


Abstract
DB289 (pafuramidine maleate; 2,5-bis[4-(N-methoxyamidino)phenyl]furan monomaleate) is a prodrug of DB75 (furamidine dihydrochloride; 2,5-bis(4-guanylphenyl)furan dihydrochloride), an aromatic dication related to pentamidine that has demonstrated good efficacy against African trypanosomiasis, Pneumocystis carinii pneumonia, and malaria, but lacks adequate oral availability. The pharmacokinetics and metabolism of 14C-DB289 have been investigated in rat and monkey after oral and intravenous administration. Oral doses were well absorbed (∼50-70%) and effectively converted to DB75 in both species but subject to first-pass metabolism and hepatic retention, limiting its systemic bioavailability to 10 to 20%. Clearance of DB289 approximated the liver plasma flow and its large volume of distribution was consistent with extensive tissue binding. Plasma protein binding of DB289 was 97 to 99% in four animal species and humans, but that of DB75 was noticeably less and more species- and concentration-dependent. Together, prodrug and active metabolite accounted for less than 20% of the plasma radioactivity after an oral dose, but DB75 was the major radiochemical component in key organs such as brain and liver and was largely responsible for the persistence of 14C in the body. The predominant route of excretion of radioactivity was via the feces, although biliary secretion was not particularly extensive. High-performance liquid chromatography and liquid chromatography-mass spectrometry investigations showed that the formation of DB75 from the prodrug involved the sequential loss of the two N-methoxy groups, either directly or by O-demethylation followed by reduction of the resulting oxime to the amidine. It was estimated that almost half of an oral dose of DB289 to rats and about one-third of that to monkeys was metabolized to DB75.
Historically, effective clinical treatment of human African trypanosomiasis, visceral leishmaniasis, and, more recently, Pneumocystis carinii pneumonia (PCP; the cause of death of many AIDS patients), has been notoriously difficult (e.g., Barrett, 1999; Walzer, 2000). Vaccination is not yet possible for these conditions, and first-line drug treatments such as pentamidine and suramin require parenteral administration with their attendant severe toxic side effects. Both drugs possess very poor oral bioavailability, and neither crosses the blood-brain barrier in sufficient concentrations to act against the organisms present in the CSF and brain. Treatment of late-stage disease requires the use of even more toxic (sometimes fatal) drug therapy. Malaria is another tropical disease that causes nearly two million deaths a year, almost all of which are caused by Plasmodium falciparum, against which there is also currently no effective vaccine (Guerrant, 1999). As resistance to first-line antimalarial treatments such as chloroquine and mefloquine increases (Sherwin, 1998), the use of combination therapies such as atovaquone/proguanil (Malarone) is becoming necessary. It is apparent, therefore, that development of new drugs for these tropical diseases with new mechanisms of action is a matter of some urgency.
Pentamidine (Fig. 1) has been used as a first-line treatment for infections such as human African trypanosomiasis and drug-resistant leishmaniasis for more than half a century, and in the treatment of AIDS-related PCP for approximately 20 years, despite its loss of activity when administered orally and its toxicity when administered intravenously. The diamidine structure of pentamidine has been shown to be essential for its antimicrobial activity, which results from binding to DNA (Das and Boykin, 1977), although the exact mechanism of action is currently unknown.

Chemical structures of synthetic reference compounds.
Investigations have been under way for some time to modify the pentamidine molecule to provide an analog that retains the antipneumocystis and antitrypanosomal activities of the original, but not its side effects, yet possesses good oral bioavailability. From the many structurally related compounds that have been synthesized and tested to date, the dicationic agent DB75 (furamidine dihydrochloride; 2,5-bis(4-guanylphenyl)furan dihydrochloride) has emerged as the most promising from the therapeutic/toxicity aspect when administered intravenously, but it lacks the desired property of good oral activity because of inadequate bioavailability. However, its bis(N-methoxy-amidoxime) derivative, DB289 (pafuramidine maleate; 2,5-bis[4-(N-methoxyamidino)phenyl]furan monomaleate), acts as a prodrug for DB75 (Boykin et al., 1998) and exhibits high oral activity against the organisms in question as well as reduced acute toxicity in the animal models of PCP and trypanosomiasis (Boykin et al., 1996). In addition, DB289 has been shown to possess good in vivo activity against malaria in the mouse and in vitro activity against various Plasmodium strains, such that it is currently undergoing clinical trials for all these disease conditions. Normal human erythrocytes are impermeable to diamidine drugs, but those infected with P. falciparum are not, by virtue of a new porous pathway induced in the host cell membrane by the parasite itself (Ginsburg et al., 1983; Stead et al., 2001).
To support existing toxicity studies on DB289 in laboratory animals and clinical trials in humans, the pharmacokinetics, absorption, distribution, biotransformation, and excretion of the 14C-radiolabeled prodrug, including its postabsorption conversion to the active metabolite, have now been investigated in the rat and cynomolgus monkey, the principal species used in its safety studies.
Materials and Methods
Chemicals. DB289 and DB75, together with the corresponding 2H8-labeled analogs used as internal standards for the LC-MS/MS bioanalytical method, were provided through Immtech Pharmaceuticals, Inc. (Vernon Hills, IL), as were synthetic samples of the following potential metabolites: DB290, DB775, and DB810 (see Fig. 7). 14C-DB289 and 14C-DB75 (Fig. 1) were obtained from commercial radiosynthesis laboratories and stored in the dark at approximately -20°C. Where necessary, 14C-DB289 was repurified as the free base to a radiochemical purity of at least 97% at Huntingdon Life Sciences (Huntingdon, UK) by flash chromatography on a column of silica gel and converted back to the monomaleate salt, before storage in methanol solution in the dark at -20° or -70°C. Ultima Gold Universal and Permafluor E+ scintillators and Carbo-Sorb E carbon dioxide absorbers were obtained from Packard Biosciences UK (Pangbourne, UK). All other chemicals and solvents were purchased from commercial suppliers and were of analytical or HPLC grade.
Animals. All procedures involving animals were performed in compliance with the relevant guidelines issued by the United Kingdom Home Office, from whom Project and Personal Licenses were obtained as specified by the Animals (Scientific Procedures) Act 1986 following the European Union Directive 86/609/EEC, which requires ethics review of the Licenses and their Amendments. Such animal studies conducted at Huntingdon Life Sciences are subject to review for approval by the Huntingdon Life Sciences Ethical Review Process Committee. All animals were obtained from accredited commercial suppliers.
Rats were housed in standard battery cages or glass metabolism cages (as appropriate) and allowed free access to standard laboratory diet and water. Body weights at the time of dosing were 166 to 220 g for nonpregnant rats, 212 to 271 g for pregnant females, and 321 to 389 g for nursing dams. Cynomolgus monkeys (Macaca fascicularis) were housed in stainless steel metabolism cages during the sample collection periods. Food was normally offered to the monkeys twice daily, but on the day of drug administration the morning offering was withheld for 2 h after dosing; drinking water was freely available at all times. Body weights at the time of dosing were in the range 2.15 to 3.65 kg.
14C-DB289 was administered as the monomaleate salt, but dose levels and radioactivity concentrations are expressed in terms of the free base. Since the test compound is light-sensitive, dose formulations were prepared and administered and biological samples were analyzed (as appropriate) under subdued (yellow) lighting conditions.
Oral doses of 14C-DB289 were prepared as suspensions in 0.5% (w/v) aqueous carboxymethylcellulose containing 0.1% Tween 80 at concentrations appropriate to the dose level and administered by oral gavage at a dose volume of 5 or 10 ml/kg. Intravenous (i.v.) doses were formulated in a “parenteral premix” prepared from Vitamin E-TPGS (d-α-tocopheryl polyethylene glycol 1000 succinate) and citric acid in a small volume of aqueous ethanol diluted suitably with 5% (w/v) dextrose, and administered by bolus injection into a lateral caudal vein (rats) or a femoral vein (monkeys) at a dose volume of 1 ml/kg. All dose formulations were freshly prepared on the day of administration. The radiochemical purity of 14C-DB289 in each dose preparation was determined by HPLC and found to be at least 97%. The total drug concentration in the formulation was calculated from its specific activity and measured radioactivity concentration. Typical radioactive doses were 20 to 30 μCi for rats and 120 to 170 μCi for monkeys.
Studies in Rats. Unless otherwise stated, rats used in these studies were Sprague-Dawley albino animals, as this strain was used in toxicity studies. Partially pigmented rats used to investigate melanin binding of the test compound were Lister Hooded animals.
Pharmacokinetics. Single 1 mg/kg i.v. and 1 and 7.7 mg/kg oral doses of 14C-DB289 were each administered to four subgroups of male and female rats (n = 3 per sex per subgroup), and then serial blood samples were taken up to 72 h postdose. Terminal blood samples were obtained from each animal in the four subgroups at 96, 120, 144, and 168 h postdose, respectively. Plasma was separated by centrifugation and aliquots were taken immediately for measurement of radioactivity; the remainder was stored in the dark at about -70°C pending analysis for DB289 and DB75. Distribution. Single 10 mg/kg oral doses of 14C-DB289 were administered to six male and six female rats; then, the animals were euthanized in male/female pairs at selected times up to 120 h after dosing. The carcasses were immediately prepared for examination by QWBA. In addition, 12 partially pigmented male rats received single 10 mg/kg oral doses of 14C-DB289, and the animals were sacrificed in pairs from 3 to 504 h after dosing, when selected tissues/organs (including the eyes and skin) were removed for radioassay following combustion. For microautoradiography, single 10 mg/kg oral doses of 14C-DB289 were administered to one male and one female rat, and both animals were euthanized at 120 h after dosing. The liver and kidneys were removed and one-half of each was fixed in formalin. The other half was snap-frozen in isopentane cooled with liquid nitrogen.
