


Abstract
CYP3A7 is a member of the human CYP3A family and a major form of P450 expressed in human fetal livers. Although CYP3A7 shares nearly 90% base sequence with CYP3A4, CYP3A7 shows striking functional differences in the catalytic preference for several substrates, such as dehydroepiandrosterone (DHEA) or dehydroepiandrosterone 3-sulfate (DHEA-3S). First, to clarify the reason for the differences between CYP3A7 and CYP3A4, a homology model of CYP3A7 was constructed using the CYP3A4 crystal structure. Because these two structures were similar, four kinds of chimeric enzymes were constructed to determine which sequences are important for exhibiting the characteristics of CYP3A7. The results of kinetic analysis of DHEA and DHEA-3S 16α-hydroxylations by CYP3A7, CYP3A4, and CYP3A chimeras suggested that the amino acid residues from Leu210 to Glu279 were important to express the specificity for substrates as CYP3A7. This region was on the F and G helices of the modeled CYP3A7. Furthermore, to assess which amino acid in this sequence is important for the substrate specificity of CYP3A7, a one-point mutation of CYP3A7 to CYP3A4 was made by site-directed mutagenesis. The mutants of K224T and K244E had lost DHEA and DHEA-3S 16α-hydroxylation activities. The mutants also greatly decreased the metabolism of testosterone, erythromycin, nevirapine, and triazolam relative to those activities of CYP3A7 wild-type enzyme. From these results, it is expected that CYP3A7 can recognize specific substrates using the lysines in F–G loops.
Cytochromes P450s (P450s) comprise a superfamily of monooxygenases that metabolize a wide variety of exogenous chemicals as well as endogenous substrates such as steroids, fatty acids, and prostaglandins. Multiple forms of P450 have been shown to be present in the liver (Nelson et al., 1996). Among the forms of P450s, CYP3A is the most abundant form in human livers (Kitada et al., 1985a; Shimada et al., 1994), and the human CYP3A subfamily consists of four isoforms: CYP3A4 (Beaune et al., 1986), CYP3A5 (Aoyama et al., 1989), CYP3A7 (Komori et al., 1989), and CYP3A43 (Domanski et al., 2000).
CYP3A7 accounts for 30 to 50% of total P450s in the fetal liver (Kitada et al., 1985b; Shimada et al., 1996). CYP3A4 is the major form of P450 expressed in the adult liver, and the expression of CYP3A4 mRNA was not seen in human fetal livers and began to increase after birth (Lacroix et al., 1997). It has been reported that a 5′-flanking region from 1 kilobase to the transcriptional start site of the CYP3A7 gene was 91% identical to that of the CYP3A4 gene (Itoh et al., 1992; Hashimoto et al., 1993). Recently, an experiment using HepG2 cells showed that Sp1 and Sp3 bound to the nuclear factor-κB-like element of the CYP3A7, but not the CYP3A4 gene, and that the expression of the CYP3A7 gene was cooperatively regulated by Sp1, Sp3, hepatocyte nuclear factor 3β, and upstream stimulatory factor 1 (Saito et al., 2001).
The nucleotide and amino acid sequences of CYP3A7 (Komori et al., 1989) share 94 and 88% identities, respectively, with those of CYP3A4 cDNA (Beaune et al., 1986; Gonzalez et al., 1988; Bork et al., 1999). CYP3A7 has been demonstrated to be responsible for the metabolism of exogenous chemicals as well as CYP3A4 (Ohmori et al., 1998). For most, but not all, substrates, the metabolic capability of CYP3A4 seems to be greater than that of CYP3A7 (Pearce et al., 2001; Williams et al., 2002). CYP3A7 shows striking functional differences in catalytic preference for endogenous substrates. For example, CYP3A4 exhibits a high level of activity for testosterone 6β-hydroxylation, whereas CYP3A7 shows a low level of activity (Ohmori et al., 1998). On the other hand, CYP3A7 catalyzed DHEA-3S 16α-hydroxylation with a high level of activity, whereas CYP3A4 is mostly unreactive (Kitada et al., 1987). It is still unclear why CYP3A7 shows substrate specificities and specific activities different from those of CYP3A4 despite the fact that the amino acid sequences of CYP3A7 and CYP3A4 are similar.
To determine the reason for the differences between substrate specificities of CYP3A4 and CYP3A7, chimeric enzymes and mutant enzymes were constructed with CYP3A7 and CYP3A4, and the sequences or amino acids that are important for exhibiting the characteristics of CYP3A7 were investigated. Furthermore, the related amino acids were examined using a homology model of CYP3A7 based on CYP3A4 crystal structure (Yano et al., 2004).
