


Abstract
Multidrug resistance protein 2 (MRP2/Mrp2) is a transporter that can influence the absorption, distribution, and elimination of many drugs. Mrp2 knockout mice are being used to study Mrp2 functions in vivo, including pharmacokinetics of drugs. To assess possible species-specific differences between human MRP2 and mouse Mrp2, we generated polarized cell lines expressing mouse Mrp2 and used these to investigate transport of clinically important agents. We also tested the ability of other drugs to modulate MRP2/Mrp2-mediated transport, a phenomenon that can lead to drug-drug interactions. In MDCK cells stably expressing human MRP2 or mouse Mrp2, saquinavir and docetaxel were more efficiently transported by mouse Mrp2, whereas vinblastine was transported better by human MRP2. MRP2/Mrp2-mediated transepithelial transport of several drugs could be stimulated by probenecid and sulfanitran, but stimulation was often more pronounced for human MRP2 than for mouse Mrp2. Interestingly, for some drugs the MRP2 modulator sulfinpyrazone had opposite effects on both transporters, stimulating human MRP2 and inhibiting mouse Mrp2 activity. In vesicular transport studies, transport of estradiol-17β-glucuronide by mouse Mrp2 showed homotropic cooperativity, as previously described for human MRP2. The MRP2 modulators again showed differential effects on estradiol-17β-glucuronide transport, most notably with sulfinpyrazone stimulating human MRP2 and profoundly inhibiting mouse Mrp2 activity. In conclusion, although human and mouse MRP2/Mrp2 have largely overlapping substrate specificities, there are important species differences in the transport efficiency of MRP2 substrates and in the modulation of transport by other compounds. These differences should be taken into account when results obtained in mice are extrapolated to humans.
Multidrug resistance protein 2 (MRP2/Mrp2; ABCC2/Abcc2) belongs to the ATP-binding cassette transporter family and transports a wide range of organic anions such as glutathione, glucuronide, and sulfate conjugates (Paulusma et al., 1999; Keppler and König, 2000). Moreover, many nonconjugated xenobiotics, including drugs, are also efficiently transported by MRP2 (Chan et al., 2004; Borst et al., 2006). MRP2 is localized in the apical membranes of polarized cells such as hepatocytes, renal epithelial cells, and the enterocytes of the small intestine (Keppler et al., 1997; Schaub et al., 1999; Mottino et al., 2000). There it reduces the uptake of substances from the intestinal lumen and, furthermore, plays an important role in the elimination process by excreting its substrates into bile, urine, and feces (Chan et al., 2004). Insight into the physiological role of this transporter has been obtained in rats naturally lacking Mrp2 (Jansen et al., 1985; Hosokawa et al., 1992) and in patients with Dubin-Johnson syndrome, who lack functional MRP2 (Kartenbeck et al., 1996; Kotal et al., 1997). The absence of functional MRP2 is associated with reduced hepatobiliary elimination of organic anions such as bilirubin glucuronides, leading to hyperbilirubinemia (Takikawa et al., 1991; Hosokawa et al., 1992). The same symptoms were also seen in recently generated Mrp2 knockout mouse strains (Chu et al., 2006; Vlaming et al., 2006).
Mrp2 knockout mice are currently used to investigate the impact of Mrp2 on the pharmacokinetics of clinically important drugs (Lagas et al., 2006; Vlaming et al., 2006). This information can lead to a better understanding of how Mrp2 is involved in the processes of drug absorption, distribution, and elimination. Furthermore, this model offers the possibility to explore how modulators of the transporter (i.e., inhibitors or stimulators) can change the pharmacokinetics of a drug, thereby giving insight into potential drug-drug interactions. However, data obtained in mice do not necessarily correlate with the human situation. The amino acid sequence identity of human MRP2 with its mouse ortholog is approximately 78% (Nies and Keppler, 2007), implying that there might be substantial differences in substrate recognition or modulation efficiency. Ninomiya et al. (2006) compared the intrinsic transport activity and kinetic profiles of mouse, monkey, dog, and rat Mrp2. They observed species differences in these parameters, although the substrate specificities were similar. To our knowledge, there is as yet no report that systematically compares human MRP2 and mouse Mrp2 transport properties.
The complex modulation (stimulation and/or inhibition) of MRP2-mediated transport by various compounds (Bakos et al., 2000; Evers et al., 2000; Huisman et al., 2002; Zelcer et al., 2003) suggests the presence of two interacting binding sites in the MRP2 protein: one site that mediates the transport of the substrate and another site modulating the affinity of the transport site (Zelcer et al., 2003; Borst et al., 2006). Moreover, the ability of modulating compounds to either stimulate or inhibit MRP2-mediated transport depends on the substrate that is transported. It therefore seems that each substrate-modulator pair has a unique interaction with the MRP2 protein (Zelcer et al., 2003). Until now, many modulators for human MRP2, which could potentially lead to drug-drug interactions, have been discovered in vitro (Bakos et al., 2000; Huisman et al., 2002; Zelcer et al., 2003; Mano et al., 2007). Evidence for such interactions occurring in vivo is still sparse, although a study in rats reported that the benzylpenicillin-induced increase of bile flow can be attributed to a stimulation of Mrp2-mediated biliary glutathione excretion (Ito et al., 2004). This finding was further substantiated by demonstration in vitro that benzylpenicillin stimulates glutathione transport in MDCKII cells expressing MRP2 (Ito et al., 2004).
Mouse models are important tools to further study the phenomenon of Mrp2 modulation and to assess whether this mechanism can lead to relevant drug-drug interactions in vivo. However, again data that address possible differences between human and mouse concerning modulation properties of this drug transporter are lacking. For these reasons, we generated polarized cell lines expressing mouse Mrp2 and we further used mouse Mrp2-expressing Sf9 membrane vesicles. These two different assay systems allowed us to compare human MRP2 (hMRP2) and mouse Mrp2 (mMrp2) regarding transport of several clinically important drugs and the influence of known MRP2 modulators on drug transport. This information gives insight into species differences in MRP2-mediated drug transport and can help in extrapolating results obtained in mice to humans.
