferring multidrug resistance in cancer therapy and in determining in vivo disposition of drugs and xenotoxins (Mao and Unadkat, 2005; van Herwaarden and Schinkel, 2006).

Transepithelial transport of 10 μM nitrofurantoin in MDCK-mock (parent) (A), MDCK-Bcrp1 (B–E), and MDCK-BCRP (F–I) monolayers. The studies were started with the addition of nitrofurantoin to one donor compartment (basolateral or apical). At 30, 60, and 90 min, the amount of nitrofurantoin appeared in the opposite compartment was measured by HPLC. BCRP inhibitor FTC (C and G) was used as the positive control. Chrysin (D, E, H, and I) was studied as the compound of interest. The data are expressed as mean ± S.D., n = 3 or 6. •, transport from the basolateral to apical compartment; ○, transport from the apical to basolateral compartment.
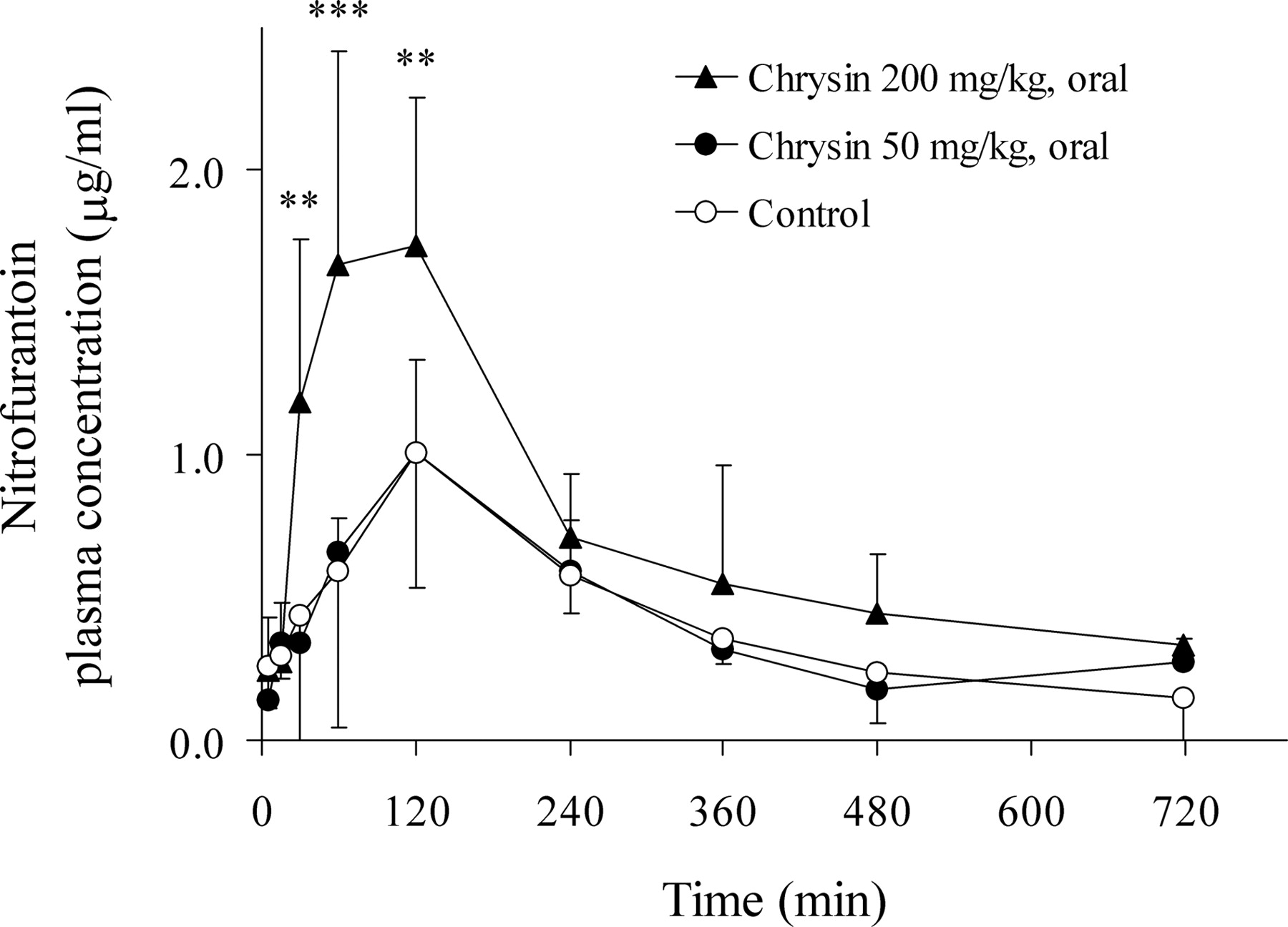
Plasma concentration versus time profile of nitrofurantoin after p.o. coadministration of nitrofurantoin (10 mg/kg) with control vehicle (○) or chrysin (50 mg/kg, •) or chrysin (200 mg/kg, ▴) in SD female rats. Plasma levels of nitrofurantoin were determined by HPLC. The data are expressed as mean ± S.D.; n = 6 for control group, n = 4 for 50 mg/kg chrysin group, and n = 5 for 200 mg/kg chrysin group (**, p < 0.01; ***, p < 0.001 compared with the control group).
The flavonoid selected for this study is chrysin, which was shown to be a potent human BCRP inhibitor (Zhang et al., 2004a). The BCRP/Bcrp1 substrate nitrofurantoin, used in this investigation, was previously reported as a specific substrate of BCRP/Bcrp1 (Merino et al., 2005). To determine whether chrysin interacts with nitrofurantoin in vivo, we first investigated the effects of chrysin on the transport of nitrofurantoin across MDCK-BCRP and MDCK-Bcrp1 cell monolayers. As shown