Materials and Methods
Materials
DHEA (5-androsten-3 β-ol-17-one), DHEA-3S (5-androsten-3 β-ol-17-one sulfate), testosterone (4-androsten-17 β-ol-3-one), and EM were purchased from Sigma Chemical (St. Louis, MO). NVP, 2-hydroxy-NVP, and 12-hydroxy-NVP were provided by Boehringer Ingelheim Pharma Co. (Ingelheim, Germany). TZM, α-hydroxy-TZM, and 4-hydroxy-TZM were donated by Kyowa Hakkou Kogyo Co. (Tokyo, Japan). All other chemicals used were of the highest grade commercially available.
Construction of Chimeric Enzymes
Designs of chimeric enzymes are illustrated in Fig. 1. CYP3A7-p2Bac and CYP3A4-p2Bac were constructed as follows. The full-length cDNAs encoding human CYP3A7 and CYP3A4 isolated from a human adult liver cDNA library were cloned to pSVL and pCI-neo (Pearce et al., 2001). CYP3A7-pSVL was treated with XhoI and BamHI. CYP3A7 cDNA was inserted into pBluescript II SK+. CYP3A4 cDNA was inserted into pBluescript II SK+ by treatment with XhoI and XbaI from CYP3A4-pCI-neo. Both CYP3A7 and CYP3A4 were cloned into the SmaI and XbaI restriction site of baculovirus shuttle vectors, p2Bac. CYP3A7(279)/4-p2Bac and CYP3A4(279)/7-p2Bac were constructed as follows. CYP3A4-pCI-neo and CYP3A7-pSVL were digested with EcoRI, and a 723-bp fragment (AA 39–AA 279) was isolated from each plasmid. The partial sequence of CYP3A4 was inserted into digested CYP3A7/pSVL [CYP3A4(279)/7-pSVL], and the partial sequence of CYP3A7 was inserted into digested CYP3A4-pCI-neo [CYP3A7(279)/4-pCI]. CYP3A7(279)/4-p2Bac and CYP3A4(279)/7-p2Bac were made from CYP3A4(279)/7-pSVL and CYP3A7(279)/4-pCI by the same procedure as that used for CYP3A7-p2Bac and CYP3A4-p2Bac. When CYP3A4(209)/7-p2Bac was constructed, CYP3A4-pBluescript II SK+ was treated with HindIII and a 543-bp fragment (AA 29–AA 209) was isolated. When CYP3A4(109)/7-p2Bac was constructed, CYP3A4-pBluescript II SK+ was treated with XhoI and StuI, and a 327-bp fragment (AA 1–AA 109) was isolated.
Site-Directed Mutagenesis
Six mutations of CYP3A7 containing N116S, H174D, N214D, K224T, K244E, and K262E were made using site-directed mutagenesis. Mutan-Super Express Km (TaKaRa, Tokyo) was used to introduce a single nucleotide change using the primers shown in Table 1. The entire coding region, including the mutated sites, was verified by sequencing using a Thermo Sequenase Cy 5.5 Dye Terminator Cycle Sequencing Kit (GE Healthcare, Buckinghamshire, UK). The entire cDNA was then excised and subcloned into a p2Bac vector.
Primers used for amplification of CYP3A7 cDNA site-directed mutagenesis
The oligonucleotides used to introduce the different substitutions are underlined with the specific change.
Expression of CYP3A7, CYP3A4, Chimeric Enzymes, and Mutated Enzymes in Baculovirus
Baculovirus shuttle vectors, p2Bac (XhoI/XbaI) wild-type, chimeras, and mutated enzymes were cotransfected to SF-9 cells with viral DNA. Recombinant virus was constructed according to the instructions provided by the manufacturer (Invitrogen, Gaithersburg, MD). TN-5 cells were infected with the recombinant viruses in the presence of hemin (2 μg/ml). After 2 to 3 days, TN-5 cells were harvested, and microsomes were prepared by two different centrifugations (Kedzie et al., 1991). The expressed CYP3A enzymes were detected by Western blot analysis (Guengerich et al., 1982). The CYP3A content was determined by the CO difference spectrum (Omura and Sato, 1964). The reductase activity was measured by the method of Phillips and Langdon (1962). The protein concentration was determined according to the method of Lowry et al. (1951) using bovine serum albumin as the standard.