To investigate blood cell binding in vivo, single 10 and 50 mg/kg oral doses and 1 mg/kg i.v. doses of 14C-DB289 were administered to separate groups of seven male and seven female rats. Serial blood samples were obtained at 6, 12, 24, and 48 h after dosing, and terminal samples were collected at 96 h. Portions of each were retained as whole blood, and plasma was harvested from the remainder. Blood/brain transfer of drug-related material was examined by administering single 7.5 mg/kg oral doses of 14C-DB289 to nine male and nine female rats and, then, after the animals were sacrificed in groups of three per sex at 2, 4, and 6 h postdose, taking samples of CSF and blood and the whole brain from each carcass.
Excretion/mass balance. Single 10 and 50 mg/kg oral doses and 1 mg/kg i.v. doses of 14C-DB289 were administered to separate groups of three male and three female rats. Urine (into solid CO2-cooled receivers) and feces were collected for 7 days after dosing. The rats were then sacrificed, and the liver and gastrointestinal tract were removed and retained. Separate groups of three male and three female rats each with the common bile duct surgically cannulated received single 10 mg/kg oral or 1 mg/kg i.v. doses of 14C-DB289. Bile, urine, and feces were collected for 48 h after dosing, during which time replacement bile salts were infused via an intraduodenal cannula. Each animal was euthanized at 48 h, and the liver and gastrointestinal tract were removed and retained.
Placental transfer/milk secretion. Single 10 mg/kg oral doses of 14C-DB289 were administered to six pregnant rats on day 15 of gestation. Animals were euthanized at selected times up to 120 h after dosing; then a terminal blood sample was taken and the carcasses were prepared for QWBA. After pregnancy, a group of 15 rats (dams with suckling pups) received single 10 mg/kg oral doses of 14C-DB289 approximately 12 days postparturition. Milk samples were collected directly from mammary tissue at five selected times up to 24 h after dosing and approximately 15 min after intraperitoneal administration of oxytocin. After collection of the milk sample, each dam was sacrificed and a terminal plasma sample obtained.
Repeat-dose studies. Repeated daily 3.8 mg/kg oral doses of 14C-DB289 were administered to 11 male and 5 female rats. Individual male rats were taken for QWBA after sacrifice at 24 h after 1, 7, and 14 daily doses and at several times after the final 21st daily dose, whereas individual female rats were taken for QWBA at 24 h after 1, 7, 14, and 21 daily doses. Urine and feces were collected from the rats terminated at 24 h after a single dose and at 24-h intervals from those scheduled for termination at 72 and 168 h after the 21st daily dose.
Studies in Monkeys. Single 1 and 5 mg/kg oral and 1 mg/kg i.v. doses of 14C-DB289 were administered to separate groups of three male and three female cynomolgus monkeys. Serial blood samples were taken at suitable times up to 96 h postdose, and plasma was harvested from all or part of each sample. Urine (into solid CO2-cooled receivers) and feces were collected at suitable intervals for 7 days after dosing. Cage debris (mainly uneaten food contaminated with excreta) was also collected on a daily basis; then the cages were rinsed with water and the washings retained.
At 43 days after dosing, each animal from all three groups of monkeys was euthanized, and the liver, kidneys, lungs, heart, and gastrointestinal tract were removed for analysis, together with samples of bile and CSF from the animals given the 1 mg/kg oral doses. Urine and feces samples were also collected for a 24-h period commencing a few days before termination. All biological samples generated during the course of this study were stored at approximately -20°C or -70°C, as indicated, apart from whole blood, which was stored at approximately +4°C.
Sample Analysis. Concentrations of DB289 and DB75 in the plasma of rats and monkeys were determined by an LC-MS/MS bioanalytical method developed and validated at Tandem Labs (Salt Lake City, UT; Trendler et al., 2000). Plasma samples were transferred from Huntingdon Life Sciences to Tandem Labs in isothermal boxes cooled with solid CO2. In brief, the analytical method comprised solid-phase extraction of plasma and simultaneous determination of the two analytes in the extracts using an API 3000 liquid chromatographtandem mass spectrometer (MDS/Sciex; Applied Biosystems, Foster City, CA) operating in the multiple reaction monitoring mode with 2H8-DB289 and 2H8-DB75 as internal standards. The lower limit of quantification was 0.25 ng/ml for both analytes.
Radioactivity concentrations were determined by liquid scintillation analysis using Wallac automatic liquid scintillation counters equipped with external standardization. Radioactivity in plasma, urine, cage washings, bile, rat carcass digests, and HPLC fractions was measured by direct counting after mixing with Ultima Gold Scintillator. Rat carcasses were solubilized at 55°C overnight in a solution of Triton X-405 and sodium hydroxide in aqueous methanol. Where necessary, tissues were homogenized with or without the prior addition of distilled water. Feces and cage debris were homogenized with distilled water (1:1 w/v). Weighed portions of whole blood and homogenized feces and tissues were burned in oxygen using a sample oxidizer. The products of combustion were absorbed in Carbosorb E and mixed with Permafluor E+ scintillator.
Pharmacokinetic Data Processing. Pharmacokinetic parameters were determined by standard noncompartmental methods using the program WinNonlin Pro (version 3.1; Pharsight Corporation, Mountain View, CA). Maximum plasma concentrations (Cmax) and their time of occurrence (Tmax) were the observed values. Areas under the mean plasma concentration-time curve up to the time of the last quantifiable sample (AUCt) were calculated using the linear trapezoidal rule. In the calculation of AUCt values after i.v. dosing, the mean concentration at time 0 (C0) was estimated from the first two sampling times by back-extrapolation using log-linear regression analysis. The area under the plasma concentration-time curve extrapolated to infinite time (AUC∞) was calculated as AUCt + (Clast/k), where Clast is the predicted concentration at the last quantifiable sampling time and k is the elimination rate constant, determined by linear regression of the terminal log-linear phase of the concentration-time curve. Terminal half-life (t½) was calculated as ln2/k, total body clearance (CL) as Dose/AUC∞, and the volume of distribution at steady state (Vss) as Dose × AUMC/AUC 2∞, where AUMC is the area under the first moment of the concentration-time curve. Oral bioavailability (F) for DB289 was obtained from [AUC(oral)/Dose(oral)]/[AUC(intravenous)/Dose(intravenous)]. When AUC∞ could not be adequately calculated, AUCt was used instead, where t was the same for both treatments.
Plasma Protein Binding. Plasma protein binding of 14C-DB289 and 14C-DB75 in mouse, rat, rabbit, cynomolgus monkey and human in vitro was determined by ultracentrifugation, since preliminary studies showed that the test compounds were both subject to unacceptable levels of nonspecific binding when analyzed by ultrafiltration or equilibrium dialysis. Each radiolabeled analyte was added to aliquots of pooled control plasma obtained from stock animals or healthy human volunteers over a range of concentrations that encompassed those found in toxicity or clinical studies. After incubating at 37°C for 10 min, duplicate 1-ml aliquots of fortified plasma in polycarbonate tubes were centrifuged in a prewarmed rotor at 150,000g at 37°C for 16 h using a Beckman Coulter (Fullerton, CA) XL-90 or L8-70M ultracentrifuge. Weighed aliquots of each supernatant, taken from halfway between the supernatant surface and the pellet, were then mixed with scintillator for measurement of radioactivity. Radioactivity was also measured in the remaining contents of the tube after sonication to resuspend the pellet and rinsing the tubes with buffer. This enabled the extent of protein binding to be corrected for nonspecific binding to the tubes, which in the event proved to be <8% for both ligands. The radiochemical purity of the two test compounds before and after the 16-h centrifugation period was >90% by HPLC.
Tissue Distribution by QWBA in the Rat. Frozen rat carcasses were embedded in a block of 2% (w/v) aqueous carboxymethylcellulose at ca. -80°C. Samples of whole blood containing different concentrations of radioactivity (14C-glucose) were placed into holes drilled into the block for construction of a calibration line (nominal 1-1000 nCi/g blood). The block was sectioned in a Bright Instruments (Huntingdon, UK) SL684 cryomicrotome, and sagittal sections (nominal 30-μm thickness) were obtained at six levels through the carcass. The freeze-dried sections were exposed to phosphor-imaging plates for 7 days and scanned using a Fuji BAS2000 Bio-image Analyser. The electronic images were quantified using Seescan densitometry software (LabLogic Systems Ltd., Sheffield, UK). Concentrations of radioactivity were calculated by reference to a calibration line constructed from the blood standards on each section. The whole-body autoradiography techniques used in this study were based on those developed by Ullberg and Larsson (1981).
