


Abstract
The widely prescribed lipid-lowering statins are considered to be relatively safe drugs. However, the risk of severe myopathy and drug interactions as a consequence of statin therapy provides a challenge for development of novel cholesterol-lowering agents, targeting enzymes other than HMG-CoA reductase. The novel pyridylethanol-(phenylethyl)amine derivative, (2-((3,4-dichlorophenethyl)(propyl)-amino)-1-(pyridin-3-yl)ethanol (LK-935), blocking lanosterol 14α-demethylase, was demonstrated to efficiently reduce cholesterol biosynthesis. The drug interaction potential of LK-935 was investigated and compared with that of atorvastatin and rosuvastatin in primary human hepatocytes. Clear evidence was provided for the induction of CYP3A4 by LK-935. LK-935 was proved to be a potent human pregnane X receptor (hPXR) activator as a prerequisite for the transcriptional activation of CYP3A4 gene; however, the rapid metabolism of LK-935 in primary hepatocytes prevented maximal CYP3A4 induction. Therefore, the induction of CYP3A4 by LK-935 may be prone to mild or negligible drug interactions. However, because CYP3A4 and also CYP2C9 play a significant role in LK-935 metabolism, the inhibition of these cytochromes P450 by coadministered drugs may lead to some increase in the LK-935 concentration required for the potent induction of CYP3A4. Rosuvastatin was found to increase human constitutive androstane receptor (hCAR)-mediated transcription of CYP3A4, CYP2C9, and CYP2B6 genes, predicting the consequent potential for drug interactions with several coadministered drugs. Activation of hCAR and hPXR by atorvastatin and the subsequent induction of not only CYP2B6 and CYP3A4 but also of CYP2C9 present an additional target by which atorvastatin, a widely used cholesterol-lowering drug, can modify the kinetics of numerous drugs.
Cardiovascular disease is one of the major causes of mortality in the developed world. The imbalance of cholesterol homeostasis and a high plasma concentration of low-density lipoprotein-cholesterol are risk factors for cardiovascular diseases (atherosclerosis, coronary heart disease, and myocardial infarction). Therefore, cholesterol-lowering therapy is essential for prevention of the progression of cholesterol-laden plaques in vessel linings (Maas and Böger, 2003; LaRosa, 2007). Lipid-modifying therapy to decrease elevated cholesterol concentrations constitutes the inhibition of de novo cholesterol biosynthesis in the liver and the decrease of the dietary cholesterol uptake from intestine (LaRosa, 2007; Bays et al., 2008; Koh et al., 2008; Sanossian and Ovbiagele, 2008). The most commonly prescribed cholesterol-lowering agents are the statins, the HMG-CoA reductase inhibitors (Rallidis et al., 2007). Competitive inhibition of HMG-CoA reductase by the statins efficiently decreases the conversion of HMG-CoA to mevalonate, the rate-limiting step of cholesterol biosynthesis (Fig. 1A). Even if the lipid-lowering statins are still considered to be relatively safe drugs that are taken by millions of patients worldwide, considerable attention has been paid recently to the statin-based risk of muscular adverse drug reactions and hepatotoxicity (Martin and Krum, 2007). Although officially only 2 to 3% of patients get muscle aches and cramps, considering that 5 to 10% of the population in developed societies are treated with statins, this occurrence represents a major health risk problem for development of severe myopathies, rhabdomyolysis, impaired cognitive function, and nephropathies and can even lead to death (Tiwari et al., 2006; Alsheikh-Ali et al., 2007; Armitage, 2007; Rallidis et al., 2007).