Assays for Enzyme Activities
The typical reaction mixture for enzyme assays consisted of 100 mM potassium phosphate buffer (pH 7.4), 0.1 mM EDTA, an NADPH-generating system (0.33 mM NADP+, 0.1 U of glucose-6-phosphate dehydrogenase, 8 mM glucose 6-phosphate, and 6 mM MgCl2), microsomes, substrate and desired amounts of purified cytochrome b5 and NADPH-P450 reductase in a final volume of 500 μl, except that 100 mM Tris-HCl buffer (pH 7.4) was used for the assay of metabolism of DHEA and its sulfate. The concentrations of DHEA, DHEA-3S, testosterone, EM, NVP, and TZM used as substrates were 100 μM. The contents of CYP3A microsomal enzymes were 10 pmol for the assay of DHEA, DHEA-3S, testosterone, EM, and NVP metabolism and 5 pmol for the assay of TZM metabolism. The reactions were started by the addition of the NADPH-generating system and were conducted for 30 min at 37°C with shaking under an aerobic condition. The reactions were linear up to 30 min when catalyzed by each CYP3A enzyme. Hydroxylations of DHEA and DHEA-3S at the 16α position were determined by the method described previously (Kitada et al., 1987; Nakamura et al., 2003). The determinations of 6β-hydroxylation of testosterone, N-demethylation of EM, 2- and 12-hydroxylations of NVP, and α- and 4-hydroxylations of TZM were performed according to the method described elsewhere (Miura et al., 1988; Nakamura et al., 2002).

Schematic representation of wild-type and chimeric CYP3As constructs used for the analysis of substrate specificity. The CYP3A7 sequence is shown in the hatched column, and the CYP3A4 sequence is shown in the white column. The numbers indicate amino acid residues.
Theoretical Calculations
Construction of the Model for Calculation. To examine the difference between CYP3A4 and CYP3A7, the three-dimensional structure of CYP3A7 was modeled on the basis of the crystal structure of CYP3A4 (PDB entry 1TQN) (Yano et al., 2004). The program used for modeling was Insight II/Homology (Accelrys Software Inc. San Diego, CA). The complex structure of CYP3A7 and chemical compounds was constructed as follows. 1) First, an O atom was perpendicularly placed on the Fe atom of the heme because the crystal structure does not contain oxygen above the heme (Fe O form) (Hata et al., 2001). Next, a substrate was docked into the active site of CYP3A7 in an orientation toward its metabolic site, and molecular mechanics (MM) calculations were performed. 2) Water model molecules (TIP3P model) (Jorgensen et al., 1983) were generated around the enzyme by the Monte Carlo method (Pearlman et al., 1995). The thickness of the water molecules was 11 Å, and there were ∼6000 water molecules in the model. After MM calculations, the interactions between substrates and amino acids in CYP3A7 were investigated.
O form) (Hata et al., 2001). Next, a substrate was docked into the active site of CYP3A7 in an orientation toward its metabolic site, and molecular mechanics (MM) calculations were performed. 2) Water model molecules (TIP3P model) (Jorgensen et al., 1983) were generated around the enzyme by the Monte Carlo method (Pearlman et al., 1995). The thickness of the water molecules was 11 Å, and there were ∼6000 water molecules in the model. After MM calculations, the interactions between substrates and amino acids in CYP3A7 were investigated.

Sequence alignment between CYP3A4 and CYP3A7. Helices are indicated by letters and β-sheets are indicated by numbers above the sequences. Asterisks show the positions of different amino acids in CYP3A4 and CYP3A7.
Computational Details. The computational program package used for MM calculations was AMBER 6 (Case et al., 1999). An all-atom force field was applied to the model structure (Weiner et al., 1986). Point charges on heme and substrates for use in MM calculations were determined by the RESP program included in AMBER 6. Electrostatic potentials and coordinates for use of the input of RESP were determined by quantum chemical calculations at the ab initio Hartree-Fock level. The basis set used was 6-31G**. The program package used for calculations was Gaussian 98 (Frisch et al., 2001). For point charges on the other residues and van der Waals parameters, the standard AMBER residue database and the AMBER Force Field Parameter File (parm94.dat), respectively, were used. The models of substrate-free and substrate-enzyme ternary complexes described in the previous section were fully minimized under the conditions shown above. The cutoff distance for non-bonded interaction was 12 Å. The energy minimization algorithm adopted was the steepest descent method (6000 steps) followed by the conjugate gradient method (2000 steps). The minimized structures are shown in Figs. 3 and 6.