in Fig. 1, chrysin, at concentrations of 20 and 100 μM, significantly decreased apically directed transport and increased basolaterally directed transport of nitrofurantoin in both MDCK-Bcrp1 and MDCK-BCRP cells, resulting in efflux ratios close to unity. These results suggested that chrysin indeed effectively inhibited both Bcrp1- and BCRP-mediated transport of nitrofurantoin in MDCK cells. Consistent with a previous report (Merino et al., 2005) in MDCK parental cells, nitrofurantoin was consistently transported somewhat more efficiently in the basolateral direction than in the apical direction, indicating the presence of low endogenous basolaterally directed transporter(s). Further study is needed to identify this unknown endogenous transporter(s). Similar to the results reported previously (Merino et al., 2005), the efflux ratio of nitrofurantoin in MDCK-Bcrp1 cells was higher than that in MDCK-BCRP cells (13.9 versus 4.02). As suggested in other studies, this is possibly because of an effectively lower expression level or reduced transport efficiency of human BCRP in the cell line used (Merino et al., 2006). Compared with the inhibitory effects of chrysin on murine Bcrp1, chrysin appeared to be a more efficient inhibitor of human BCRP. At the concentrations of 20 and 100 μM, chrysin almost completely inhibited the activity of human BCRP, resulting in vectorial transport patterns similar to that of the MDCK parental cells.

Plasma concentration versus time profile of nitrofurantoin after i.v. injection of nitrofurantoin (2 mg/kg), 5 min after the i.p. administration of control vehicle (○) or chrysin (50 mg/kg, •) in SD female rats. Plasma levels of nitrofurantoin were determined by HPLC. The data are expressed as mean ± S.D.; n = 7 for both control and 50 mg/kg chrysin group.