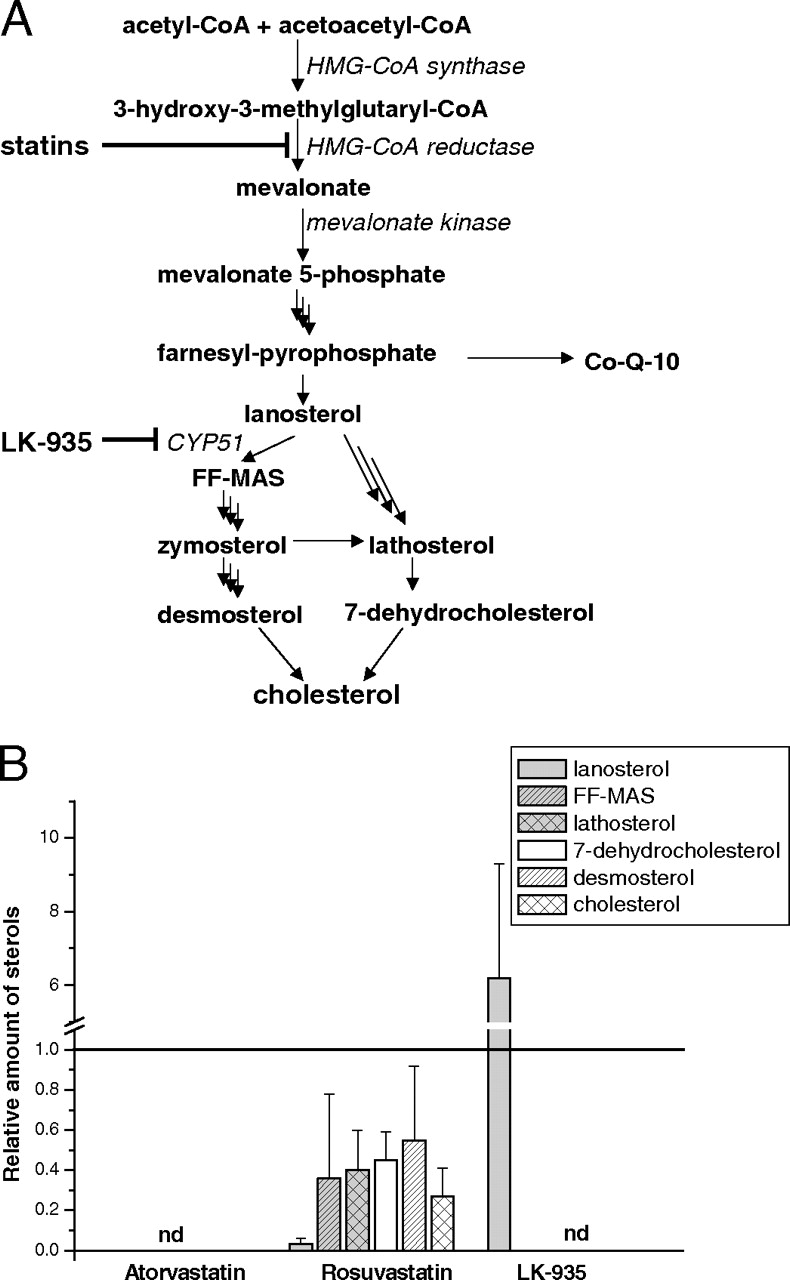
The action of hypolipidemic statins and LK-935 in de novo biosynthesis of cholesterol. A, the main intermediates and the sites of inhibition by statins and LK-935 are shown in the cholesterol biosynthesis pathway. B, HepG2 cells were treated for 24 h with statins (10 μM) and LK-935 (10 μM). The amounts of sterol intermediates were determined after a 4-h incubation with [3H]acetate. Controls for each independent experiment were assigned values of 1 and the results of treatments are expressed relative to the controls. CoQ-10, coenzyme Q; nd, not detectable.
Almost all statins used clinically have been reported to interact with the cytochrome P450 (P450) enzymes, leading to pharmacokinetic interactions with coadministered drugs (Shitara and Sugiyama, 2006). Lovastatin, simvastatin, and atorvastatin are both substrates and inhibitors of human CYP3A4, the main drug-metabolizing P450 enzyme, whereas CYP2C9 is the target of drug interactions caused by fluvastatin (Beaird, 2000; Shitara and Sugiyama, 2006). Approximately 60% of cases of statin-related rhabdomyolysis have been attributed to drug-drug interactions (Muscari et al., 2002; Sica and Gehr, 2002; Neuvonen et al., 2006; Tiwari et al., 2006). On the other hand, many compounds that are substrates for P450s also induce the transcription of P450 genes, resulting in elevated enzyme levels and metabolic functions. HMG-CoA reductase inhibitors have been proved to increase the expression of several P450s. Cerivastatin, simvastatin, atorvastatin, and fluvastatin have been shown to activate human constitutive androstane receptor (hCAR) and pregnane X receptor (PXR) and consequently to increase the transcription of the target genes, CYP2B6 and CYP3A4 (El-Sankary et al., 2001; Kocarek et al., 2002; Kobayashi et al., 2005). In contrast to these statins, pravastatin does not seem to interact with the P450 system, does not inhibit it substantially, and does not induce the transcription of P450 genes, possibly because of its hydrophilic nature (Beaird, 2000; El-Sankary et al., 2001; Schachter, 2004; Neuvonen et al., 2006).
It has been reported that derangements in mevalonate kinase but not in more distal enzymes of cholesterologenesis are associated with the skeletal myopathy provoked by statins (Buhaescu and Izzedine, 2007). By inhibition of the major regulatory enzyme of cholesterol biosynthesis, HMG-CoA reductase, statins also decrease the level of an essential metabolite, coenzyme Q (Fig. 1A), which probably contributes to muscle-related complications. Thus, the development of novel cholesterol-lowering agents, targeting enzymes below the farnesyl pyrophosphate branchpoint of cholesterol biosynthesis, offers the possibility for the elimination of some adverse effects. The lanosterol 14α-demethylase CYP51 is a potential anticholesterolemic drug target. CYP51 is responsible for the removal of the 14α-methyl of lanosterol and 24,25-dihydrolanosterol to produce follicular fluid meiosis activating sterol (FF-MAS) (Stromstedt et al., 1996; Debeljak et al., 2003). A novel pyridylethanol(phenylethyl)amine derivative, LK-935 developed by Lek Pharmaceuticals d.d. (Ljubljana, Slovenia) has been identified as a postlanosterol cholesterol biosynthesis inhibitor. Structure-activity relationship studies of the binding with overexpressed human CYP51 indicate that the pyridine binds within the heme pocket in an analogy with azoles (Korosec et al., 2008).