Results
CYP3A7 Computer Modeling.Figure 2 shows different amino acids in CYP3A7 and CYP3A4. There are 56 different amino acids in all sequences, and the percentage of different amino acids is especially high in the helix A and F–G regions. We constructed the proposed structure of CYP3A7 on the basis of the CYP3A4 crystal structure (Fig. 3) to determine the reason for the metabolic differences between these two enzymes. The structure of model CYP3A7 was topologically similar to that of crystallized CYP3A4 (Williams et al., 2004; Yano et al., 2004). As in CYP3A4, helices F and G did not pass over the active site cavity in CYP3A7 (Fig. 3) because these helices are shorter. The model of CYP3A7 also proposed longer sequences between helices F and G that generally exhibit two additional helices F′ and G′. This region is located above and perpendicular to helix I in CYP3A7, and the outer surfaces of helices F′ and G′ are hydrophobic.
In addition to the structural similarity of CYP3A7 and CYP3A4, the proposed important amino acids in CYP3A7 (Ser180, Arg212, Phe304, Tyr307, Glu308, Thr309, Thr310, Ser311, and Asn451), which are present <5 Å from the substrate docked into the active site of CYP3A7 described under Materials and Methods, were the same as those in CYP3A4. That is, the difference between CYP3A7 and CYP3A4 could not be explained using only a homology model of CYP3A7. To clarify which sequence is more important for exhibiting the characteristics of CYP3A7, we made a chimeric enzyme constructed with CYP3A4 and CYP3A7 as shown in Fig. 1.
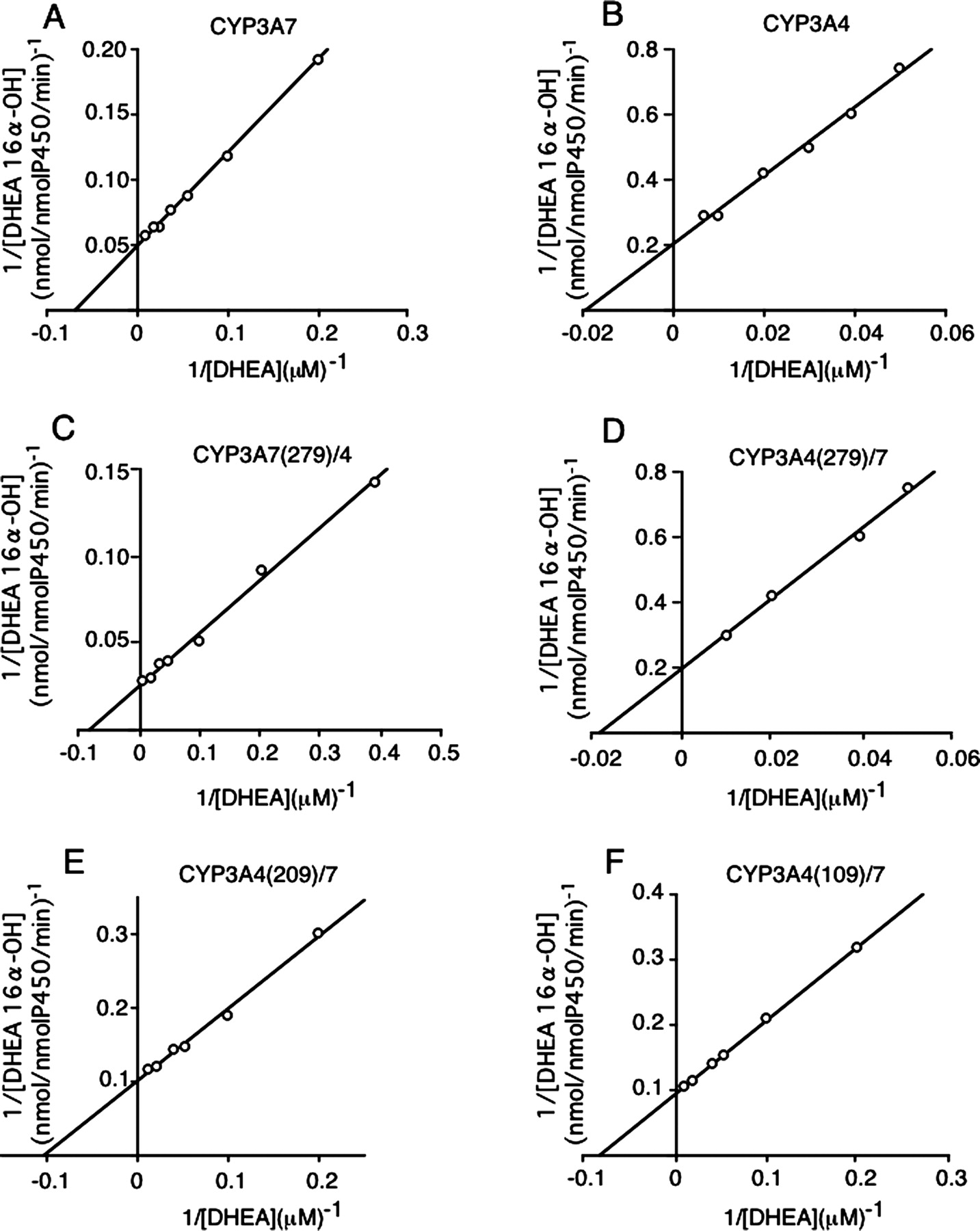
Kinetic Analysis of DHEA and DHEA-3S 16α-Hydroxylation by Chimeric CYP3A Enzymes. Kinetic parameters of DHEA and DHEA-3S 16α-hydroxylation by CYP3A7, CYP3A4, and chimeric CYP3A enzymes are shown in Fig. 4 and Table 2 and Fig. 5 and Table 3, respectively. In each assay, the relative ratios of CYP/reductase/b5 were 1:50:20 for CYP3A4 and CYP3A7 and 1:100:20 for chimeric enzymes, respectively. For DHEA and DHEA-3S 16α-hydroxylation by CYP3A7, Km values were almost the same. However, the Vmax of DHEA 16α-hydroxylation was 3 times greater than that for DHEA-3S 16α-hydroxylation. On the other hand, Vmax/Km values for DHEA and DHEA-3S 16α-hydroxylation by CYP3A4 were 10 times lower than those for 16α-hydroxylation by CYP3A7.