Materials and Methods
Chemicals. [3H]Vinblastine, [3H]etoposide, [3H]inulin, and [14C]inulin were from GE Healthcare (Little Chalfont, Buckinghamshire, UK). [14C]Saquinavir and saquinavir were provided by Roche Discovery Welwyn (Welwyn Garden City, UK). [3H]Docetaxel was from Sankyo (Tokyo, Japan). [3H]Estradiol-17β-glucuronide (E217βG) was from PerkinElmer Life and Analytical Sciences (Waltham, MA). GF120918 was kindly provided by GlaxoSmithKline (Uxbridge, UK). MultiscreenHTS-FB 96-well filter plates were from Millipore Corporation (Bedford, MA). Creatine kinase was from Roche (Basel, Switzerland). Probenecid, sulfinpyrazone, sulfanitran, and all other chemicals and reagents were from Sigma-Aldrich (St. Louis, MO). As primary antibodies we used a mouse anti-rat Mrp2 antibody that reacts with human and mouse MRP2 (M2III-5; from G. L. Scheffer, Free University Hospital, Amsterdam, The Netherlands) and a rabbit anti-mouse Mrp2 antibody that recognizes only mouse Mrp2 (from J. M. Fritschy, University of Zurich, Institute of Pharmacology and Toxicology, Zurich, Switzerland).
Generation of MDCKII Cells Expressing Mouse Mrp2. Generation of MDCKII-MRP2 and MDCKII-Neo cells has been described (Evers et al., 1998). For the transduction of mouse Mrp2 we used parental MDCKII cells. All cells were cultured in Dulbecco's modified Eagle's medium with Glutamax (Life Technologies, Breda, The Netherlands) supplemented with 50 U/ml penicillin, 50 mg/ml streptomycin, and 10% (v/v) fetal calf serum (complete medium; Life Technologies). Culturing conditions were 37°C in 5% CO2. Cells were tested to be mycoplasma-free. The full-length mouse Mrp2 cDNA clone derived from BALB/c mouse liver and inserted in pFASTBAC1 (Ninomiya et al., 2006) was a kind gift of K. Ito (Graduate School of Pharmaceutical Sciences, Chiba University, Chiba, Japan). The cDNA was excised using HindIII and AfeI for insertion into the LZRS-IRES-GFP vector. This retroviral expression vector (Kinsella and Nolan, 1996) provides expression of the gene of interest, along with the enhanced green fluorescence protein linked via an internal ribosome entry site, by transcription from the viral 3′-long terminal repeat promoter. The mouse Mrp2 cDNA clone fragment was inserted into the LZRS vector at the SnaBI restriction site by blunt-end ligation. The resulting construct was checked by sequencing using the ABI Prism Big Dye Terminator kit (PerkinElmer Life and Analytical Sciences) confirming the cDNA sequence of mouse Mrp2. Then the construct was transfected into the amphotropic Phoenix producer cell line using the calcium phosphate precipitation method. Virus-containing supernatants were harvested 48 h after transfection and used to transduce MDCKII cells in the presence of 5 μg/ml Polybrene. MDCKII cells with high levels of green fluorescent protein expression were sorted individually into 96-well plates with a FACSTAR Plus sorter (BD Biosciences, Franklin Lakes, NJ). Single-cell clone lines were then established and analyzed for Mrp2 mRNA expression using real-time reverse transcriptase-polymerase chain reaction (mouse Mrp2 QuantiTect Primer Assay; QIAGEN, Valencia, CA). The expression of Mrp2 protein was verified in selected clones by Western blot analysis.
Preparation of Sf9 Membrane Vesicles. Membrane vesicles from Sf9 insect cells were obtained after infection with a human MRP2 (Bakos et al., 2000) or mouse Mrp2 (Ninomiya et al., 2006) cDNA-containing baculovirus at a multiplicity of infection of 1. After incubation for 3 days, cells were harvested by centrifugation at 500g at 4°C for 5 min. The pellet was resuspended in ice-cold hypotonic buffer (0.5 mM sodium phosphate and 0.1 mM EDTA, pH 7.4) supplemented with a protease inhibitor cocktail (Roche) and incubated at 4°C for 90 min. The suspension was centrifuged at 100,000g for 40 min, and the pellet was homogenized in ice-cold TS buffer (50 mM Tris-HCl and 250 mM sucrose, pH 7.4) using a tightly fitting Dounce homogenizer. After centrifugation at 500g at 4°C for 10 min, the supernatant was collected and centrifuged at 4°C at 100,000g for 40 min. The pellet was resuspended in TS buffer and passed through a 27-gauge needle 25 times. The vesicles were dispensed in aliquots, snap-frozen in liquid nitrogen, and stored at –80°C until use.
Western Blot Analysis. Cells were trypsinized, washed with ice-cold PBS, and resuspended in TD buffer (10 mM Tris-HCl, pH 8.0, 0.1% Triton X-100, 10 mM magnesium sulfate, 2 mM CaCl2, 40 U/ml DNase, 1 mM dithiothreitol, and protease inhibitor cocktail from Roche). Cell suspensions were subjected to three freeze-thaw cycles and were then incubated at 37°C for 10 min. After centrifugation (14,000 rpm for 5 min) the protein concentration in the supernatant was determined using a BCA Protein Assay Kit (Pierce Chemical, Rockford, IL). Ten micrograms of protein of the MDCKII cells and 0.5 μgof protein of the Sf9 vesicles were separated on a 8% polyacrylamide gel and transferred to a nitrocellulose membrane (Amersham Biosciences UK, Ltd.). After blocking overnight at 4°C (PBS with 1% bovine serum albumin, 1% milk powder, and 0.05% Tween 20), the membranes were incubated with a primary mouse monoclonal antibody to MRP2 (M2III-5) diluted 1:1000 in blocking buffer for 1 h. As a secondary antibody a rabbit anti-mouse antibody conjugated to horseradish peroxidase (Dako Denmark A/S, Denmark) was used in a 1:1000 dilution for 1 h. A second staining was performed as described above but using a rabbit anti-mouse Mrp2 primary antibody (Soontornmalai et al., 2006) diluted 1:1000 and goat anti-rabbit secondary antibody diluted 1:2000 (Dako Denmark A/S). The bands were visualized using an ECL detection kit (GE Healthcare).