The goal of the present study was to investigate whether the drug interaction potential of the statin-based cholesterol-lowering therapy could be eliminated by using the CYP51-targeted LK-935. The P450-inducing capacity of LK-935 was examined in primary cultures of human hepatocytes and compared with that of atorvastatin and rosuvastatin. We provided evidence for a role of hPXR in the induction of the CYP3A4 gene by LK-935. We also attempted to explain the contradiction of the strong PXR activation potential and the weak CYP3A4-inducing capacity exerted by LK-935.
Materials and Methods
Chemicals. LK-935, atorvastatin, and rosuvastatin were provided by Lek Pharmaceuticals d.d. Rifampicin and dimethyl sulfoxide were the products of Sigma Chemie GmbH (Deisenhofen, Germany). Chemicals for hepatocyte isolation, cell culture media, and supplements were purchased from Sigma Chemie GmbH and Merck (Darmstadt, Germany). Commercial and laboratory sterol standards, lanosterol, lathosterol, 7-dehydrocholesterol, desmosterol, cholesterol, and FF-MAS, were obtained from Steraloids (Newport, RI) and Laboratory of Reproductive Biology (The Juliane Marie Center for Children, Women and Reproduction, University Hospital of Copenhagen, Copenhagen, Denmark). [3H]Acetate was purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA).
Sterol Profile Analysis in HepG2 Cells. Human hepatoma cells (HepG2, No. HB-8065; American Type Culture Collection, Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal calf serum and 1 mM glutamine. After 24 h, the medium was changed, and the cells were treated for 24 h with LK-935 (10 μM) and atorvastatin (10 μM) or rosuvastatin (10 μM) as the reference controls. The growth medium was replaced, and [3H]acetate was added in a concentration of 40 μCi/ml. After a 4-h incubation, the cells were harvested, washed, and homogenized. The internal standard ergosterol (0.25 mg/ml) was added before the extraction procedure. After addition of 0.3 M NaH2PO4 (pH 1.0), sterols were extracted in n-heptane-isopropanol (75:25 v/v) from cell homogenates. The organic phases were dried and dissolved in n-heptane. The samples were analyzed by high-performance liquid chromatography (radiodetection) for quantitation of lanosterol, FF-MAS, lathosterol, 7-dehydrocholesterol, desmosterol, and cholesterol using a Prism-RPN (250 × 4.6 mm, 5 μm column; Thermo Fisher Scientific Inc., Waltham, MA) and the mobile phase of 100% acetonitrile. The column was eluted at a rate of 1 ml/min at 40°C, and the effluent was analyzed by flow scintillation using an on-line radioactivity detector. The amounts of various sterols were normalized on ergosterol quantity and protein concentration. Three separate experiments with two replicas were performed. The results were expressed as the amounts of sterols in treated cells relative to untreated cells.
Isolation and Culture of Human Hepatocytes. Human livers (HH-089, HH-114, HH-129, and HH-272) were obtained from kidney transplant donors or from lobectomy segments (HH-269, HH-270, and HH-271) resected from adult patients for medical reasons unrelated to our research program (Transplantation and Surgical Clinic, Semmelweis University, Budapest, Hungary, and Centre Hospitalier Universitaire, Saint-Eloi, Montpellier, France). Permission to use the human tissues was obtained from the Hungarian and French Regional Committee of Science and Research Ethics. Clinical histories of the donors are shown in Table 1. Liver cells were isolated by the method of Bayliss and Skett (1996). Hepatocytes having viability better than 90%, as determined by trypan blue exclusion, were used in the experiments. The cells were plated at a density of 1.7 × 105 cells/cm2 in plastic dishes precoated with collagen in medium described by Ferrini et al. (1998). After overnight culture, the medium was replaced by serum-free medium. Forty-eight hours after serum deprivation, cells were cultured in the presence or absence of inducers for 12, 24, or 48 h. Hepatocytes were treated with rifampicin (5 μM), atorvastatin (10 μM), rosuvastatin (10 μM), or LK-935 (10 μM).