Kinetic parameters of DHEA 16α-hydroxylation by CYP3As and CYP3A chimeras expressed in TN-5 cells
Maximal velocity (Vmax) and apparent affinity constant (Km) were determined by Lineweaver-Burk plot analysis. DHEA 16α-hydroxylation was determined in three experiments over a DHEA concentration range of 2.5 ot 100 μM. S.D was <10% of the mean value.
Kinetic parameters of DHEA-3S 16α-hydroxylation by CYP3As and CYP3A chimeras expressed in TN-5 cells
Maximal velocity (Vmax) and apparent affinity constant (Km) were determined by Lineweaver-Burk plot analysis. DHEA-3S 16α-hydroxylation was determined in three experiments over a DHEA concentration range of 2.5 to 100 μM.

Three-dimensional structure of CYP3A7 determined by computer modeling. A, whole three-dimensional structure of CYP3A7. This structure is illustrated from the distal side of heme. B, active site structure of CYP3A7. Letters indicate helices. The heme group is shown in red. Helix F is shown in cyan, helix G is shown in green, and helix I is shown in magenta. Asn116 and Hid174 are shown in orange, Lys224, Lys244, and Lys262 are shown in yellow, Asn214 is shown in blue, and Phe304 is shown in black. The figures were produced using RasMol 2.7.1 (http://www.umass.edu/microbio/rasmol/).
When Vmax/Km of DHEA 16α-hydroxylation by CYP3A7 was assumed to be 1, Vmax/Km values of DHEA 16α-hydroxylation by CYP3A4, CYP3A7(279)/4, CYP3A4(279)/7, CYP3A4(209)/7, and CYP3A4(109)/7 were 0.06, 2.2, 0.06, 0.7, and 0.7, respectively. For DHEA-3S 16α-hydroxylation, when Vmax/Km by CYP3A7 was assumed to be 1, Vmax/Km values by CYP3A4, CYP3A7(279)/4, CYP3A4(279)/7, CYP3A4(209)/7, and CYP3A4(109)/7 were 0.02, 1.5, 0.02, 0.43, and 0.64, respectively. These results suggested that amino acids from Leu210 to Glu279 are important for the DHEA and DHEA-3S 16α-hydroxylation activities of CYP3A7.