Microautoradiography. To investigate the cellular distribution of radioactivity in the liver and kidneys of treated rats, frozen sections (8 μm thick) obtained using a Bright Instruments SL684 cryomicrotome were transferred to microscope slides pretreated with Ilford K5 photographic emulsion. The slides were then stored in light-free boxes at about -20°C for 9 (liver) or 30 days (kidney). The slides were developed and fixed, then dried in air and counter-stained with 1% nuclear fast red for 1 min, dehydrated, cleared, and mounted.
The formalin-fixed samples of liver and kidney were processed to paraffin wax on a Shandon Hypercenter. Sections (4 μm thick) obtained using a Leica-Jung M2035 cryomicrotome were transferred to microscope slides and allowed to dry overnight. The slides were immersed in Ilford K5 photographic emulsion, which was allowed to gel, and then stored in light-free boxes at about +4°C for 83 (liver) or 36 days (kidney), before development.
Centrifugal Fractionation of Liver and Kidney Cells. The subcellular localization of radioactivity within the livers and kidneys of animals used for metabolite profiling (see below) was investigated by a centrifugal fractionation technique. A known weight of tissue (ca. 2-3 g) was homogenized in Tris/sucrose buffer (4 vol), then centrifuged at 260g for 10 min. The supernatant was removed from the nuclear pellet and centrifuged at 12,500g for 7 min. This supernatant was removed from the pellet and centrifuged at 105,000g for 60 min, to provide the cytosol supernatant and microsomal pellet. The pellet and supernatant were weighed at each stage and their radioactivity concentrations measured. Microsomal protein concentrations were determined using the method of Lowry et al. (1951).
Metabolite Profiles. Urine, feces, bile, plasma, liver, kidney, brain, and lung from rats and/or monkeys administered oral or i.v. doses of 14C-DB289 were analyzed by HPLC, and the separated radiometabolites were quantified using an on-line radioactivity detector or by fraction collection/liquid scintillation counting (depending on the sample radioactivity concentration). The liquid chromatograph comprised a Thermo Fisher Scientific (Waltham, MA) P4000 Quaternary Gradient pump coupled to an SCM1000 Vacuum Membrane Degasser and a UV2000 variable wavelength UV absorbance detector (λ 362 nm), in series with a β-RAM radioactivity detector (LabLogic), which incorporated a solid scintillant flow-through cell. Simultaneous UV and radio-chromatogram data were acquired and processed using on-line Laura software (v1.4a; LabLogic). Chromatography was performed in the reversed-phase mode using a Zorbax Bonus RP analytical column (15 cm × 4.6 mm i.d.; Agilent Technologies, Palo Alto, CA) with a Zorbax Bonus RP guard column. Gradient elution conditions were employed with solvent A [aqueous ammonium formate (15 mM)/formic acid (35 mM)] and solvent B [80% acetonitrile and 20% aqueous ammonium formate (15 mM)/formic acid (35 mM)]. Solvent A was programmed from 80% to 70% over 15 min, then to 55% during the next 15 min, to 30% during the next 9 min, and finally to 0% during 1 min. After holding at these final conditions for 5 min, the proportion of solvent A was returned to its original value of 80% during a further 5 min. Under these conditions and with a constant flow rate of 1 ml/min, DB289 and DB75 eluted with typical retention times of 6 and 41 min, respectively.
Urine was analyzed directly, i.e., after centrifugation to remove any particulate matter. Feces, liver, kidney, brain, and lung samples were homogenized with distilled water (1 vol), then extracted three times with methanol and three times with methanol containing 1M HCl (8:1, v/v), centrifuging after each extraction. The combined supernatants were reduced in volume under nitrogen at room temperature and an aliquot was mixed with mobile phase A before analysis. Plasma samples were extracted three times with acetonitrile and the combined extracts reduced in volume before mixing with mobile phase A for analysis. Aliquots of bile were mixed with mobile phase A and applied to preconditioned Nexus solid-phase extraction cartridges. The cartridges were washed with mobile phase A, and the radioactivity eluted first with mobile phase B, then with methanol/1 M HCl (99:1 v/v). The combined extracts were taken to dryness and the residue dissolved in mobile phase A before analysis. Selected biological samples were also analyzed after dilution with acetate buffer (0.2M; pH 5) and incubation with β-glucuronidase/arylsulfatase at 37°C for approximately 16 h. In general, sample extraction efficiencies were greater than 85% and column recoveries were at least 90%.
Metabolite Identification. Initial identification of the radioactive components present in the tissues and excreta of rats and monkeys dosed with 14C-DB289 was performed by HPLC with cochromatographic comparison with the available synthetic reference compounds. Confirmation of the principal structures was subsequently achieved by tandem liquid chromatography-mass spectrometry (LC-MS and/or LC-MS/MS). The chromatography conditions used for these analyses were the same as those for metabolite profiling, but Shimadzu (Kyoto, Japan) hardware was used (i.e., two LC-10AD pumps, an SCL10A controller, and an SIL-10A autosampler). The mass spectrometer was a Thermo Fisher Scientific TSQ7000 operating in conjunction with a Gateway E-4200 data system and Xcalibur software (v2; Thermo Fisher Scientific). The HPLC eluate first passed through a Shimadzu UV detector and then a splitter, where the majority was diverted into a LabLogic β-RAM radioactivity detector and the remainder into the mass spectrometer for electrospray ionization in the positive ion mode (ESI+). The spray voltage and the capillary temperature of the ESI source were set at 4.5 kV and 250°C, respectively. Nitrogen was used as sheath gas at a head pressure of 66 psi and as auxiliary gas at ca. 10 units.
Candidate molecular ions in the Q1 region were identified from the LC-MS data, and particular ions were selected for MS/MS analysis in product ion scan mode. Each precursor ion chosen was fragmented by collision-induced dissociation in the Q2 region with argon gas at ca. 2 mTorr. The product ion spectrum was recorded by scanning the Q3 region across a mass range from m/z 10 to several mass units above the precursor ion mass at a scan rate of 1 scan/s.
Results
Pharmacokinetics. It is important to note that, due to the study designs, i.e., the small number (three) and different animals used in each group of monkeys and the composite nature of the rat data resulting in single AUC values for each group, it was considered that there were insufficient animals to provide sufficient power for a meaningful statistical analysis of the plasma pharmacokinetic parameters to be performed. Basic pharmacokinetic data for the prodrug DB289 and its active metabolite DB75 in male and female rats and monkeys (including standard deviations, where appropriate) are given in Table 1, and plasma concentration-time curves for male animals only after single 1 mg/kg i.v. and oral doses of 14C-DB289 are illustrated in Figs. 2 (rats) and 3 (monkeys).
Plasma pharmacokinetic parameters (mean ± S.D.) of the prodrug DB289 and its active metabolite DB75 after single intravenous and oral doses of 14C-DB289 to rats and cynomolgus monkeys
Concentration and AUC data for DB289 and DB75 refer to the free bases.
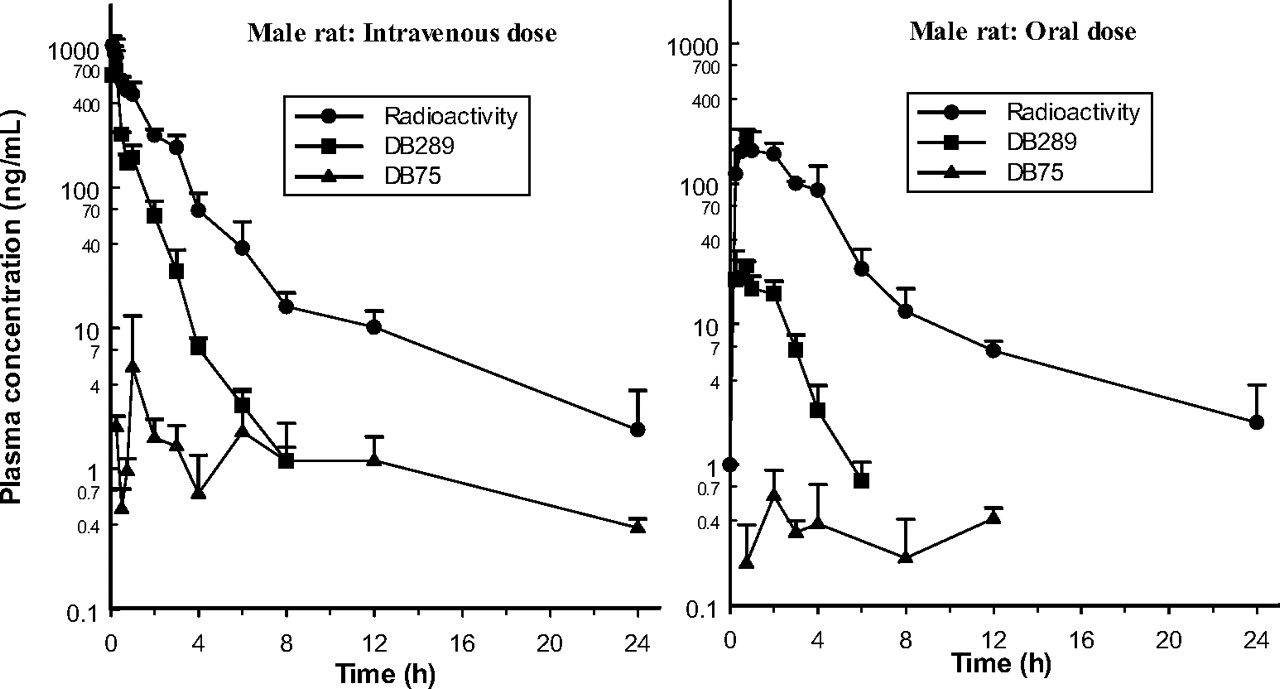
Plasma DB289 levels declined rapidly in rats after the i.v. dose, the mean terminal half-life being 1.5 h in males and 1.3 h in females. In monkeys, the rate of decline was noticeably slower (t½ = 10.9 h in both sexes). Total plasma clearance of DB289, which lay in the approximate range 20 to 30 ml/min/kg for male and female rats and monkeys, was comparable to the average hepatic and renal plasma flows of 26 to 30 and 16 to 20 ml/min/kg in rat and monkey, respectively, but much lower than their average cardiac plasma outputs (160 and 128 ml/min/kg, respectively; Davies and Morris, 1993). However, definitive interpretation of total body clearance and calculation of tissue extraction ratios were not possible from the available data. The volume of distribution at steady state for the prodrug was 1.8 and 1.5 l/kg in male and female rats, respectively, and approximately 9.1 and 9.4 l/kg in male and female monkeys, respectively. These values are well in excess of the total body water for each species (0.67 and 0.69 l/kg for the rat and monkey, respectively; Davies and Morris, 1993) and indicate that tissue binding of DB289 exceeded plasma binding.