Transepithelial Transport Assay. One million cells per well were seeded on microporous polycarbonate membrane filters (Transwell 3414; Corning Life Sciences, Cambridge, MA). After 3 days of culturing, including daily medium changes, the cells were washed with prewarmed PBS and then preincubated for 2 h with Optimem medium (Invitrogen, Grand Island, NY). During both the preincubation and the transport experiment 1 μM GF120918 and the appropriate MRP2 stimulator (probenecid, sulfanitran, or sulfinpyrazone) were present in the apical and the basolateral compartments. GF120918 was added to block the activity of endogenous P-glycoprotein and breast cancer resistance protein. The transport experiment was started by adding the MRP2 substrate to the donor compartment (either basolateral or apical) and fresh Optimem medium to the acceptor compartment. The MRP2 substrates (saquinavir, docetaxel, etoposide, and vinblastine) were applied at a concentration of 5 μM traced with radiolabeled drug (0.09 μCi/well). Furthermore, the drug solution contained radiolabeled inulin (0.09 μCi/well) to check for leakage of the monolayer. The transwell plates were then incubated at 37°C in 5% CO2 for 4 h. Every hour a 50-μl sample was drawn from the acceptor compartment. Samples were mixed with 4 ml of scintillation fluid (Ultima Gold; Packard, Meriden, CT) and counted in a scintillation counter. Results are expressed as the percentage of transported drug relative to the initially applied amount of drug. This relative drug transport was plotted versus time. The transport ratio R is calculated as basolateral-to-apical transported drug divided by apical-to-basolateral transported drug after 4 h. This value reflects the efficiency of a cell line to promote apically directed transport. After the last sample was taken, the wells were washed with cold PBS, and the filters were excised and also measured for radioactivity. The relative cellular drug uptake was determined by dividing the amount of radioactivity in the filters by the initially applied amount of radioactivity.
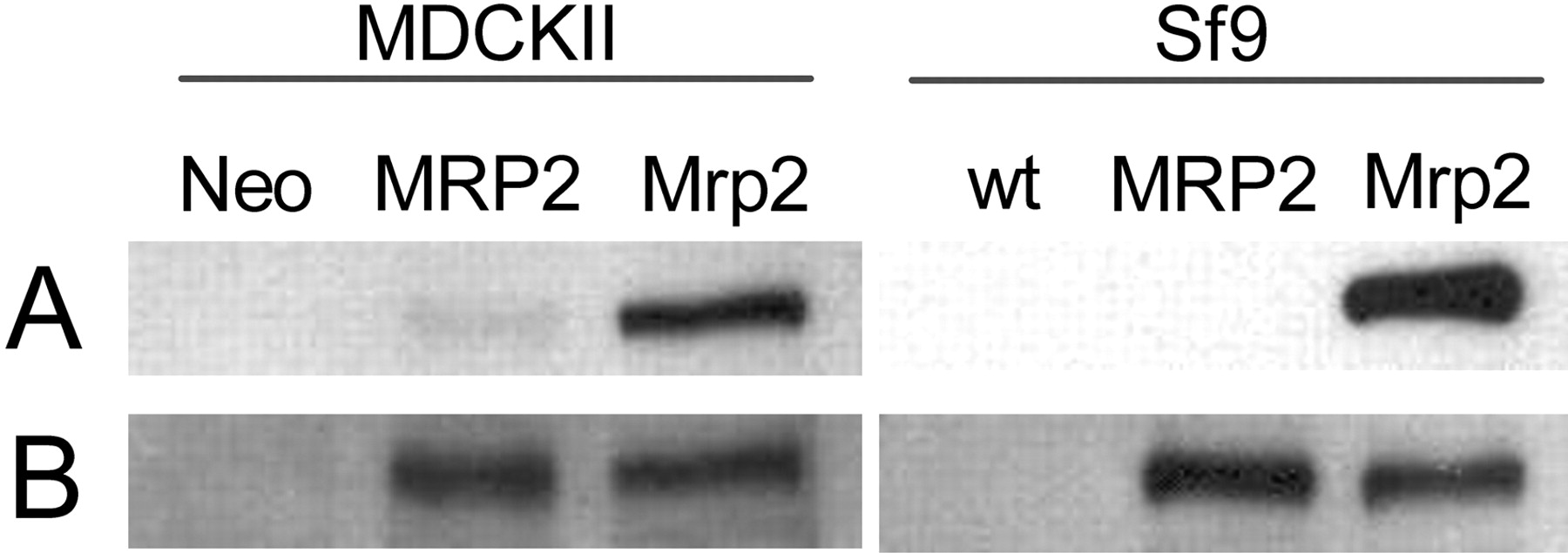
Protein expression of MRP2/Mrp2 in the MDCKII cell lines and in Sf9 membrane vesicles as determined by Western blot analysis. MDCKII-Neo and Sf9 wilt-type (wt) are negative controls. The MRP2 and Mrp2 clones were stably transduced with human MRP2 or mouse Mrp2, respectively. Ten micrograms of protein for the MDCKII cells and 0.5 μg of protein for the Sf9 membrane vesicles were loaded per lane and size fractionated on a 8% SDS-polyacrylamide gel. A, rabbit polyclonal anti-mouse Mrp2 antibody was used (1:1000). B, mouse monoclonal anti-human MRP2 antibody (M2III-5) that also recognizes rat and mouse Mrp2 was used (1:1000).
Vesicular Transport Assays. Vesicular transport assays were performed at 37°C in buffer consisting of 100 mM KCl, 50 mM Hepes/KOH, 10 mM MgCl, 10 mM creatine phosphate, and 100 μg/ml creatine, pH 7.4, in the presence or absence of 4 mM ATP. Uptake of E217βG into membrane vesicles was studied following the rapid filtration method as described previously (van de Wetering et al., 2007). We chose an uptake time of 2 min that appeared to be within the linear phase of the time-dependent uptake (data not shown). ATP-dependent transport was calculated by subtracting the transported amount of E217βGin the absence of ATP from that in its presence. Dose-response data were evaluated by nonlinear regression analysis (Origin software, version 7.5; OriginLab Corp., Northampton, MA) using a sigmoidal Emax model to estimate the maximum transport rate (Vmax), the substrate concentration yielding half-maximal transport (Km), and the sigmoidicity parameter n (Hill coefficient).
Results
Generation of MDCKII Cell Lines Expressing Mouse Mrp2. MDCKII parental cells were stably transduced with a retroviral expression construct in which we had cloned the mouse Mrp2 cDNA. Expression of mouse Mrp2 mRNA in the MDCKII clones obtained was verified by real-time PCR, and in selected clones protein expression was quantified by Western blotting (data not shown). We chose a MDCKII-mMrp2 clone with an intermediate protein level, apparently comparable with the previously established MDCKII cell line expressing human MRP2 (Evers et al., 1998) (Fig. 1B). For this purpose we used the M2III-5 antibody that recognizes both orthologs (Vlaming et al., 2006). It is unknown whether this antibody has exactly the same affinity for both proteins, but similar detection of human MRP2 and mouse Mrp2 in Sf9 membrane vesicles (Fig. 1B) suggests that there are no pronounced differences in affinity. In the selected MDCKII clone and in Sf9 membrane vesicles, the presence of mouse Mrp2 protein was additionally verified with an antibody specific for mouse Mrp2 (Fig. 1A).