To determine the reason for differences between substrate affinities and between metabolizing velocities of CYP3A7 and CYP3A4, DHEA and DHEA-3S were docked into the active site of the CYP3A7 model (Fig. 6, A and B). Both DHEA and DHEA-3S were stably kept by Ser180 and Phe304 in the active site. The distance between the O atom above the heme and the 16α site of DHEA or DHEA-3S was almost the same, suggesting that the distance between the O atom and both hydroxylation sites of the substrate might determine Km values for both reactions. However, the reason that the Km values of DHEA and DHEA-3S were almost the same despite a high polar site of DHEA-3S is still unclear. The sequences from Leu210 to Glu279, which were thought to be important for substrate specificity, were located on the position of the F–G helices, and this region was on the surface of CYP3A4. There are 16 different amino acids in CYP3A4 and CYP3A7 in this region (Fig. 2). We then made some one-point mutants converted from CYP3A7 to the corresponding CYP3A4 type amino acid by site-directed mutagenesis.
Activity of CYP3A7 Mutants. The sequence from Leu210 to Gln279 has frequent substitution of amino acids and includes 16 amino acid replacements (Fig. 2). In the sequence, amino acids showing the change in charge were chosen because change of charge might induce a variation in the affinity to substrates. Asn214, Lys224, Lys244, and Lys262 in CYP3A7 indicating uncharged (Asn214) and positive charge (Lys224, Lys244, and Lys262) show a big difference from Asp214, Thr224, Glu244, and Glu262 in CYP3A4 indicating negative charge (Asn214, Glu244, and Gly262) and uncharged (Thr224). Substitutions for each of the four polar residues to the corresponding amino acids found in CYP3A4 were made by mutagenesis of CYP3A7. The N116S mutant was used as a negative control because Asn116 does not locate in the sequence from Leu210 to Gln279 and N116S was not accompanied by a change of charge. H174D was prepared because a change of charge from positive to negative was included, although His174 does not locate in the sequence from Leu210 to Gln279. Whereas T250R and Q254R changed from uncharged to positive charged, mutants were not prepared successfully.
To determine whether the DHEA and DHEA-3S 16α-hydroxylation activities of the six CYP3A7 mutants were changed, the activities of the mutants were compared with those of CYP3A7 and CYP3A4 (Fig. 7). The DHEA 16α-hydroxylation activities of the mutants K224T and K244E were 23 and 17%, respectively, of the activity of CYP3A7 and were similar to the CYP3A4 activity. In contrast, the activities of the mutants H174D and N214D were increased to 151 and 133%, respectively, of the activity of CYP3A7. The activities of the mutants N116S and K262E were almost the same as the activity of CYP3A7. Km values of K224T and K244E were 39.2 and 48.6 μM, respectively, and Vmax values were 26.0 and 9.9 nmol/nmol of P450/min, respectively. These results indicate that Km values of the mutants were increased compared with that of CYP3A7. That is, the mutants had converted to an enzyme showing low affinity for DHEA metabolism.
DHEA-3S 16α-hydroxylation activity of the mutant K244E was 8% of that of CYP3A7, and the activity of the mutant K224T was not detectable. The activities of the mutants N116S, H174D, N214D, and K262E had not conspicuously changed. Km and Vmax were not determined because the activities of the mutants were too low or not detectable.
Because the mutated amino acid of CYP3A7 was designed to change the amino acid of CYP3A4, it was possibly expected that the characteristics of CYP3A4 may express in place of the characteristics of CYP3A7. We measured testosterone 6β-hydroxylation, EM N-demethylation, NVP 2- and 12-hydroxylation, and TZM α- and 4-hydroxylation activities of CYP3A4 (Fig. 8). Figure 8 shows that the metabolic activities of each of the four substrates by CYP3A7 were lower than those activities by CYP3A4. Metabolic activities of all substrates of both K224T and K244E mutants were greatly decreased from 51% of those of CYP3A7 wild type to “not detectable.” The activities of mutants (N116S, H174D, N214D, and K262E) were not markedly changed. These results suggested that Lys224 and Lys244 were important amino acids to indicate the function as monooxygenases of CYP3A7.

Lineweaver-Burk plot of DHEA 16α-hydroxylation by CYP3As and CYP3A chimeras expressed in TN-5 cells. A, CYP3A7; B, CYP3A4; C, CYP3A7(279)/4; D, CYP3A4(279)/7; E, CYP3A4(209)/7; F, CYP3A4(109)/7. DHEA 16α-hydroxylation was determined in three experiments over a DHEA concentration range of 2.5 to 100 μM.