Systemic bioavailability of the 1 mg/kg oral dose of DB289 was only 11% and 18% in male and female rats, respectively, and 17% and 22% in male and female monkeys, respectively: values that are consistent with limited absorption and/or extensive presystemic elimination. It is interesting to note that increasing the oral dose level from 1 to 7.7 mg/kg for rats or from 1 to 5 mg/kg for monkeys resulted in an apparent increase in bioavailability to ca. 40% in the former species, but a decrease to about 11% in the latter.

Plasma concentration-time curves after single 1 mg/kg intravenous and oral doses of 14C-DB289 to male rats (mean ± S.D.; four subgroups of three rats sampled in turn per dose).
Absorption. The rate of absorption of radioactivity after single oral doses of 14C-DB289 was more rapid in the rat (7.7 mg/kg) than in the monkey (5 mg/kg), and the systemic availability was twice as great (Table 2). There were no marked gender-related differences in the pharmacokinetics of radioactivity in the rat or monkey after oral (or i.v.) doses. In particular, the rate and extent of systemic exposure to total drug-derived material, as reflected by radioactivity Cmax and AUC, respectively, of female rats and monkeys were similar to those of their male counterparts. However, the proportion of the plasma radioactivity in the form of DB289 was higher in female rats than in males, whereas that of DB75 was lower; i.e., conversion of the prodrug to the active metabolite was more extensive in male rats. No such sex differences were apparent in monkeys.
Plasma pharmacokinetic parameters (mean ± S.D.) of total radioactivity after single intravenous and oral doses of 14C-DB289 to rats and cynomolgus monkeys
Comparison of total radioactivity measurements with those of unchanged DB289 and DB75 showed that at 2 h after single 1 mg/kg oral doses to rats and monkeys, 10 to 22% and 16 to 22% of the plasma radioactivity, respectively, was associated with the intact prodrug, but only <1% and 3 to 5%, respectively, with the active metabolite. In most cases, the t½ of DB289 was shorter than that of total radioactivity in both species after each route of administration, particularly in the case of the monkey (Table 2). These results therefore indicate the rapid and extensive formation of metabolites with longer half-lives than that of parent drug. Accordingly, the AUC for unchanged DB289 was ca. 6 to 7% of that for total radioactivity in both the rat at 1 mg/kg and the monkey at 5 mg/kg, whereas the AUC for the metabolite DB75 represented <1% and 5%, respectively. Despite its limited presence in plasma, however, metabolic conversion of DB289 to DB75 was extensive, as a large proportion of an oral dose of the prodrug was retained by the liver as the active metabolite (see below).

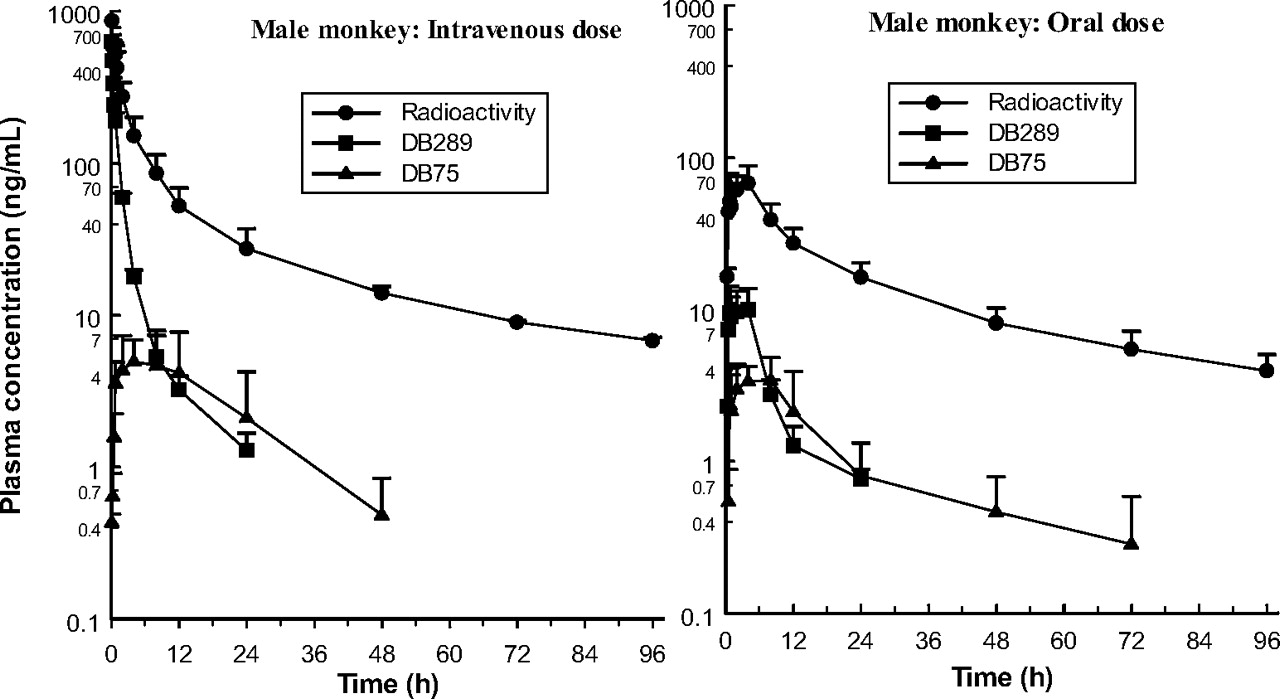
Plasma concentration-time curves after single 1 mg/kg intravenous and oral doses of 14C-DB289 to male cynomolgus monkeys (mean ± S.D.; n = 3 per dose).
The rat data obtained in this study suggest that either the radioactive dose was more rapidly absorbed than it was in the monkey (cf. Cmax and/or Tmax values) or DB289 was subjected to less presystemic elimination (cf. bioavailability values). The rat studies performed using oral dose levels of 1 and 7.7 mg/kg indicated approximate proportionality for radioactivity, but nonproportionality for both drug and active metabolite. DB289 also represented a smaller fraction of the total radioactivity after the low dose. In addition, the fraction of the total plasma radioactivity represented by DB75 was less after the low dose compared with the high dose. Thus, bioavailability of the prodrug was greater after the high dose than after the low dose, and greater than expected from a linear relationship, suggesting saturation of DB289 metabolism. In monkeys administered single 1 and 5 mg/kg oral doses of 14C-DB289, the pharmacokinetic data for radioactivity of DB289 and DB75 were not dose-proportional, and in this case, bioavailability was less after the high dose. Because Tmax for DB289 and DB75 was noticeably later after the higher dose in the monkey, absorption at this dose level may have been dissolution rate-limited.
Distribution. After single 10 mg/kg oral doses of 14C-DB289 to albino rats, the greatest concentrations of radioactivity in most tissues reached a peak at 6 h, albeit earlier for a few (e.g., adrenal at 3 h) and later for others (e.g., liver at 24 h). Concentrations in almost all tissues exceeded those in blood and declined more slowly (Table 3) such that they were undetectable in blood at 72 h but present in all tissues at 120 h.
Concentrations of radioactivity in tissues after single oral doses of 14C-DB289 (10 mg/kg) to rats
Results are expressed as μg-Eq DB289 free base/g tissue.
The greatest concentrations in albino rats were found in the liver at every termination time, and radioactivity was apparently cleared more slowly from the liver of females than from that of males. Thus, at 6 h postdose, tissue concentrations in male rats could be ranked as follows: liver > nasal mucosa > small intestine > adrenal > kidney (medulla) > urinary bladder > pancreas > thyroid. Microhistoauto-radiography of the liver and kidney did not show regional distribution within the tissue. The relatively high levels of radioactivity in the nasal mucosa of rats after the oral dose of 14C-DB289 were not reflected in others administered 14C-DB75 intravenously (data not shown), possibly suggesting that the prodrug, but not metabolite, was involved in this secretion. Sex differences in tissue concentrations of radioactivity in albino rats were not evident. However, whole-blood/plasma radioactivity concentration ratios in rats after oral dosing tended to increase with time (from about 1:1 or less during 24 h to up to 5:1 at 96 h; data not shown), reflecting the slower clearance of DB289 and/or its metabolites from the blood cells, and this tendency was noticeably more pronounced in males.