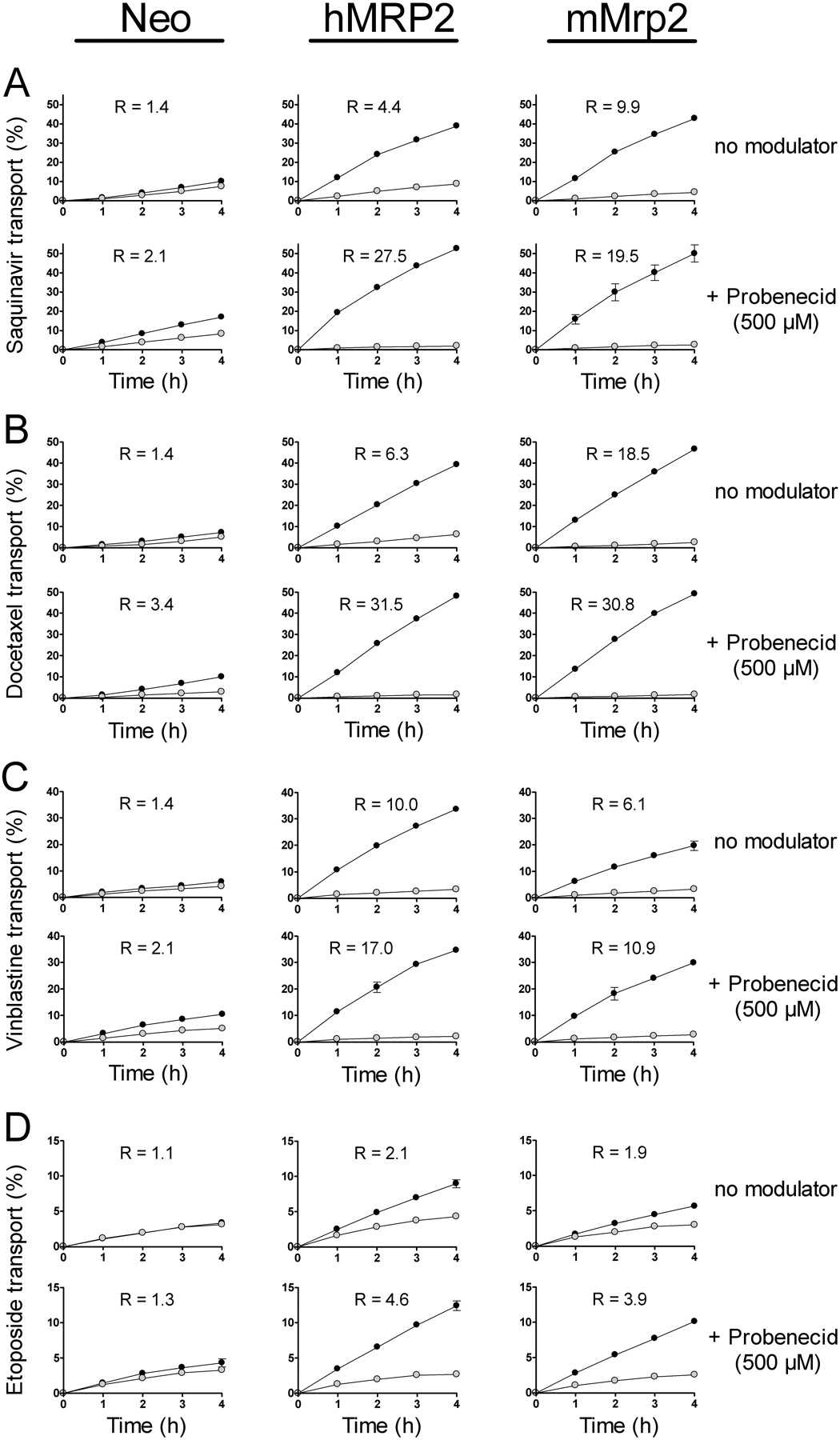
Transepithelial Transport of Typical MRP2 Substrates. Transport studies were performed using MDCKII-mMrp2 cells, previously generated MDCKII-hMRP2 cells, and as a control MDCKII-Neo cells that express only very low levels of endogenous canine Mrp2 (Evers et al., 1998). Saquinavir, docetaxel, vinblastine, and etoposide are known human MRP2 substrates and were applied at a concentration of 5 μM (Huisman et al., 2002, 2005). For all substrates, apically directed transport by human MRP2 and mouse Mrp2 was observed (Fig. 2, A–D, upper rows in each panel). In all cases transport ratios (R), defined as apically directed transport values divided by basolaterally directed transport values, were >1. This result demonstrates that all these drugs are substrates of both transporters. The transport efficiency, however, differed between the cell lines. For saquinavir and docetaxel the transport ratios were 2- and 3-fold higher in the MDCKII-mMrp2 cells compared with the MDCKII-hMRP2 cells, whereas vinblastine was more efficiently transported by human MRP2 (transport ratio of 10.0 versus 6.1). Etoposide was found to be a poor substrate for both human MRP2 and mouse Mrp2, with transport ratios of 2.1 and 1.9, respectively. Consequently, the order of transport efficiency (at a 5 μM drug concentration) for human MRP2 was vinblastine > docetaxel > saquinavir > etoposide and for mouse Mrp2 was docetaxel > saquinavir > vinblastine > etoposide. The very low apically directed transport of the substrates studied in the Neo cell line may be caused by low levels of endogenous canine Mrp2.
Measuring drug accumulation in the cell layers provided further evidence for the differential transport efficiency of human and mouse MRP2 (Fig. 3,  ). Cellular drug accumulation was determined 4 h after addition of the drug to the apical compartment. Similar results were observed when the drug was added to the basolateral compartment (data not shown). Cellular accumulation of saquinavir and docetaxel was lower in the MDCKII-mMrp2 cells than in the MDCKII-hMRP2 cells, in agreement with the higher transport by mouse Mrp2. Cellular vinblastine accumulation was lower in the MDCKII-hMRP2 cells, consistent with the higher vectorial transport rate for this compound found in this cell line. For etoposide drug accumulation was extremely low in all cell lines (approximately 0.1%), but significantly lower in the MDCKII-mMrp2 cells despite equal transport ratios. Taken together, these data demonstrate species-dependent differences in transport efficiencies for most of the drugs tested.