Change in Distances between Amino Acids by Mutations. To determine the reason that K224T and K244E mutants lost the metabolic activities of all substrates used in this study (DHEA, DHEA-3S, testosterone, erythromycin, nevirapine, and triazolam), the distance between two atoms in amino acids of CYP3A7 wild type and in amino acids of mutants were determined by theoretical calculations (Table 4). In the wild type, the Lys224 residue did not make a hydrogen bond, but in the K224T mutant, the Thr224 residue made hydrogen bonds with the carbonyl O atom of the Val220 residue. In the K244E mutant, the distances between the carboxyl O atom of the Glu244 residue and Arg243 or Ser247 residues were shorter than those distances in the wild type. These results suggested that the three-dimensional structure could be changed by mutation.
Changes in distance between CYP3A7 wild-type and mutants
Homology models of K224T, K244E, and K262E were constructed by replacing Lys224, Lys244, and Lys262 in CYP3A7 wild-type (WT) with Thr224, Glu244, and Glu262. These models were fully minimized under conditions described under Materials and Methods.
Discussion
To clarify the differences between CYP3A7 and CYP3A4, chimeric enzymes were constructed from CYP3A7 and CYP3A4. The kinetic parameters of DHEA and DHEA-3S 16α-hydroxylation by CYP3A7, CYP3A7(279)/4, CYP3A4(209)/7, and CYP3A4(109)/7 were similar. That is, P450 chimeras including the sequences from Leu210 to Glu279 amino acids of CYP3A7 showed high levels of specific activity for DHEA and DHEA-3S 16α-hydroxylation compared with CYP3A4(279)/7, indicating that the amino acid residues from Leu210 to Glu279 may be important to express the specificity for substrates as CYP3A7.
The three-dimensional structure of CYP3A7 determined by computer modeling was almost the same as that of CYP3A4. Helix I in CYP3A7 was also closest to heme. Phe304 is one of the compartments of the active site and contributed to maintenance of DHEA and DHEA-3S (Fig. 6, A and B). It has been reported that Phe304 in CYP3A4 also contributed to maintenance of nevirapine and carbamazepine (Torimoto et al., 2003). The benzene ring of Phe304 may behave to cap the active site of both CYP3A7 and CYP3A4 for maintaining substrate. From these results, the topology of the active site did not explain the difference between CYP3A7 and CYP3A4.

Lineweaver-Burk plot of DHEA-3S 16α-hydroxylation by CYP3As and CYP3A chimeras expressed in TN-5 cells. A, CYP3A7; B, CYP3A4; C, CYP3A7(279)/4; D, CYP3A4(279)/7; E, CYP3A4(209)/7; F, CYP3A4(109)/7. DHEA-S 16α-hydroxylation was determined in three experiments over a DHEA concentration range of 2.5 to 100 μM.
The sequence from Leu210 to Glu279 was present on the surface of the F–G loop in the three-dimensional structure of CYP3A7. Furthermore, the space of the active site and the expected entrance of the active site of CYP3A7 were almost the same sizes as those of CYP3A4. Taken together, the results suggest that CYP3A7 could recognize specific substrates using the amino acid sequence of the surface structure such as the strict gate for entering into the active site. It has been reported in CYP2C8 (Schoch et al., 2004) and CYP3A4 (Williams et al., 2004) that those conformations of the F–G loop would contribute for entering substrates into the active site by X-ray crystallography. Ekroos and Sjögren (2006) reported that ketoconazole and erythromycin differentially alter the conformation of the F–G helices of CYP3A4 and shift the coordinates of amino acid residues. Amino acids from Leu210 to Glu279 of CYP3A7 correspond to the helix F–G region (Fig. 2), suggesting that ligand binding could alter the conformation of this region in CYP3A7. As a result, amino acid differences between CYP3A4 and CYP3A7 in the helix F–G region could contribute to the substrate specificity of CYP3A7. Generally, typical substrates for CYP3A4 can also be metabolized by CYP3A7, but the affinity to CYP3A7 was much lower than that to CYP3A4. In other words, although the sequence from Leu210 to Glu279 of CYP3A7 could not recognize specific substrates for CYP3A4, CYP3A7 could catalyze the substrates when the substrates enter the active site by chance.

DHEA (A) and DHEA-3S (B) docked into the active site of CYP3A7 in an orientation conductive to their 16α-hydroxylation. The heme group is shown in red. DHEA and DHEA-3S are shown in blue. Ser180 and Phe304 are shown in green.