Clinical histories of human donors
P450 Enzyme Assays. Microsomal fraction from cultured human hepatocytes was prepared by differential centrifugation (van der Hoeven and Coon, 1974). Protein content of microsomes was determined by the method of Lowry et al. (1951), with bovine serum albumin as the standard. Published methods were followed to determine selective P450 enzyme activities: (S)-mephenytoin N-demethylation for CYP2B6 (Heyn et al., 1996), tolbutamide 4-hydroxylation for CYP2C9 (Miners and Birkett, 1996), (S)-mephenytoin 4′-hydroxylation for CYP2C19 (Srivastava et al., 1991), and nifedipine oxidation (Guengerich et al., 1986) for CYP3A4/5. The incubation mixture contained an NADPH-generating system (1 mM NADPH, 10 mM glucose 6-phosphate, 5 mM MgCl2, and 2 units/ml glucose-6-phosphate dehydrogenase), microsomes, and various selective substrates for P450 forms [(S)-mephenytoin, tolbutamide, or nifedipine]. The rates of enzyme activities were linearly dependent on the amount of microsomal protein added for the 10- to 30-min incubation period. The metabolic extraction procedures and high-performance liquid chromatography analyses were performed according to published methods (Guengerich et al., 1986; Srivastava et al., 1991; Heyn et al., 1996; Miners and Birkett, 1996). P450 enzyme assays were performed in triplicate and means ± S.D. were calculated. For comparison among untreated and treated groups, statistical analysis of the results was carried out using a two-tailed t test with p < 0.05 as the criterion for significance. Because of the high variation in basic P450 expression of human hepatocytes, the entire experiment was repeated in hepatocytes isolated from four to seven donors to confirm the results.
RNA Extraction and Quantitative RT-PCR. Total RNA was isolated from human hepatocytes using TRIzol reagent (Invitrogen, Carlsbad, CA). Ten million liver cells were homogenized in 1 ml of TRIzol reagent, and total RNA was extracted according to the manufacturer's instructions. The RNA was precipitated using ethanol and stored at –80°C for further analyses. RNA (3 μg) was reverse-transcribed into single-stranded cDNA using a Transcriptor First Strand cDNA synthesis kit (Roche Diagnostics GmbH, Mannheim, Germany), and then real-time PCR with human cDNA was performed using FastStart Taq DNA polymerase (LightCycler TaqMan Master; Roche Diagnostics GmbH) and probes for CYP2B6, CYP2C9, CYP2C19, and CYP3A4 (Universal ProbeLibrary kits; Roche Diagnostics GmbH). The quantity of target RNA relative to that of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase was determined. P450 mRNA levels were quantified by RT-PCR measurements in the same human hepatocytes in which P450 activities were measured. Statistical analysis of the results obtained by RT-PCR was preformed similarly to that of P450 activities.
Western Blot Analysis. Microsomal proteins prepared from human hepatocytes (2.5 μg/lane) were separated by SDS-polyacrylamide gel electrophoresis using 12% acrylamide. The gels were shaken in transfer buffer [40 mM glycine, 50 mM Tris (pH 8.3), 0.04% SDS, and 20% methanol] for 20 min before electroblotting onto a nitrocellulose membrane (GE Healthcare, Freiburg, Germany). For blocking, the membrane was incubated for 1 h in Tris-buffered solution (TBS) containing 5% nonfat dried milk and 0.1% Tween 20. Incubation with monoclonal mouse anti-human CYP3A (BD Gentest, Heidelberg, Germany) diluted 1:5000 in TBS containing 0.1% Tween 20 was performed for 2 h. After the blots were washed three times for 30 min in phosphate-buffered saline, they were exposed to the horseradish peroxidase-conjugated rabbit anti-mouse antibody (Dako Deutschland GmbH, Hamburg, Germany) in a dilution of 1:1000 in TBS containing 0.1% Tween 20. Three washing steps for 30 min in phosphate-buffered saline were done before the blots were exposed to Hyperfilm ECL (GE Healthcare) and the ECL Plus Western blotting reagents (GE Healthcare) following the manufacturer's instructions.
Cell Culture and Transfection. HepG2 cells were maintained in DMEM supplemented with 10% fetal calf serum, 1 mM glutamine, 10 mM sodium pyruvate, and 100 μg/ml penicillin and streptomycin. Transient transfections with 10 ng of expression plasmid containing hPXR or hCAR together with 100 ng of luciferase reporter constructs pGL3(CYP3A4/XREM[–7800/–7200]/–263/+11)LUC were performed in batches on 5 × 104 cells seeded into 24-well tissue culture plates using FuGENE 6 transfection agent (Roche Diagnostics, Basel, Switzerland) as described previously (Pascussi et al., 2003a). After 16 h, the medium was changed, and the cells were treated with atorvastatin (10 μM), rosuvastatin (10 μM), LK-935 (10 μM), rifampicin (3 μM), or solvent [0.1% dimethyl sulfoxide (DMSO)] in DMEM containing 5% delipidated and charcoal-treated calf serum. After a 24-h incubation, luciferase activities were measured as described previously (Pascussi et al., 2003a). Values are expressed as the mean ± S.D. for three experiments measured in triplicate. Reporter gene activity of the DMSO-treated cells served as a control and was set to unity.