Analysis of key tissues in pigmented male rats after single oral doses of 14C-DB289 at 10 mg/kg reiterated the albino rat data obtained and emphasized the extensive uptake into the liver. Thus, in the pigmented rat killed at 24 h postadministration, the liver/plasma concentration ratio was 1300:1 and 28% of the radioactive dose was present in this organ. Radioactivity levels in the eyes of pigmented rats were about 10-fold greater than in those of albino rats given similar doses (data not shown), reflecting the basic nature of the drug (Ings, 1984). Concentrations in the whole blood of these animals (like those in the whole blood of pregnant and nonpregnant female rats) during 24 h after a single oral dose of 14C-DB289 were generally less than those in plasma.
Tissue concentrations of radioactivity in pregnant rats mirrored those in their nonpregnant counterparts and were also greatest in liver (Table 3). Radioactivity was distributed throughout the fetuses, but at concentrations lower than those in most maternal tissues. These levels declined steadily and were undetectable at 120 h. In lactating rats with suckling pups, radioactivity concentrations in milk reached peak values at 6 h after a 10 mg/kg oral dose of 14C-DB289 on day 12 postpartum, before declining at similar rates to plasma levels. It could be calculated that during the 24-h period after dosing, an individual neonate would receive less than 0.5% of the administered dose to the dam by ingestion of the milk.
At 2 h after single oral doses of 7.5 mg/kg 14C-DB289 to male rats in a separate study (data not shown), concentrations of radioactivity in the brain were initially similar to those in plasma. However, radioactivity was then cleared from brain more slowly than from plasma, such that levels in the former were still detectable at 168 h but not in the latter after 24 h. Concentrations of radioactivity in the cerebrospinal fluid remained below the limit of detection throughout the 168-h experimental period.
After repeated daily oral doses of 14C-DB289 (3.8 mg/kg/day) to rats for up to 21 days, QWBA showed that, as was the situation after a single dose, concentrations of radioactivity at 24 h after the 1st, 7th, 14th, and 21st doses were greatest in the liver, and relatively high in the nasal mucosa, adrenal gland, kidney, and brown fat (data not shown). Concentrations in the liver increased with increasing number of doses administered, but this increase was considerably greater in female rats than in males. Thus, after a single dose, liver concentrations were similar for the two sexes, but after multiple doses, the female/male radioactivity concentration ratio increased to between 3.0 and 3.5 after 7, 14, and 21 doses. In the male rats, most of the liver and kidney radioactivity was located in the mitochondrial and microsomal fractions, and little with the nuclear pellet and cytosolic fraction for the former tissue, whereas distribution was more even between the fractions in the latter. In the livers of female rats, however, noticeably less radioactivity was present in the microsomes and more with the nuclear pellet and mitochondria, and this effect was particularly evident for the mitochondrial fraction after repeated drug administration. When expressed in terms of microgram-equivalents of DB289 per milligram of protein, the association of liver radioactivity with mitochondrial protein was some 16-fold greater after 21 daily doses than after a single dose in female rats, compared with only a 5.5-fold increase in males. Essentially all of the radioactivity in liver was in the form of the active metabolite DB75 by HPLC (see below); thus, it would appear that the greater concentration of this metabolite in the mitochondrial fraction contributed to the greater hepatic binding of radioactivity in female rats.
Plasma Protein Binding. The in vitro binding of 14C-DB289 to the plasma proteins of mouse, rat, rabbit, monkey, and human was similar and extensive (97.3-98.8%) over the concentration range 30 to 1000 ng/ml (Table 4). By contrast, the binding of 14C-DB75 was less (75.2-94.5%), and it exhibited concentration dependence in the rabbit and monkey and species differences in the extent of binding, which was greatest in the monkey.
Plasma protein binding of 14C-DB289 and 14C-DB75 in vitro
Results represent the mean of duplicate determinations.
Excretion. After single 10 and 50 mg/kg oral doses of 14C-DB289 to rats, excretion of radioactivity was relatively slow and incomplete at 7 days postadministration (Table 5). Thus, only ca. 70 to 80% of the dose was recovered in the excreta during this time, all but 3 to 5% of which was present in the feces. A considerable proportion of the dose was retained in the liver at 7 days, an effect that was more marked in the female animal (16-21% dose) than in the male (6-13%). After i.v. administration of a 1 mg/kg dose, retention in the liver amounted to 22% of the dose in the male rat and 41% dose in the female, and again, urinary excretion only amounted to about 5% of the dose. During 0 to 48 h, 14CO2 was not detected in the expired air of rats, indicating that the radiocarbons were located in metabolically stable positions of the drug molecule.
Cumulative excretion and retention of radioactivity after single intravenous and oral doses of 14C-DB289 to rats and cynomolgus monkeys
Results are expressed as mean percentage radioactive dose.
In the monkey, totals of only 33% of an i.v. dose and 72% of an oral dose of 14C-DB289 were excreted during 7 days, fecal excretion accounting for about 24% of the former and 70% of the latter (Table 5). Retention in the liver was also markedly more extensive in this species after intravenous administration of 14C-DB289 (ca. 40% dose) than after oral administration (ca. 11%) but was the same for males and females.
In rats with cannulated bile ducts, 26% of a 10 mg/kg oral dose of 14C-DB289 to males and 14% to females was excreted in bile during 48 h, but biliary excretion was relatively slow: only 5 to 6% of the dose was excreted by this route in the first 6 h (Table 6). Fecal excretion accounted for 29% of the dose in males and 25% in females, whereas in both sexes renal excretion accounted for only 4% of the dose. As was the case with the intact rats, hepatic retention was markedly greater in female rats (42% dose) than in males (29%), and about 8% of the dose was present in the remaining carcasses. After i.v. administration, biliary and urinary excretion of radioactivity was similar to that after oral dosing, but fecal excretion amounted to only 4% of the dose. Retention in the liver was about 40% of the dose in male rats and 50% in females.
Cumulative excretion and retention of radioactivity after single intravenous and oral doses of 14C-DB289 to bile duct-cannulated rats
Results are expressed as mean percentage radioactive dose.
Metabolite Profiles. Metabolite profiles of 14C-DB289, generated using HPLC with on-line or off-line measurement of radioactivity, were evaluated in the liver, urine, and feces of both rat and monkey, and in the plasma, milk, brain, and bile of the rat only.
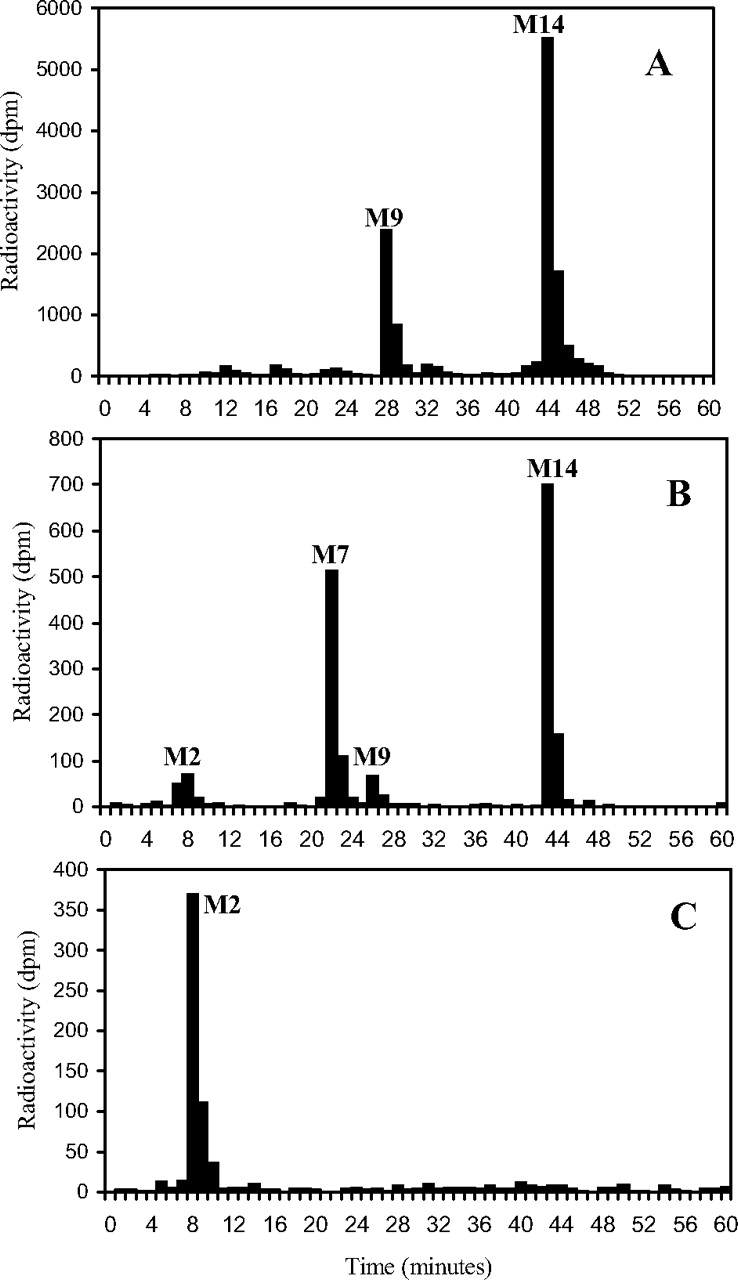
Plasma. During 2 to 6 h after single oral doses (7.5 mg/kg) to rats, approximately 55% of the plasma radioactivity was in the form of unchanged prodrug DB289 (M14), whereas the active metabolite DB75 only accounted for about 2% (Fig. 4). Most of the other radioactive components were of minor significance and only two correspond to a reference compound, one of which, DB810 (M7), was prominent in excreta (approximately 10% of dose), whereas the other, DB775 (M9), accounted for up to 24% of the plasma radioactivity, but was a minor metabolite in excreta (ca. 1% dose). There was no evidence for the presence of β-glucuronidase/sulfatase-hydrolyzable conjugates in rat plasma.