DHEA (A) and DHEA-3S (B) 16α-hydroxylation activities of CYP3A7, CYP3A4, and CYP3A7 mutants. The concentrations of DHEA and DHEA-3S used in this study were 100 μM. The columns represent the average values obtained from three determinations. Significantly different from the value of the activity of CYP3A7 at *p < 0.05 and ***p < 0.005, respectively. N.D., not detectable.
The mutants of K224T and K244E had lost DHEA and DHEA-3S 16α-hydroxylation activities. These mutants are located on the F–G helix of CYP3A7. The calculated distances between mutated amino acids and the closest atoms in the three-dimensional structure of CYP3A7 were suggested to be changeable (Table 4). In the K224T mutant, the shape of the turn between the helices F and G might be changed by hydrogen bonds between Thr224 and Val220 residues. In the K244E mutant, the distance between Glu244 and Arg243 or Ser247 was closer, with the result that the structure of the helix G could be a conformational change. It is expected that the structural change of the entrance of the active site may contribute to the loss of the activity.
The Km of DHEA-3S was almost the same as that of DHEA, although DHEA-3S was hydrophilic because of the presence of a sulfate group. How can DHEA and DHEA-3S interact with CYP3A7? Our study shows by computer modeling that the region of helices F–G must be important for indicating enzyme activity and that the F–G loop passed over helix I. Then, we assumed that the F–G loop is important for affinity of substrates for CYP3A7. It is expected that a mutation in the F–G region might alter substrate binding affinity. One possibility is a direct change in the affinity, which means interaction between side chains of amino acid and substrates. For example, it is expected that the side chain of lysine might interact with the sulfate of DHEA-3S because the side chain of lysine shows a positive charge and the sulfate shows a negative charge. We expected that the mutants would lose their affinity to DHEA-3S and then the activity of DHEA-3S 16α-hydroxylation might be lost (Fig. 7B) as K224T and K244E lost the positive charge. Furthermore, it has been reported that pregnenolone-3-sulfate, 17α-hydroxypregnenolone-3-sulfate, and DHEA-3S increased the activity of carbamazepine 10,11-epoxidation by expressed CYP3A7 (Nakamura et al., 2003). Because lysine is polar and positively charged, it is expected that the positive charge on the F–G helix could interact with the negative charge of sulfate. These findings suggested that lysine residues in the F–G region might induce direct interaction with substrates in CYP3A7.
Another possibility for a change in enzymatic activity by mutation is an indirect effect in mutants. We proposed that conformation of the F–G loop including lysine in CYP3A7 could recognize the structure of DHEA, although DHEA does not have a negative charge like DHEA-3S. Because the conformation of the F–G loop might change the three-dimensional structure by mutation, the mutants decreased the activity of DHEA 16α-hydroxylation. The change in distance between Thr224 and Val220 in the K224T mutant (Table 4) might support this idea. On the other hand, affinity of testosterone to CYP3A4 is much higher than that to CYP3A7 (Ohmori et al., 1998), and Thr224 in K224T and Glu244 in K244E did not contribute to testosterone 6β-hydroxylation. These data also suggested that conformation of the F–G loop in CYP3A4 could recognize the testosterone structure, although the topology of the F–G loop in CYP3A4 is not greatly different from the topology of the F–G loop in CYP3A7 by computer modeling. It has been reported that the conformation of the F–G loop might be flexible (Williams et al., 2004; Ekroos and Sjögren, 2006). These data suggested that the amino acid differences in the F–G region between 3A4 and 3A7 result in different conformations of this region, leading to differences in substrate selectivity.

Testosterone 6β-hydroxylation (A), EM N-demethylation (B), NVP 2- or 12-hydroxylation (C), and TZM α- or 4-hydroxylation (D) activities of CYP3A7, CYP3A4, and CYP3A7 mutants. The concentrations of substrates used in this experiment were 100 μM. The columns represent the average values obtained from duplicate determinations.
Unfortunately, the actual structure of CYP3A7 as determined by crystallography has not been reported. However, we will try to clarify this question in our next study.
Acknowledgments
The computations were carried out by a DRIA system at the Graduate School of Pharmaceutical Sciences, Chiba University.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.011304.
-
ABBREVIATIONS: P450, cytochrome P450; DHEA-S, dehydroepiandrosterone 3-sulfate; DHEA, dehydroepiandrosterone; EM, erythromycin; NVP, nevirapine; TZM, triazolam; bp, base pair(s); AA, amino acids; MM, molecular mechanics.
- Received June 3, 2006.
- Accepted December 15, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}