In Vitro Kinetics of LK-935. Biotransformation of LK-935 was performed in a cell suspension (1.7 × 106 cells/ml) at 37°C in a humid atmosphere containing 5% CO2. The parent compound was added directly to the medium (final concentration of LK-935: 1 and 10 μM). Aliquots (0.5 ml) of the incubation mixtures were terminated by the addition of 0.5 ml of ice-cold acetonitrile at 0, 2, 4, 6, 8, 10, 15, 20, and 25 min, and the samples were stored at –80°C. Cell debris was separated by centrifugation, and 10 μlofthe supernatant was analyzed by LC-MS/MS for quantitation of the parent compound. For this purpose, an ODS-AQ 20 × 2.1 mm (5 μm) column (YMC Europe GmbH, Dimslaken, Germany) was used. Gradient elution was applied with mobile phase A consisting of 10 mM ammonium-formate and with acetonitrile as mobile phase B. The column was eluted at a rate of 0.4 ml/min at 40°C, and the effluent was analyzed by mass spectrometry. MS/MS measurements were obtained on a 3200 Qtrap hybrid (quadrupole-linear ion trap) tandem mass spectrometer (Applied Biosystems, Foster City, CA) equipped with a TurboV ion source. The ionization mode was electrospray in positive mode. The instrument was scanned in enhanced product ion (EPI) mode for structure identification and in multiple reaction monitoring (MRM) mode for quantitation. The source conditions were as follows: curtain gas, 35 units; spray voltage, 5000 V; source temperature, 350°C; nebulizing gas GS1, 50 units; drying gas GS2, 40 units; and declustering potential, 30 V. Two MRM transitions (the first transition as the quantifier and the second as the qualifier channel), 353/173 (collision energy: 37 eV) and 353/137 (collision energy: 49 eV), were monitored for LK-935 with dwell time of 150 ms.
Estimation of Clearance. The intrinsic clearance for hepatocytes (Clint) (ml/min/1.7 × 106 cells) was calculated from the decrease in the concentration of LK-935 as follows: Clint = β = (ln2/t1/2) (Houston, 1994). For scaling up the Clint value to obtain Clint per whole liver (g)/bw (kg), the following parameters were used: cell concentration in liver, 1.07 × 108 cells/1 g of liver (Wilson et al., 2003); average human liver weight, 1660 g; and average human body weight, 70 kg. The value for hepatic clearance (ClH) was calculated as follows:  where Qplasma = QH · plasma/blood ratio. To calculate ClH, QH = 19 ml of blood/min/kg, plasma/blood ratio = 0.57, and fu = 1 values were used, where QH is the hepatic blood flow, and fu is the unbound fraction of the compound. The hepatic extraction ratio was defined as E = ClH · QH.
where Qplasma = QH · plasma/blood ratio. To calculate ClH, QH = 19 ml of blood/min/kg, plasma/blood ratio = 0.57, and fu = 1 values were used, where QH is the hepatic blood flow, and fu is the unbound fraction of the compound. The hepatic extraction ratio was defined as E = ClH · QH.
Identification of LK-935 Metabolites. The chemical structures of LK-935 metabolites were determined in the incubation mixture using human hepatocytes. The analytical conditions were the same as those described for the parent compound. Metabolites were initially characterized by LC-MS/MS in full scan mode by comparing 0-min samples with 60-min samples. To improve sensitivity, the metabolites were also characterized using the MRM mode in which the MRM transitions of the parent drug were modified according to the mass shifts of the metabolites comparing with LK-935.
Inhibition Studies. Metabolite formation from LK-935 was determined in human liver microsomes. The incubation mixture contained the NADPH-generating system (1 mM NADPH, 10 mM glucose 6-phosphate, 5 mM MgCl2, and 2 units/ml glucose-6-phosphate dehydrogenase), human liver microsomes (1 mg of protein/ml), and LK-935 at various concentrations. After a 20-min incubation, reactions were terminated by the addition of ice-cold acetonitrile. The amounts of LK-935 metabolites produced were determined by LC-MS/MS. The analytical conditions were the same as those described for the parent compound except for the MRM transitions and collision energies, which were 311/173 (37 eV) and 311/137 (49 eV) for M1, 232/137 (45 eV) and 232/173 (45 eV) for M2, and 369/173 (45 eV) and 369/189 (45 eV) for M3. Inhibition of metabolite formation from LK-935 was carried out in the absence and presence of various P450 inhibitors (pentoxyresorufin for CYP2B6, sulfaphenazole for CYP2C9, tranylcypromine for CYP2C19, and ketoconazole for CYP3A4/5) (Bourrié et al., 1996; Ono et al., 1996; Salsali et al., 2004). Ki values (inhibition constants) were determined by using different concentrations of LK-935 (5, 10, and 20 μM) and inhibitors (0.2–10 μM). Ki values were calculated from Dixon plots of velocity–1 versus inhibitor concentration at the three LK-935 concentrations. The type of inhibition and the apparent Ki values were estimated from the intercept of three lines of Dixon plots and expressed as the mean ± S.D. of the intercepts.