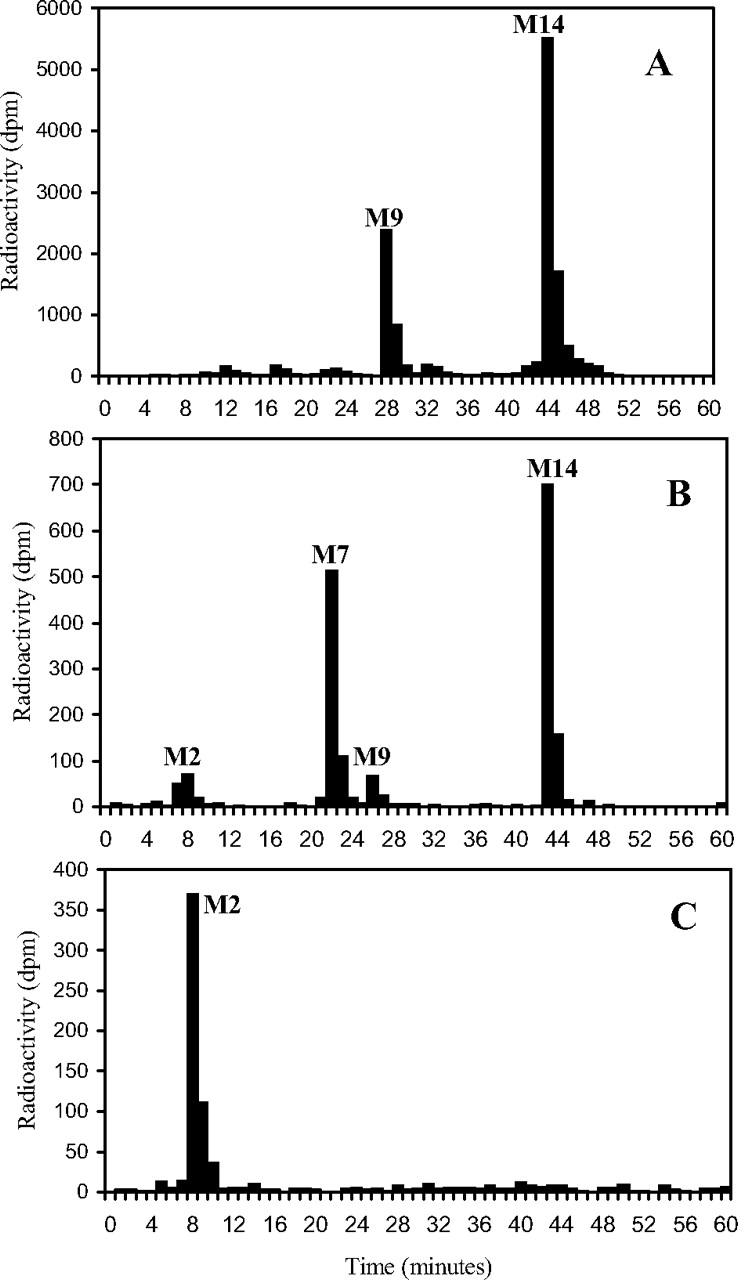
HPLC radiohistograms of rat plasma (2 h; 7.5 mg/kg oral dose) (A), rat brain (2 h; 7.5 mg/kg oral dose) (B), and rat brain (96 h; 7.7 mg/kg oral dose) (C).

HPLC radiochromatograms of rat feces (0-24 h; 10 mg/kg oral dose) (A) and rat liver (168 h; 10 mg/kg oral dose) (B).
Metabolite profiles in the plasma of monkeys were not evaluated because of limited sample availability; instead, the concentrations of the prodrug DB289 and its active metabolite, DB75, were measured by LC-MS/MS. The resultant data showed that up to 28% and 9% of the plasma radioactivity represented DB289 and DB75, respectively. Similar data obtained after i.v. administration of 14C-DB289 indicated that its metabolism was, at least initially, relatively rapid, since in the first (5-min) plasma sample only 73% of the radioactivity corresponded to DB289, and DB75 was not detected.
Brain. Only four radioactive components in brain accounted for more than about 2% of the sample radioactivity during 2 to 96 h after a single oral dose, and they corresponded to DB75 (M2; from 7% to 67% during this period), DB810 (34% to 0), DB775 (5% to 0), and unchanged DB289 (46% to 3%) (Fig. 4). These results do not reflect the corresponding plasma chromatograms, which contained essentially no DB75, suggesting that biotransformation occurred within the brain tissue.
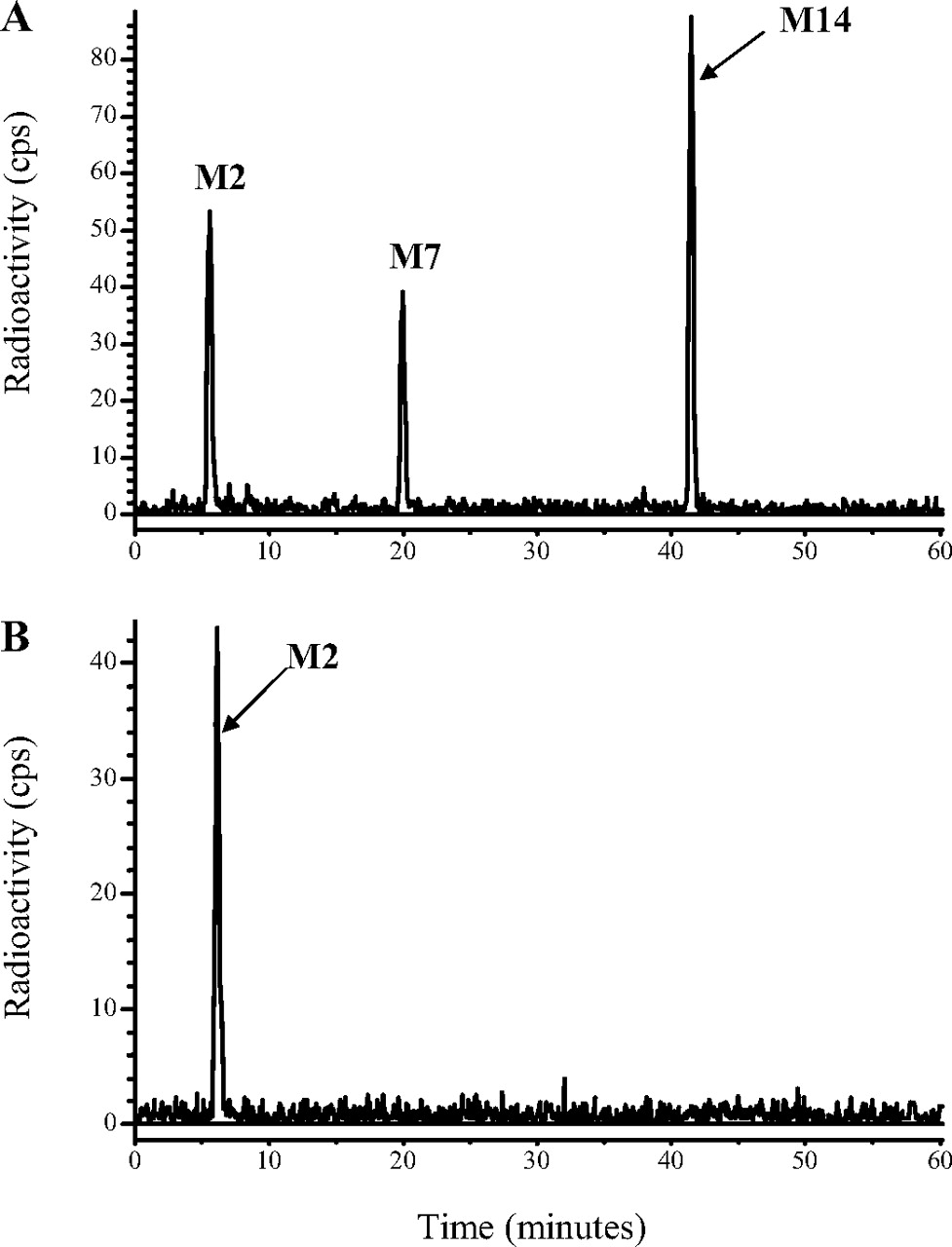
Liver. The livers of rats (taken at 7 days; Fig. 5) and monkeys (taken at 43 days; Fig. 6) after single oral or intravenous doses contained essentially only one detectable component, which corresponded chromatographically to the active drug DB75.