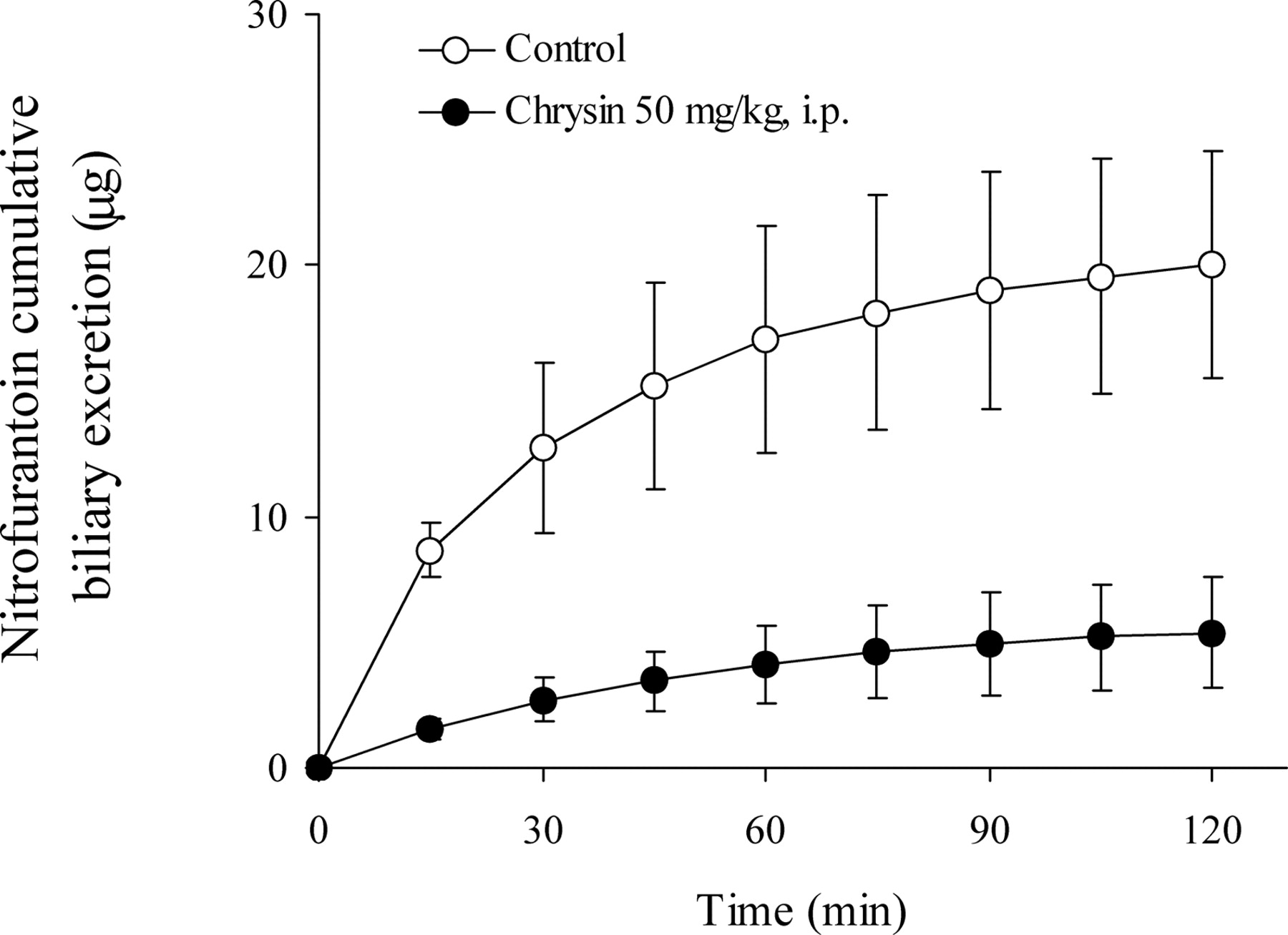
Cumulative biliary excretion of nitrofurantoin after tail vein injection of nitrofurantoin (1.5 mg/kg), 5 min after the i.p. administration of control vehicle (○) or chrysin (50 mg/kg, •) in SD female rats. Levels of nitrofurantoin were determined in bile fractions by HPLC. The data are expressed as mean ± S.D.; n = 4 for control group, n = 5 for 50 mg/kg chrysin group. At all the time points p < 0.001, except at 15 min p < 0.01.