Statistical Analysis. For comparison among several groups, statistical analysis of the results obtained in the experiments with human hepatocytes and in cell line transfection was carried out using a paired two-tailed t test with p < 0.05 as the criterion for significance (GraphPad InStat version 3.0; GraphPad Software, San Diego, CA).
Results
Sterol Profile of HepG2 Cells Treated with LK-935. De novo biosynthesis of cholesterol was determined in HepG2 cells after incubation with [3H]acetate. 3H-labeled sterol components of the postlanosterol part of cholesterol biosynthesis were isolated from the cells treated with LK-935 and two well known statins, atorvastatin and rosuvastatin. De novo synthesis of lanosterol, FF-MAS, lathosterol, 7-dehydrocholesterol, desmosterol, and cholesterol in treated cells was compared with that in untreated cells. Although the chemical structures of these sterols are similar, they can be separated and unequivocally identified using the reference standards. De novo produced cholesterol represented approximately 20% of the radiolabeled sterol pool, whereas lanosterol and lathosterol were the two major sterol intermediates with 25% of each. LK-935, being an efficient inhibitor of CYP51 (Korosec et al., 2008), caused a substantial accumulation of lanosterol and completely blocked the formation of cholesterol and postlanosterol intermediates (Fig. 1B). As was expected, neither cholesterol nor the sterol intermediates were produced in the cells treated with atorvastatin, the potent HMG-CoA reductase inhibitor. Rosuvastatin treatment also decreased the formation of lanosterol, cholesterol, and the intermediates between them; however, rosuvastatin did not seem to be as efficient a HMG-CoA reductase inhibitor as atorvastatin possibly because of its more hydrophilic character.
P450 Induction by LK-935 and Reference Statins in Human Hepatocytes. The P450-inducing potential of statin-type cholesterol-lowering agents is one of the sources of clinically relevant drug-drug interactions. Atorvastatin has been reported to increase the expression of CYP2B6 and CYP3A4 (Kocarek et al., 2002), whereas the capability of rosuvastatin for P450 induction has been poorly described. To determine whether LK-935 treatment results in changes in the expression of the main drug-metabolizing P450s, we investigated the effect of LK-935 in primary culture of human hepatocytes. We wished to extend the studies on CYP2C9 and CYP2C19 in addition to CYP2B6 and CYP3A4. P450 enzyme activities and mRNA levels were determined in primary hepatocytes isolated from several donors (as indicated in Figs. 2 and 3) because of high individual variance in basic activities or P450 expression of the cells. To improve our knowledge about the P450 inducibility of human liver cells by the most frequently used antihyperlipidemic drugs, atorvastatin and rosuvastatin were also involved in our experiments. Because both of these statins and LK-935 efficiently inhibited de novo cholesterol biosynthesis at 10 μM, we applied this concentration in further experiments. Rifampicin was used as a reference compound at the concentration of 5 μM. Rifampicin binds to and strongly activates PXR, leading to induction of P450s such as CYP3A4 (Luo et al., 2002). To evaluate the significant inducibility of human hepatocytes by the statins and LK-935, a paired t test was performed with p < 0.05 as the criterion for significance. Figures 2 and 3 present mRNA levels and P450 activities relative to control hepatocytes (0.1% DMSO-treated cells).
In each human hepatocyte preparation, treatments with atorvastatin and rosuvastatin, but not with LK-935, increased the expression of CYP2B6 and CYP2C9 even at the 12-h time point (Fig. 2, A and B). The statistically significant induction as a result of atorvastatin treatment was similar or somewhat lower than that observed in rifampicin-treated hepatocytes. The elevation of mephenytoin N-demethylation of CYP2B6 and tolbutamide hydroxylation activity of CYP2C9 was nearly concomitant with the fold changes in mRNA levels of atorvastatin-treated cells (Fig. 3A). The CYP2B6- and CYP2C9-inducing potential of rosuvastatin seemed to be milder than that observed in rifampicin- or atorvastatin-treated cells. Although rosuvastatin elevated the levels of CYP2B6 and CYP2C9 mRNAs by approximately 3- to 4-fold relative to the control, mephenytoin N-demethylation and tolbutamide hydroxylation did not seem to be substantially induced.
Neither the statins nor LK-935 produced any consistent effect on CYP2C19 expression and mephenytoin 4-hydroxylation activity. Rifampicin was the only agent that was able to increase mRNA level and activity of CYP2C19 (Figs. 2C and 3A).