). Cellular drug accumulation was determined 4 h after addition of the drug to the apical compartment. Similar results were observed when the drug was added to the basolateral compartment (data not shown). Cellular accumulation of saquinavir and docetaxel was lower in the MDCKII-mMrp2 cells than in the MDCKII-hMRP2 cells, in agreement with the higher transport by mouse Mrp2. Cellular vinblastine accumulation was lower in the MDCKII-hMRP2 cells, consistent with the higher vectorial transport rate for this compound found in this cell line. For etoposide drug accumulation was extremely low in all cell lines (approximately 0.1%), but significantly lower in the MDCKII-mMrp2 cells despite equal transport ratios. Taken together, these data demonstrate species-dependent differences in transport efficiencies for most of the drugs tested.
Stimulation of Transepithelial Transport by Probenecid. Several reports showed that probenecid is able to considerably increase the human MRP2-mediated transport of many neutral or basic lipophilic drugs (Huisman et al., 2002, 2005; Zelcer et al., 2003). Here we investigated whether mouse Mrp2 is stimulated by probenecid in a similar manner. In agreement with the literature, we found that 500 μM probenecid stimulated human MRP2-mediated transport (Fig. 2, lower rows in each panel). Transport ratios increased 6-fold for saquinavir, 5-fold for docetaxel, and approximately 2-fold for both vinblastine and etoposide. In the MDCKII-mMrp2 cells, probenecid also stimulated transport of these substrates, but to a lesser extent, resulting in an increase of approximately 2-fold for all substrates tested. The extent of probenecid stimulation was mirrored by the decreases in cellular drug accumulation (Fig. 3, ▪). In the MDCKII-hMRP2 cells, probenecid treatment resulted in a more pronounced reduction of intracellular concentrations for saquinavir and docetaxel compared with vinblastine and etoposide. For MDCKII-mMrp2 cells stimulation with probenecid only led to modest decreases of intracellular drug concentration for all substrates. This analysis confirmed the quantitative difference in the effect of probenecid treatment on drug transport by human MRP2 and mouse Mrp2 and the fact that the extent of stimulation can be highly dependent on the transported substrate.

Transepithelial transport of the MRP2 substrates saquinavir (A), docetaxel (B), vinblastine (C), and etoposide (D) through MDCKII-Neo, -hMRP2, and -mMrp2 monolayers. Transport was measured in the absence of a MRP2 modulator (upper rows) and in the presence of probenecid (lower rows). The substrates were used at a concentration of 5 μM and probenecid was used at 500 μM. Substrates were applied to either the apical or the basolateral side of the monolayers and the relative amount appearing on the other side was determined. •, translocation from the basolateral to the apical side; ○, apical-to-basolateral translocation. Results are expressed as mean values (n = 3) of relative transport ± S.D. (error bars are frequently within the symbols). The transport ratio (R) was calculated as the quotient of apically directed transport and basolaterally directed transport at 4 h.

Cellular accumulation of the MRP2 substrates saquinavir (A), docetaxel (B), vinblastine (C), and etoposide (D) in MDCKII-Neo, -hMRP2, and -mMrp2 monolayers and the effect of probenecid. The radioactive substrates were applied on the apical side of the monolayer at a concentration of 5 μM. After 4 h cells were washed and the radioactivity in the filters was determined. Results are expressed as mean relative radioactivity (n = 3) ± S.D.  , drug uptake in the absence of MRP2 modulators; ▪, drug uptake in the presence of probenecid (500 μM). *, significant difference (p < 0.05, using Student's unpaired, two-tailed t test). Lines with asterisks indicate whether drug uptake without modulator is significantly different between human and mouse MRP2/Mrp2-expressing cell lines.
, drug uptake in the absence of MRP2 modulators; ▪, drug uptake in the presence of probenecid (500 μM). *, significant difference (p < 0.05, using Student's unpaired, two-tailed t test). Lines with asterisks indicate whether drug uptake without modulator is significantly different between human and mouse MRP2/Mrp2-expressing cell lines.
We also compared the probenecid concentration dependence for stimulation of saquinavir transport by human MRP2 and mouse Mrp2. As displayed in Fig. 4A, maximal stimulation was found at 500 μM probenecid for both human MRP2 and mouse Mrp2. Also in this experiment the saquinavir transport ratio increased up to 6-fold for the human and only up to 2-fold for the mouse ortholog. At higher probenecid concentrations transport rates declined again in both cell lines.
Modulation of Transepithelial Transport by Sulfanitran and Sulfinpyrazone. Sulfanitran and sulfinpyrazone have also been shown to stimulate human MRP2 transport activity (Huisman et al., 2002; Zelcer et al., 2003). We therefore assessed the effect of these drugs (at 500 μM) on the transport activity of human MRP2 and mouse Mrp2 in a pilot experiment (see Supplemental Data Table 1). For human MRP2 the results obtained with sulfanitran and sulfinpyrazone were roughly similar to the results obtained with probenecid, albeit less pronounced: sulfanitran and sulfinpyrazone also stimulated saquinavir, docetaxel, and, to a lesser extent, vinblastine and etoposide transport. In contrast, in the cells expressing mouse Mrp2, sulfanitran showed at best only a weak stimulation of transepithelial transport for all of the compounds tested. Sulfinpyrazone hardly stimulated vinblastine or etoposide transport in these cells, whereas saquinavir and docetaxel transport was even inhibited.
These results led us to investigate the effect of a broader concentration range of the modulators on saquinavir transport. Figure 4, B and C, shows that human MRP2 can be stimulated by sulfanitran and sulfinpyrazone with maximum stimulation at 500 and 1000 μM, respectively. Mouse Mrp2 was at best only slightly stimulated by sulfanitran at concentrations up to 500 μM, and transport was inhibited at higher concentrations. In contrast to its effect on human MRP2, sulfinpyrazone only inhibited mouse Mrp2-mediated transport in a dose-dependent manner. At 1000 μM sulfinpyrazone, which maximally stimulated human MRP2, transport in MDCKII-mMrp2 cells was nearly abolished, with a transport ratio close to that found in the Neo control cells. Sulfinpyrazone thus has opposite effects on saquinavir transport by human MRP2 and mouse Mrp2.