Milk. The profiles of radioactive components in milk samples collected directly from mammary tissue in lactating rats with suckling pups during 24 h after a 10 mg/kg oral dose of 14C-DB289 were similar to the corresponding plasma samples, which contained mainly unchanged prodrug, initially, and then more DB810 and DB775, but no DB75.
Bile. The 0- to 48-h bile of male rats dosed orally at 10 mg/kg contained about 26% of the dose, most of which was in the form of six main metabolites. One of these accounted for about 10% of the dose and the others up to 3%. The former (M3) appeared to be a conjugate of DB75, based solely on its behavior and that of the corresponding aglycone (M2) toward β-glucuronidase/arylsulfatase. Thus, the proportion of M3 in untreated bile and of M2 in the enzyme-treated sample increased from about 10% at 0 to 6 h to nearly 80% at 24 to 48 h. Relatively smaller amounts of both metabolites were present in the bile of female rats, where biliary excretion was only half that in males. Metabolite profiles of bile after i.v. dosing were similar to those after oral dosing.
Feces. The 0- to 72-h feces of male rats dosed orally at 10 mg/kg contained about 62% of the dose and approximately 14 radioactive components, but only three of these accounted for more than 2% of the dose (Fig. 5). These were the prodrug DB289 (about 20% dose), the active drug DB75 (about 22% dose), and a metabolite (DB810, about 10% dose) that was also present in plasma and urine.
Similar metabolite profiles were obtained after 50 mg/kg oral and 1 mg/kg i.v. doses to rats, except that after the latter, proportions of unchanged DB289 were low (<0.5% dose), indicating that after oral doses it was unabsorbed prodrug excreted in the feces, rather than absorbed prodrug, which was excreted via the bile. Furthermore, after the higher (50 mg/kg) oral dose, there was a larger proportion (about 42%) of the prodrug DB289 and a lower proportion (about 17%) of the active drug in feces, implying that absorption of DB289 was not dose-proportional in the rat.
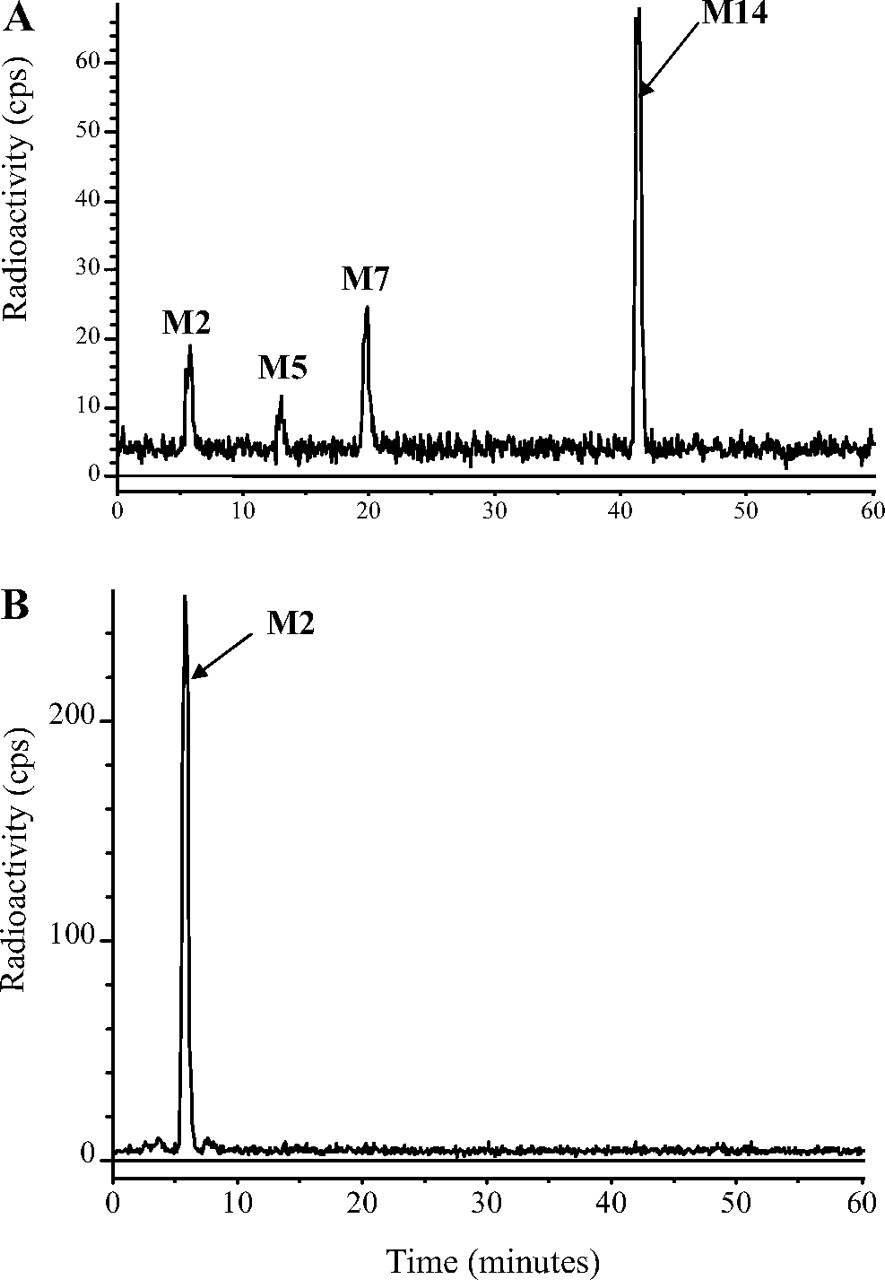
HPLC radiochromatograms of monkey feces (0-24 h; 1 mg/kg oral dose) (A) and monkey liver (43 days; 1 mg/kg oral dose) (B).
The 0- to 72-h feces of male monkeys dosed orally at 5 mg/kg contained about 67% of the dose and approximately eight radioactive components, but only three accounted for more than 1% of the dose (Fig. 6). These were the prodrug DB289 (ca. 49% dose), the active metabolite DB75 (ca. 5% dose), and a metabolite (DB810, ca. 11% dose) that was also present in plasma. Similar profiles were obtained after i.v. doses, except that there were more radioactive components and the proportions of DB289 were much lower (<1%), indicating that after oral administration, it was unabsorbed, rather than absorbed, prodrug that was excreted in the feces.
Urine. The 0- to 24-h urine of rats dosed orally at 10 mg/kg contained only about 3% of the dose and about a dozen radioactive components, none of which accounted for more than 1% of the dose. At least two conjugates, hydrolyzable by β-glucuronidase/sulfatase, were present, one of which corresponded to the biliary conjugate of DB75, which was itself also present in small quantities in urine as the aglycone. Other radioactive components present in urine corresponded to a metabolite in plasma (DB810), to the di-oxime of DB289 (DB290), and to DB289 itself. Metabolite profiles after a higher oral dose (50 mg/kg) or an i.v. dose (1 mg/kg) were similar to those obtained after the lower oral dose.
The 0- to 72-h urine of monkeys dosed orally contained only 1 to 2% of the dose and approximately 15 radioactive components, none of which accounted for more than 0.5% of the dose. At least one conjugate (<0.1% dose), hydrolyzable by β-glucuronidase/sulfatase, was present that appeared to be a conjugate of DB290. Small amounts of DB775 and the conjugate of DB75 also appeared to be present. Metabolite profiles after single intravenous doses to monkeys were similar to those obtained after single oral doses.
The metabolite profiling data indicate, therefore, that in the rat and monkey approximately 20 and 50% of the oral doses, respectively, were not absorbed from the gastrointestinal tract. These results are in agreement with the extent of absorption estimated from the radioactivity excretion data (Table 5) and are consistent with the conclusions drawn from the plasma radioactivity concentration-time data (Table 3). There did not appear to be gender-related differences in metabolite profiles, except that in the rat it was evident that DB75 was cleared more slowly from the liver of the female rat than from the male.
After 21 daily oral doses of 14C-DB289 to male rats at a dose level of 3.8 mg/kg/day, urinary and fecal metabolite profiles were generally similar to those obtained after a single oral administration. In the liver, kidney, and lung of these animals, DB75 was the only noteworthy radioactive component, again reflecting the inability of this (charged) metabolite to exit certain tissues after being either generated in or transported to them.
Metabolite Identification. LC-MS analysis of biological samples collected from rats and monkeys dosed orally with 14C-DB289 confirmed the presence of the active metabolite DB75 in rat bile and monkey liver and feces. While these samples were being analyzed, plasma and urine became available from healthy human subjects dosed orally with nonradiolabeled DB289 during clinical trials with the test compound; therefore, these samples were also analyzed by LC-MS. The identities of all monkey, rat, and human metabolites of DB289 characterized conclusively by mass spectrometric comparison with reference compounds (where available) are summarized in Table 7, and postulated biotransformation pathways are depicted in Fig. 7.
LC-MS (ESI+) data for DB289 and metabolites in biological matrices
Human samples were collected during clinical trials with nonradiolabeled DB289.