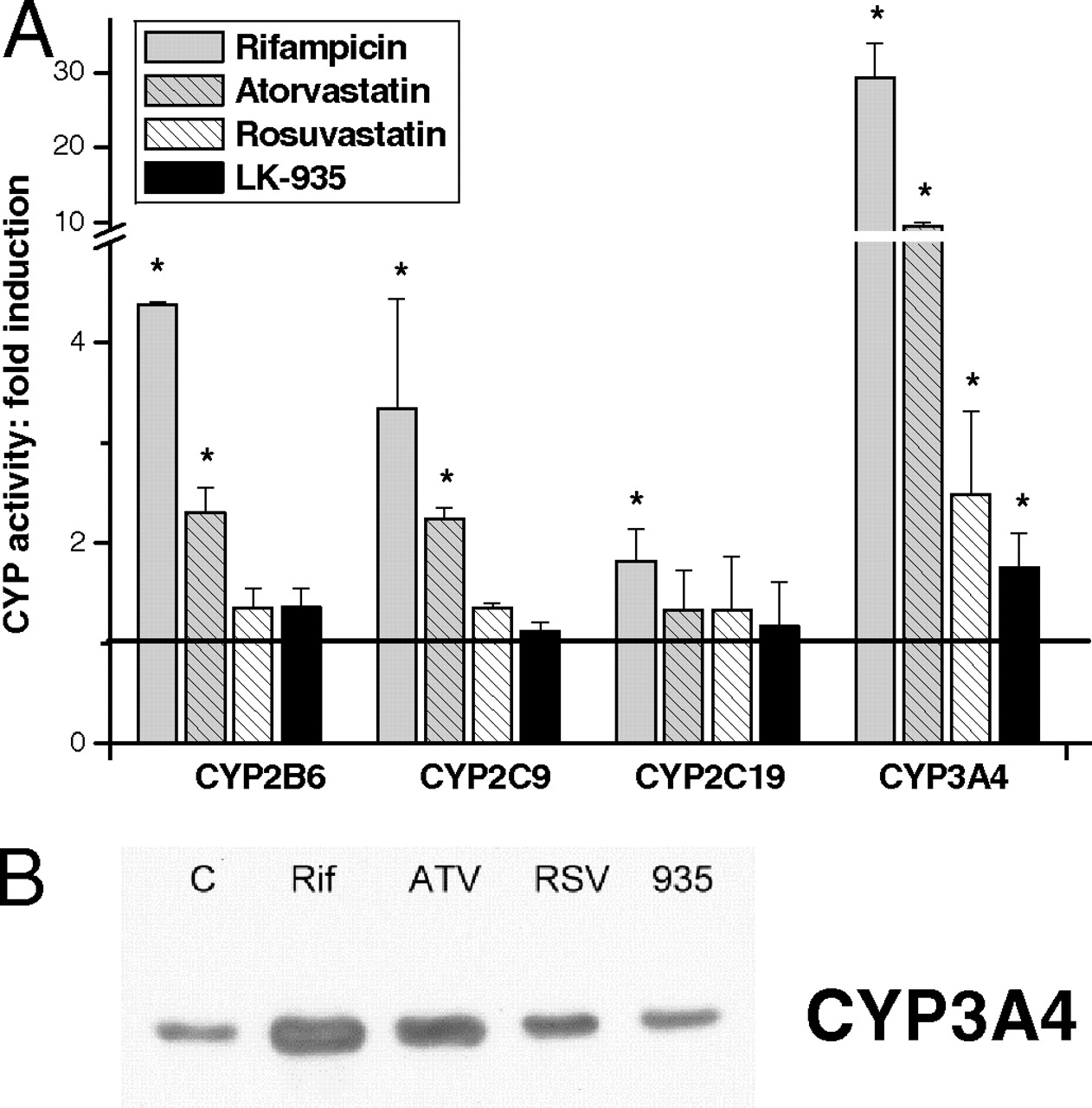
Induction of CYP2B6 (A), CYP2C9 (B), CYP2C19 (C), and CYP3A4 (D) mRNA expression in primary human hepatocytes. Primary human hepatocytes were treated for 12, 24, and 48 h with rifampicin (RIF; 5 μM), atorvastatin (ATV; 10 μM), rosuvastatin (RSV; 10 μM), and LK-935 (10 μM). mRNA levels in human hepatocytes (n = 7 donors) were quantified and normalized to glyceraldehyde-3-phosphate dehydrogenase as described under Materials and Methods. Error bars represent means ± S.D. of seven donors.