Modulation of E217βG Transport by Human MRP2 and Mouse Mrp2 in Sf9 Vesicular Transport Experiments. To further increase insight into differential transport and modulation properties of human MRP2 and mouse Mrp2, we also studied transport of the glucuronide conjugate E217βG in vesicular uptake assays and its modulation by probenecid, sulfanitran, and sulfinpyrazone. Sf9 vesicles contained either human MRP2 or mouse Mrp2 in their membranes, and ATP-dependent uptake of the amphipathic anion E217βG was measured. Using a fixed substrate concentration (1 μME217βG) and increasing modulator concentrations (10–1000 μM), we found that probenecid stimulated mouse Mrp2 more efficiently than human MRP2 (Fig. 5A), a clear contrast to the modulation of saquinavir transport seen in Fig. 4A. At these low substrate concentrations, E217βG uptake was increased up to 6-fold for mouse Mrp2 and only up to 2.5-fold for human MRP2. Sulfanitran, on the other hand, stimulated human MRP2 more than mouse Mrp2, at least at concentrations greater than 100 μM (maximum stimulation of 15-fold versus 7-fold, respectively) (Fig. 5B). Note that sulfanitran had hardly stimulated saquinavir transport by mouse Mrp2 in the transwell system (Fig. 4B). Sulfinpyrazone was a potent stimulator of human MRP2 (8-fold increased transport at a concentration of 500 μM) (Fig. 5C). Mouse Mrp2 could be stimulated only to some extent at medium modulator concentrations (approximately 2-fold up to 100 μM), but then transport decreased again at higher sulfinpyrazone concentrations. Together, these data illustrate that modulator effects for mouse and human MRP2 can be strongly dependent on the species and on the transported substrate used.

Dose-dependent effect of MRP2 modulators on the transport of saquinavir in MDCKII-Neo, -hMRP2, and -mMrp2 cells. Probenecid (A), sulfanitran (B), and sulfinpyrazone (C) were applied with increasing concentrations (only concentrations at which no cell toxicity was observed are displayed). Saquinavir (5 μM) was added to the apical or basolateral side of the monolayers (n = 1). After 4 h transport to the opposite compartment was measured and the transport ratio (apically directed transport divided by basolaterally directed transport) was calculated and plotted against the modulator concentration. Note the different axis scales for transport ratios between panels.
We next compared the concentration-dependent transport of E217βG mediated by the two transporters (Fig. 6, A and B). As described before, under control conditions (no modulator) human MRP2 displayed a typical S-shaped curve with a Hill coefficient of 1.6, which is indicative of positive homotropic cooperativity (Bodo et al., 2003; Zelcer et al., 2003). The mouse Mrp2 control curve also displayed a sigmoidal shape, albeit slightly less pronounced than that for human MRP2 (Hill coefficient of 1.4) (Table 1). Clearly, the transport of E217βG by both human and mouse MRP2 did not display Michaelis-Menten kinetics (which would yield a Hill coefficient of 1). The apparent difference in the maximal transport rates (Vmax) (Table 1) might be caused by species differences, although some effect of different expression levels of the transporters cannot be excluded.
Kinetic parameters of E217βG transport by human MRP2 and mouse Mrp2 in the presence of probenecid, sulfanitran, and sulfinpyrazone
Nonlinear regression results from the data shown in Fig. 7 are presented. Data were fitted to the Hill equation (unweighted). Modulators are used at a concentration of 500 μM.
The results in Fig. 5 show that in most cases the MRP2 modulators reached their maximal stimulatory effect at approximately 500 μM when 1 μME217βG was used as a substrate. We therefore investigated how this concentration of the modulators influenced the concentration-dependent transport of E217βG. First we tested the effect of probenecid on transport by human MRP2 and mouse Mrp2 (Fig. 7A). Because this drug modestly stimulated human MRP2-mediated E217βG transport over the whole concentration range of the substrate, we observed a modest increase of Vmax and a slight decrease of Km, whereas the shape of the curve remained sigmoidal with no alteration of the Hill coefficient (Table 1). In contrast, mouse Mrp2 could be stimulated much more prominently at low E217βG concentrations (as also seen in Fig. 5A). Consequently, the mouse Mrp2 curve changed from a sigmoidal to a more hyperbolic (Michaelis-Menten) type, illustrated by a clear decrease of the Hill coefficient to 1.1 (Fig. 7A; Table 1). The Vmax was also clearly increased. Hence, E217βG transport by mouse Mrp2 was also enhanced by probenecid over the whole substrate concentration range tested and in particular at low concentrations.
Sulfanitran, on the other hand, was able to strongly stimulate both transporters at low concentrations of E217βG, with the most pronounced effect seen for human MRP2-mediated transport (Figs. 5B and 7B). Accordingly, both curves changed from sigmoidal to a more Michaelis-Menten type with a marked decrease in the Hill coefficient and in the Km (Table 1). The decrease in Km was more pronounced for human MRP2, whereas the Vmax decreased for both mouse Mrp2 and human MRP2 by 20 to 25%.
Sulfinpyrazone showed the most striking difference between the human and mouse MRP2 orthologs (Fig. 7C). E217βG transport by human MRP2 was stimulated, especially at low substrate concentrations and more modestly at high substrate concentrations, resulting in a slight decrease in the Km and a modestly higher Vmax value (Table 1). The transport curve changed to a Michaelis-Menten type with a Hill coefficient of approximately 1. In contrast, mouse Mrp2 transport of E217βG was strongly inhibited compared with the control curve at all concentrations greater than 2.5 μM. Note, though, that there still was a basic, albeit very low, transport at all concentrations of E217βG (Fig. 7C). This finding suggests that the applied concentration of sulfinpyrazone (500 μM) caused primarily a dramatic decrease in the maximal transport rate of E217βG.
Discussion
The recent generation of Mrp2 knockout mouse strains (Chu et al., 2006; Vlaming et al., 2006) has yielded important tools to study in vivo MRP2 functions. To properly translate results obtained in these mice to the human situation, it is essential to be able to systematically compare the transport and modulation properties of human MRP2 and mouse Mrp2. For this purpose we generated and characterized polarized cell lines expressing mouse Mrp2 and used these in parallel with previously generated mouse Mrp2-expressing Sf9 vesicles and the analogous systems expressing human MRP2. This set of tools allowed us to study the species-specific behavior of mouse Mrp2 and human MRP2 for both lipophilic drug substrates, which are best studied in the polarized cell system, and for anionic substrates, which are best studied in the vesicle transport model. Note that our recent work in Mrp2 knockout mice revealed that Mrp2 has a more pronounced impact on the pharmacokinetics of a lipophilic drug substrate than was previously expected (Lagas et al., 2006).