Discussion
Because of its (computed) physicochemical properties (log D +4.28 at pH 7.4; pKa 3.97, 4.57), DB289 would be expected to be better absorbed from the gastrointestinal tract than the active metabolite, DB75 (log D -2.63 at pH 7.4; pKa 9.78, 10.39). Furthermore, since the free compound is basic and possesses a molecular weight of 364, DB289 (and/or its metabolites) would be excreted in the bile of rats and, after appropriate conjugation, in the bile of monkeys and even of humans (Dobrinska, 1989). The studies reported here have confirmed that biliary secretion plays a role in the disposition of DB289 in the rat, that the prodrug is well absorbed from the gastrointestinal tract by rats and monkeys, and that it delivers the active metabolite to key organs such as liver (a major organ of distribution) and brain (an important therapeutic target organ). The predominant route of excretion of radioactivity in every case was in the feces, where DB75 was a major metabolite, whereas urinary excretion was <10% for all doses.
Up to two-thirds of an oral dose of 14C-DB289 was absorbed by rats and about one-half to cynomolgus monkeys, but both were subject to presystemic metabolism and considerable hepatic retention. As a consequence, the bioavailability of a 1 mg/kg oral dose of 14C-DB289 was only ca. 10 to 20% in the two species, being marginally higher in the monkey. The only significant drug-derived compound detected by HPLC in any of the liver samples was DB75. If the (reasonable) assumption is made that all the radioactivity remaining unexcreted at 7 days postadministration was in the form of DB75, it may be calculated that up to one-half of a 10 mg/kg oral dose of 14C-DB289 to rats and approximately one-third of a 5 mg/kg oral dose to monkeys was converted to the active metabolite.

Proposed biotransformation pathways of DB289 in rat and monkey
Systemic clearance of DB289 (20-30 ml/min/kg) in rat and monkey was notably less than the typical cardiac plasma output in these species, but similar to the average liver and kidney plasma flows (Davies and Morris, 1993). Clearance was lower and systemic exposure higher in female rats compared with males, whereas in monkeys there was little difference between sexes. The finding that plasma drug concentrations were less in male rats than in females is a common observation and occurs primarily with compounds that are cleared by cytochrome P450-mediated metabolism (Lin et al., 1996; Thompson et al., 1997). Such sex differences are usually due to hormonally mediated differential expression of the sex-dependent P450 enzymes in the rat (Waxman et al., 1990; Legraverend et al., 1992) and are rarely predictive of sex differences in other species, which in our experience tend to be much less common.
The volume of distribution of the prodrug was well in excess of the total body water volume, suggesting that tissue binding was extensive and greater than plasma binding. It was confirmed experimentally that DB289 is extensively (i.e., 97-99%) bound to the plasma proteins of mouse, rat, rabbit, monkey, and human in vitro, whereas the active metabolite DB75 was less extensively protein bound (75-97%), and its binding and concentration dependence varied between species.
During 6 h after an oral dose of 14C-DB289 to rats, unchanged prodrug accounted for about half the plasma radioactivity, whereas DB75 represented much less. The AUC data indicated that metabolites other than DB75 were present in the systemic circulation, since less than 20% of the total plasma radioactivity was in the form of prodrug and active metabolite. Accordingly, the profiling and LC-MS identification studies were able to confirm the presence of the intermediary metabolites DB775, DB290, and DB810 in biological samples collected from the treated animals and/or healthy human subjects participating in clinical trials. The principal radiolabeled components in the brains of treated rats were DB75, unchanged DB289, and DB810; thus, it may be speculated, after comparison with the corresponding plasma profiles, that DB75 formed during the first pass was sequestered in the liver, whereas DB289 that escaped metabolism was taken up by the brain and metabolized there to the active metabolite. Drug-related material was not detected in the cerebrospinal fluid of rats.
Biotransformation of DB289 appears to involve successive O- demethylation reactions and reduction of the resulting amidoximes to the amidines, leading eventually to DB75, although direct N-demethoxylation may also have occurred since the mono- and di-desmethoxy metabolites (DB810 and DB75, respectively) were major radiochemical components in feces. It should be noted that the reverse biotransformations are also possible, at least in vitro, since the dicationic amidine, diminazene (which also possesses antitrypanocidal properties) is known to undergo two successive N-oxidation reactions: first to the mono-oxime and then to the di-oxime (Clement et al., 1992). The metabolic conversion of DB289 to DB75 in vitro using isolated rat hepatocytes consists of sequential oxidative O-demethylation and reductive N-dehydroxylation reactions (Zhou et al., 2004), and it has been demonstrated recently that hepatic CYP4F enzymes, including CYP4F2 and CYP4F3B, catalyze the initial O-demethylation of DB289 in humans (Wang et al., 2006). Pentamidine is metabolized in vitro mainly by multiple phase I cytochrome P450-induced side-chain C-hydroxylation reactions, but its minor biotransformation pathways involve N-hydroxylation to the oximes (Berger et al., 1990a,b, 1991, 1992; Clement and Jung, 1994). On the other hand, oximes may be hydrolyzed in vivo to the corresponding ketone (e.g., Mayo et al., 1990). Perhaps surprisingly, therefore, but in accord with the above in vitro studies, no C-oxidized metabolites of DB289 (e.g., those possessing a hydroxylated aromatic ring) were identified in the present study, although they may nevertheless have been present as minor biotransformation products.
Placental transfer and fetal exposure to DB289 and/or its metabolites occurred in the rat, albeit at relatively low levels, and the presence of radioactivity in the milk of lactating rats confirmed that suckling neonates would be exposed to the drug. However, it could be calculated that this exposure would have amounted to less than 0.5% of the dose to the maternal rat during 24 h postadministration. In pigmented rats, notable uptake of DB289-derived material into the eye occurred, reflecting the expected binding of the basic drug and/or its metabolites to ocular melanin (Ings, 1984).
It is also evident that during the 24-h dose regimen common to toxicity studies, DB289 and/or its metabolites would accumulate in the tissues, thus ensuring their continual exposure in the animal throughout the duration of such a study. As noted above, particularly significant was the extensive uptake of radioactivity (in the form of DB75) in rat and monkey liver. This material accumulated most in the mitochondrial fraction, an effect that was markedly greater in female rats than in males. In the latter sex, most was present in the mitochondrial and microsomal fractions, and little with the nuclear pellet and cytosolic fraction, whereas in the former, noticeably less was found in the microsomes and more with the nuclear pellet and mitochondria, an effect that was particularly evident for the latter fraction after repeated drug administration. The association of liver radioactivity with protein in the mitochondrial fraction was some 16-fold greater after 21 daily doses than after a single dose in female rats, compared with only a 5.5-fold increase in males. The greater concentration of DB75 in the mitochondria may therefore have contributed to the more extensive hepatic binding of radioactivity in female rats. It may be postulated that either this (charged) metabolite was transported into the cells by protein, but was subsequently unable to exit, or, after transport into the liver cells, parent drug was metabolized to DB75 there and, being unable to exit, the metabolite accumulated. As was the situation with brain, the lack of correspondence between tissue and plasma metabolite profiles suggests that the metabolite was formed within the tissue.
These studies have therefore shown that oral doses of DB289 were well absorbed by rats and monkeys and converted effectively to the active metabolite DB75, which was retained in key tissues, particularly the liver, where parasites such as Plasmodium reproduce and develop immunity. Assuming that the proportion retained is therapeutically relevant and that toxicity is not an issue, these would be highly desirable features for effective chemotherapeutic treatment of protozoan diseases. The drugs used currently for these conditions require parenteral administration with the attendant severe toxicity and logistical difficulties of dosing in a tropical environment, with the consequence that many patients fail to complete the course of treatment or are even killed by it. Thus, although it is likely that a proportion of the dose(s) would be retained in the liver for some time by human patients, a short course of oral DB289 would appear to offer a superior alternative.
Acknowledgments
We express gratitude to Drs. T. Winwick and N. Eggleton of Huntingdon Life Sciences Ltd. for repurification of the various batches of 14C-DB289, and to Dr. J. E. Hall and colleagues at the University of North Carolina for helpful discussions during the preparation of the manuscript.
Footnotes
-
This work was supported by the Bill & Melinda Gates Foundation (Grant 3436: Development of Novel Drug Candidates for the Treatment of Human African Trypanosomiasis and Leishmaniasis).
-
This article is dedicated to the memory of Dr. James L. Allen of Immtech Pharmaceuticals, Inc., a highly respected colleague who was closely involved in the development of DB289, but who tragically passed away during preparation of the manuscript.
-
doi:10.1124/dmd.106.013391.
-
ABBREVIATIONS: PCP, Pneumocystis carinii pneumonia; CSF, cerebrospinal fluid; DB75, 2,5-bis(4-guanylphenyl)furan dihydrochloride; DB289, 2,5-bis[4-(N-methoxyamidino)phenyl]furan monomaleate; HPLC, high-performance liquid chromatography; LC-MS, liquid chromatography-mass spectrometry; LC-MS/MS, liquid chromatography-tandem mass spectrometry; QWBA, quantitative whole-body autoradiography; AUC, area under the curve; CL, clearance; AUMC, area under the first moment curve; ESI, electrospray ionization.
-
↵1 Current affiliation: NPS Pharmaceuticals, Salt Lake City, Utah.
- Received November 1, 2006.
- Accepted March 12, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}