To further investigate the interactions of chrysin with nitrofurantoin in vivo, pharmacokinetic studies of nitrofurantoin after p.o. and i.v. administration were performed in rats with and without the coadministration of chrysin. After the p.o. administration of nitrofurantoin, chrysin (200 mg/kg) significantly increased the AUC and Cmax and decreased the apparent oral clearance (CL/F) of nitrofurantoin. Compared with a 4-fold increase in nitrofurantoin AUC after p.o. administration in Bcrp1 knockout mice (Merino et al., 2005), a 1.76-fold increase in AUC observed in this study suggested that the in vivo Bcrp1 inhibition by chrysin (200 mg/kg) might not have been complete, if we assume the similar contribution of Bcrp1 to the bioavailability and clearance of nitrofurantoin in both rats and mice. However, previous pharmacokinetic studies in rats and mice using topotecan as a probe substrate have shown that the contribution of Bcrp1 in mice is relatively larger than that in rats (>1.5-fold) (Jonker et al., 2000; Zhang et al., 2005). Therefore, we cannot exclude the possibility that a 1.76-fold increase in nitrofurantoin AUC in rats may still represent the nearly complete Bcrp1 inhibition by chrysin, possibly attributed to the enhanced intestinal absorption and/or decreased systemic elimination of nitrofurantoin. Considering the potentially high intestinal concentrations of chrysin after p.o. administration and low bioavailability of chrysin (Walle et al., 2001), the altered p.o. pharmacokinetics of nitrofurantoin is more likely because of the local interactions of chrysin with nitrofurantoin in the intestine. At a lower p.o. dose of 50 mg/kg, chrysin had no significant effects on nitrofurantoin pharmacokinetics. Given the fact that chrysin potently inhibited nitrofurantoin transport in vitro, the lack of in vivo effects of chrysin at a lower dose might be possibly because of its extensive metabolic degradation in the intestine (Galijatovic et al., 1999). To determine whether chrysin also affects the systemic elimination of nitrofurantoin, we administered chrysin via i.p. injection to ensure some of the dose of chrysin would reach the systemic circulation. Nitrofurantoin was quickly eliminated in rats after i.v. administration with t1/2 values of approximately 25 min, consistent with the results in a previous report (Buzard et al., 1961). It should be noted that the t1/2 values of nitrofurantoin following i.v. and p.o. administration were different (∼25 and 160 min, respectively), indicating the likely flip-flop oral kinetics of nitrofurantoin. Administration of chrysin (50 mg/kg) i.p. significantly increased the AUC of nitrofurantoin, indicating chrysin might also inhibit nitrofurantoin elimination. Taking the AUC of i.v. administered nitrofurantoin in control rats as 100%, the bioavailability of nitrofurantoin administered p.o. was significantly increased from 60.2 ± 15.3% in control rats to 114 ± 17.9% in 200 mg/kg chrysin-treated rats (p < 0.01). The apparent nitrofurantoin bioavailability value of greater than 100% may indicate that the increase in nitrofurantoin AUC resulted not only from the increased intestinal absorption but also from the decreased systemic elimination, consistent with the mdr1a/1b (–/–) mouse study and our previous topotecan study (Jonker et al., 2000; Zhang et al., 2005).
A previous study has shown that Bcrp1 plays a predominant role (>98%) in nitrofurantoin hepatobiliary excretion (Merino et al., 2005). In our study, consistent with this result, the administration of chrysin remarkably inhibited nitrofurantoin hepatobiliary excretion by 75% (Fig. 4). This inhibition is likely caused by the inhibition of Bcrp1-mediated excretion of nitrofurantoin by chrysin because nitrofurantoin is not transported by other major efflux transporters present on the hepatic canalicular membrane, such as P-glycoprotein and MRP1/2. It has been reported that nitrofurantoin undergoes enterohepatic cycling (Conklin et al., 1973). Therefore, inhibiting Bcrp1 by chrysin might also possibly interfere with nitrofurantoin enterohepatic circulation.