Dose-dependent effect of MRP2 modulators on the transport of E217βG(1 μM, for 2 min at 37°C) by hMRP2 and mMrp2 in Sf9 membrane vesicles. Vesicles containing hMRP2 or mMrp2 were incubated with increasing concentrations of probenecid (A), sulfanitran (B), and sulfinpyrazone (C). Each data point represents the mean of an experiment in triplicate ± S.D. Note the different axis scales for transport rates between panels.

Concentration-dependent transport of E217βG by hMRP2 and mMrp2. Vesicles containing hMRP2 or mMrp2 were incubated with various concentrations of E217βG at 37°C for 2 min. ATP-dependent transport is plotted for the low concentration range of 1 to 25 μM (A) and for the entire concentration range of 1 to 200 μM (B). Each data point represents the mean of an experiment in triplicate ± S.D. Note that the Vmax could not be accurately determined experimentally because of E217βG solubility problems at concentrations >200 μM.
Our results demonstrate that there can be pronounced differences in transport and modulation properties between human MRP2 and mouse Mrp2, despite the very extensive overlap in substrate specificity. Whereas human MRP2 transported a panel of lipophilic anticancer and anti-human immunodeficiency virus drugs (at 5 μM) in the order of efficiency of vinblastine > docetaxel > saquinavir > etoposide, for mouse Mrp2 this order was docetaxel > saquinavir > vinblastine > etoposide (Fig. 2). Moreover, several MRP2-modulating compounds showed much better stimulation of human MRP2 in the transport of lipophilic drugs than of mouse Mrp2. Sulfinpyrazone even strongly inhibited saquinavir transport by mouse Mrp2, whereas it stimulated human MRP2 (Fig. 4). As described before with various MRP2 isoforms (Bodo et al., 2003; Zelcer et al., 2003; Ninomiya et al., 2005, 2006), we found that the modulation behavior is also strongly dependent on the transported substrate used and on its concentration. For example, probenecid stimulated transport of E217βG much better for mouse Mrp2 than for human MRP2, whereas the opposite was true for the stimulation of saquinavir transport (Figs. 4A and 5A). Similarly, whereas sulfanitran hardly stimulated or, at higher concentrations, even inhibited saquinavir transport by mouse Mrp2, E217βG transport was strongly stimulated (Figs. 4B and 5B). Among the modulators we tested, sulfinpyrazone demonstrated the most pronounced species dependence, stimulating transport of both lipophilic saquinavir and anionic E217βG for human MRP2 and strongly inhibiting this transport for mouse Mrp2 (Figs. 4C, 5C, and 7C). Nevertheless, it remains to be seen whether this inhibition of mouse Mrp2 by sulfinpyrazone would be true for all transported substrates, given the variation in behavior observed with various MRP2 isoforms. Taken together, these extensive variations in behavior illustrate that each substrate and each substrate-modulator combination should be studied in its own right and with the proper MRP2/Mrp2 species isoforms to allow meaningful translation from mouse to human. The newly generated MDCKII-mMrp2 cell lines provide a useful additional tool for this purpose.
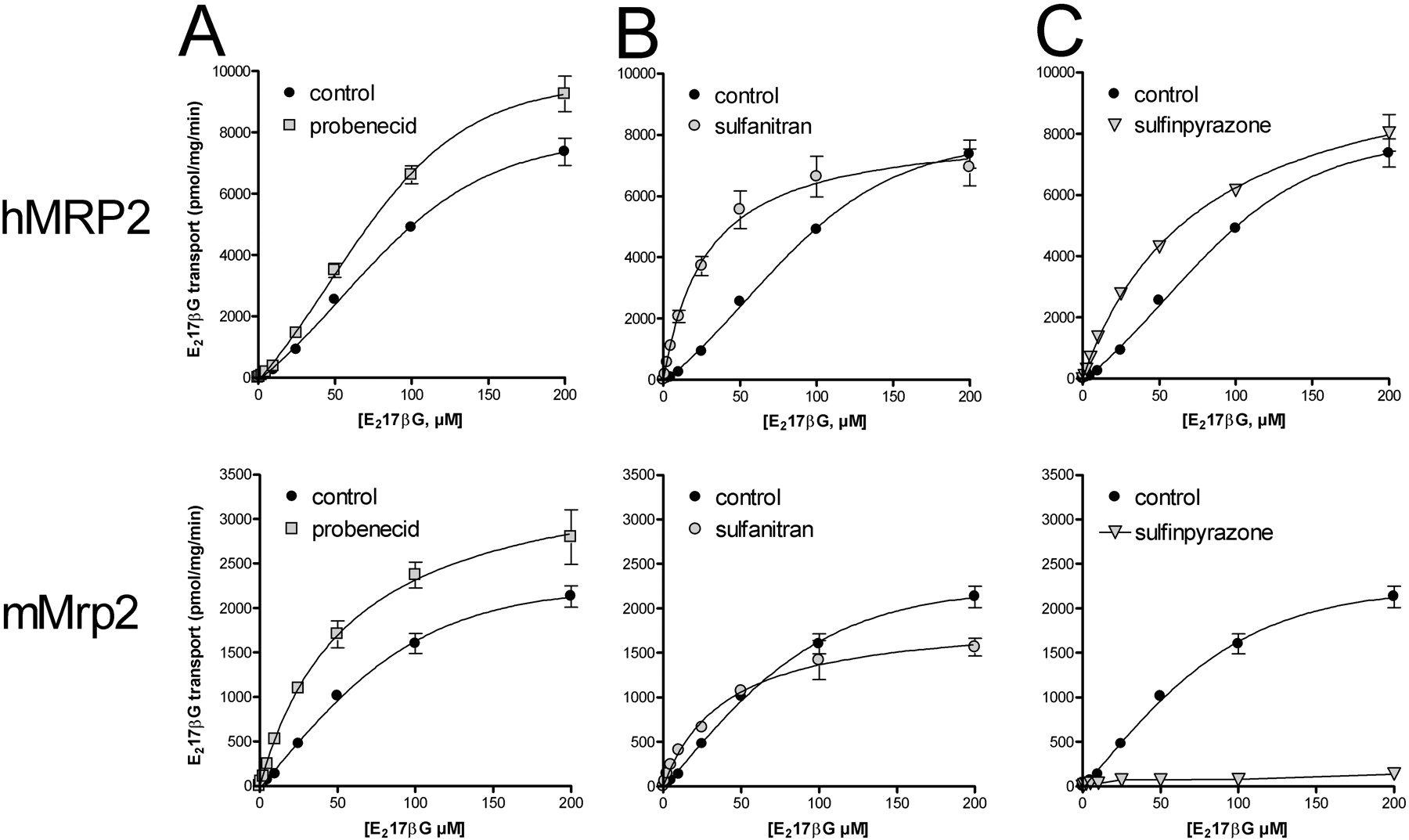
Effect of probenecid (A), sulfanitran (B), and sulfinpyrazone (C) on the concentration-dependent transport of E217βG by hMRP2 and mMrp2. Vesicles containing hMRP2 (upper row) or mMrp2 (lower row) were incubated with various concentrations of E217βG at 37°C for 2 min in the presence and absence of MRP2 modulators (each at 500 μM). Each data point represents the mean of an experiment in triplicate ± S.D.
Highly individually specific behavior has also been seen in previous comparisons of Mrp2 orthologs from different species, including mouse, rat, dog, monkey, and human (Cui et al., 1999; Ninomiya et al., 2005, 2006), notwithstanding the very extensive overlap in transported substrates. This is especially true for the effect of modulators. For instance, Ninomiya et al. (2005, 2006) showed different intrinsic transport activities for several substrates by mouse, rat, dog, and monkey Mrp2. They further reported that 4-methylumbelliferone glucuronide stimulated the transport of E217βG by mouse and monkey Mrp2 but inhibited the transport by rat and dog Mrp2. As discussed extensively by Borst et al. (2006), it seems most likely that the complex interactions of MRP2 with transported substrates and modulators are related to the presumably large size and promiscuity of the drug binding and transport site. This site needs to accommodate a large variety of compounds of different structures and sizes and can be expected to be structurally quite flexible, in analogy to the equally promiscuous binding site of cytochrome P450 3A4 (Ekroos and Sjogren, 2006). It is not difficult to imagine that such a binding site can simultaneously bind many different combinations of two or more compounds (transported substrate and modulator), which may either stimulate the transport process, delay it, or even completely inhibit it. Many different amino acids, mostly positioned in or near transmembrane segments, contribute to the binding and transport properties of these complex binding sites of multidrug transporters such as MRP2, MRP1, and P-glycoprotein. Even single amino acid substitutions can dramatically affect the substrate preference of MRP2, as demonstrated by the abrogation of methotrexate transport by a single amino acid change in the putative last transmembrane segment of the protein (Ito et al., 2001). The amino acid identity between mouse Mrp2 and human MRP2 is 78% (Nies and Keppler, 2007) and amino acid substitutions occur throughout the protein. It is therefore not surprising that there are many differences between mouse Mrp2 and human MRP2 in detailed interactions with transported substrates and modulators.
It should be noted that there can be polymorphic variations in mouse Mrp2 between mouse strains. For instance, comparison of the inbred strains BALB/c, AKR/J, C57L/J, and C57BL/6J revealed three to four amino acid substitutions in Mrp2 (Ninomiya et al., 2006). Although these differences (4/1544, i.e., 0.26%) are small compared with the human/mouse differences (22%), we cannot exclude the possibility that they might sometimes affect the behavior of the mouse Mrp2 in vivo. This possibility should be taken into account when one is choosing the optimal background strain of the Mrp2 knockout allele for detailed analyses of in vivo Mrp2 function.
An interesting aspect in the modulation of E217βG transport is the sigmoidal kinetics that we observed for both mouse Mrp2 and human MRP2 (Fig. 6), suggesting positive homotropic cooperativity. The shift from sigmoidal to hyperbolic (Michaelis-Menten-type) kinetics is the most important factor in explaining the stimulation of E217βG transport by various modulators (Fig. 7) (Bodo et al., 2003; Zelcer et al., 2003), although changes in Vmax can also contribute. In this respect it is remarkable that Ninomiya et al. (2006) did not observe sigmoidal kinetics for mouse Mrp2-mediated E217βG transport, even though we are using the same expression construct that they provided to us. The only obvious difference concerned the time of uptake measured, which was 30 s in their assay system and 2 min in ours. However, because E217βG uptake in our hands was completely linear over 2 min (not shown), we do not understand why this difference would result in qualitatively different kinetics. We note that this same group also did not find clear sigmoidal kinetics for E217βG transport by rat Mrp2, in contrast to another group (Gerk et al., 2004). As suggested by Borst et al. (2006) and Ninomiya et al. (2006), there may be subtle differences in experimental conditions between the laboratories. In any case, Ninomiya et al. (2006) also observed clear stimulation of mouse Mrp2-mediated transport of E217βG by modulators such as indomethacin and taurocholate, suggesting that modulation as such does not appear to be critically dependent on the experimental conditions. Whether this stimulation resulted from a decrease in Km or an increase in Vmax or both could not be assessed from the data presented in that study.
In summary, although human MRP2 and mouse Mrp2 have similar substrate specificities, we observed marked differences regarding their transport efficiencies and modulation properties. This result means that each substrate and each substrate-modulator combination of interest should be studied individually using the proper MRP2/Mrp2 species isoforms. The data and models described in this study will facilitate the interpretation and design of future experiments in mice with respect to Mrp2 functionality and will help to extrapolate the results to the human situation.
Acknowledgments
We are indebted to Drs. K. Ito and T. Horie for providing the mouse Mrp2 cDNA and baculovirus expression construct and to Dr. G. L. Scheffer and Professor J. M. Fritschy for providing the primary antibodies used in this study. We acknowledge Professor J. Drewe for his help with the regression analysis. For technical assistance and helpful discussions we thank M. Vlaming, C. van der Kruijssen, and J. Lagas. We further thank Professor P. Borst for critical reading of the manuscript.
Footnotes
-
We gratefully acknowledge the Swiss National Science Foundation and the Novartis Foundation for funding the research fellowship of C.Z. at The Netherlands Cancer Institute.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.019620.
-
ABBREVIATIONS: MRP2/Mrp2, multidrug-resistance protein 2; ABCC/Abcc, ATP-binding cassette transporter family C; MDCK, Madin-Darby canine kidney; Sf9, Spodoptera frugiperda; hMRP2, human MRP2; mMrp2, mouse Mrp2; E217βG, estradiol-17-β-d-glucuronide; GF120918, N-(4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide; PBS, phosphate-buffered saline.
-
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.

- Received November 2, 2007.
- Accepted January 2, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}