It has been reported that Bcrp1 is present at high levels in the kidney in rats, suggesting that Bcrp1 might play an important role in the renal excretion of substrate drugs (Tanaka et al., 2005). Therefore, administration of chrysin with nitrofurantoin would be expected to affect the urinary excretion of nitrofurantoin. In the control rats of this study, the urinary excretion of unchanged nitrofurantoin after i.v. administration was 39.0 ± 10.2% of dose, which is close to the results reported in the literature (Paul et al., 1960; Veronese et al., 1974). However, different from the results we have seen in the hepatobiliary excretion study, chrysin had no significant effects on nitrofurantoin urinary excretion (35.8 ± 8.79% of dose in chrysin-treated rats versus 39.0 ± 10.2% in control rats, p > 0.05). The exact mechanisms for this still remain to be elucidated. Interestingly, similar results were reported in other studies, in which Bcrp1 was shown to play a predominant role in determining the plasma concentration and hepatobiliary, but not urinary, excretion of topotecan and nitrofurantoin (Jonker et al., 2000; Merino et al., 2005). A possible explanation to this might be because of the relatively large variability in urinary excretion data or possibly as a result of the involvement of other potential transport mechanisms in the urinary excretion of nitrofurantoin (Moller and Sheikh, 1982).
It was previously shown that nitrofurantoin is likely a CYP1A substrate and chrysin is a CYP1A inhibitor with a Ki value in the low micromolar range (Jonen et al., 1980; Moon et al., 1998). Therefore, inhibition of CYP1A enzymes by chrysin might also be responsible for the increased plasma concentrations of nitrofurantoin. However, a previous report has shown that CYP1A-mediated metabolism of nitrofurantoin does not represent a major metabolic pathway in CYP1A-uninduced rats (Jonen et al., 1980). Moreover, our liquid chromatography/mass spectrometry analysis of the samples from pharmacokinetic studies indicated that the levels of 4-hydroxy-nitrofurantoin (the CYP1A metabolite of nitrofurantoin) were much lower than those of nitrofurantoin (<3% of parent drug) (data not shown). Therefore, the contribution of CYP1A-mediated metabolism of nitrofurantoin in our rat studies seems minor, and inhibition of CYP1A by chrysin might not have a significant role in nitrofurantoin pharmacokinetics.
In conclusion, we have shown in this study that the flavonoid chrysin significantly inhibited nitrofurantoin transport mediated by human BCRP and murine Bcrp1 in vitro. More importantly, chrysin and nitrofurantoin pharmacokinetic interactions were observed in vivo, likely because of the Bcrp1 inhibition by chrysin. These findings provide proof-of-concept for in vivo inhibition of Bcrp1 by flavonoids and lend insight into the potential clinical interactions following concomitant use of these agents.
Acknowledgments
We thank Dr. Alfred H. Schinkel from Netherlands Cancer Institute for providing MDCK-Bcrp1 and MDCK-BCRP cells.
Footnotes
-
Financial support for this study was provided by grants from the Susan G. Komen Breast Cancer and Cancer Research and Prevention Foundations.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.011684.
-
ABBREVIATIONS: BCRP, breast cancer resistance protein; GF120918, N-(4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide; MRP, multidrug resistance-associated protein; SD, Sprague-Dawley; HBSS, Hanks' buffered salt solution; FTC, fumitremorgin C; HPLC, high-performance liquid chromatography; MDCK, Madin-Darby canine kidney; AUC, area under the plasma concentration-time curve.
- Received June 27, 2006.
- Accepted November 3, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}