Nifedipine oxidation of CYP3A4 increased by more than 30-fold in rifampicin-treated cells (Fig. 3A). Rifampicin was also demonstrated to produce strong transcriptional activation of the CYP3A4 gene (more than 60-fold after a 48-h exposure) (Fig. 2D), involving the action of nuclear receptor PXR. Treatments with atorvastatin resulted in an approximately 10-fold induction of CYP3A4 activity, whereas rosuvastatin caused a 2- to 4-fold increase in nifedipine oxidation in hepatocytes isolated from various donors (Fig. 3A). The elevation in enzyme activities was confirmed by the changes in levels of CYP3A4 mRNA as a consequence of statin treatments (Fig. 2D). The maximal increase in the transcription of the CYP3A4 gene was generated after a 24-h exposure to atorvastatin or rosuvastatin. Nifedipine oxidation activity and expression of CYP3A4 were significantly induced by LK-935 (Figs. 2D and 3A). However, the degree of the elevation of CYP3A4 activity and mRNA levels relative to the control cell population (1.75- and 2.5-fold, respectively) was far from those produced by statins or rifampicin. The results of Western blot analysis of microsomal proteins isolated from the hepatocytes treated with statins, LK-935 or rifampicin, confirmed our findings regarding CYP3A4 induction (as a representative example, see Fig. 3B). As expected, rifampicin treatment resulted in an abundant expression of CYP3A protein. The statin-treated cells showed a marked increase in the amount of CYP3A immunoreactive protein, whereas LK-935 treatment resulted in a mild, but definite, increase in CYP3A protein.
Activation of hPXR and hCAR by the Statins and LK-935. Atorvastatin is known to induce the expression of CYP3A4 through the activation of PXR. Atorvastatin is also an activator of CAR that mediates the induction of CYP2B6 expression (Kocarek et al., 2002; Kobayashi et al., 2005). Both PXR and CAR have been demonstrated to be involved in regulation of several P450s (CYP2B6, CYP2C9, CYP2C19, and CYP3A4) in humans (Chen et al., 2003; Pascussi et al., 2003b). Our results demonstrate the induction of both the activities and the expression of CYP2B6, CYP2C9, and CYP3A4 by atorvastatin in human hepatocytes, thus confirming the findings of Kocarek et al. (2002) and of Kobayashi et al. (2005). The P450-inducing profile of rosuvastatin-treated hepatocytes suggested that rosuvastatin involved the action of nuclear receptor(s) in transcriptional activation of P450 genes. We also attempted to elucidate the role of nuclear receptors, PXR and CAR, in the induction exerted by LK-935.
To assess whether rosuvastatin and LK-935 are able to activate human PXR or CAR, transient cotransfection assays were performed in HepG2 cells. The effect on transcriptional activation of hPXR and hCAR was investigated using a reporter plasmid [pGL3(CYP3A4/XREM[–7800/–7200]/–263/+11)LUC] containing the distal xenobiotic-responsive element module (XREM, –7800/–7200) linked to the proximal CYP3A4 promoter (–263/+11). The reporter plasmid was cotransfected with hPXR or hCAR. Atorvastatin was able to markedly activate the reporter plasmid cotransfected with hPXR, but the extent of PXR activation was lower (5.5-fold) than that observed in the cells exposed to rifampicin (9-fold) (Fig. 4). Rosuvastatin did not seem to activate hPXR, supposing the involvement of other nuclear receptors in P450 induction. LK-935 treatment resulted in a potent increase in ligand-induced transactivation of the reporter construct by hPXR. The 9-fold induction observed at 10 μM LK-935 was identical to the increase in luciferase activity caused by the well known PXR agonist rifampicin (Luo et al., 2002). From the fact that LK-935 was as potent a PXR agonist as rifampicin, we may assume strong induction of several P450s, especially of CYP3A4. In contrast to this finding, we observed only a mild increase in the activity and expression of CYP3A4 in LK-935-treated human hepatocytes.

Induction of CYP2B6, CYP2C9, CYP2C19, and CYP3A4 enzymes in primary human hepatocytes. A, primary human hepatocytes were treated for 48 h with rifampicin (RIF; 5 μM), atorvastatin (ATV; 10 μM), rosuvastatin (RSV; 10 μM), and LK-935 (10 μM). P450 activities were determined in microsomes prepared from hepatocytes isolated from four donors. Controls (C) for each independent experiment were assigned values of 1, and results of treatments are expressed relative to the controls. B, CYP3A4 enzyme protein was determined by Western blot analysis as described under Materials and Methods. Representative examples for CYP3A4 protein content in HH-089 cells are presented.
Atorvastatin, rosuvastatin, and LK-935 were also screened in a HepG2 cell-based reporter gene assay transfected with hCAR and a CYP3A4/XREM[–7800/–7200]/–263/+11 reporter gene construct. As shown in Fig. 4, both statins increased hCAR-mediated transcriptional activity (atorvastatin by 2.8-fold and rosuvastatin by 1.8-fold). The effects of LK-935 and rifampicin on the CAR-mediated transcriptional activity were found to be negligible and not significant. It is consistent with our knowledge that rifampicin is a potent hPXR agonist but does not activate hCAR.
Metabolism of LK-935 by Primary Human Hepatocytes. In HepG2 cell-based reporter gene assays, LK-935 was demonstrated to be as potent an hPXR activator as rifampicin. On the other hand, the degree of CYP3A4 induction by LK-935 in primary human hepatocytes was far from the effect of rifampicin. From these findings and the fact that the metabolic capacity of HepG2 cells is less than that of primary hepatocytes, we may assume rapid metabolism of LK-935 in primary liver cells and, consequently, rapid reduction of the effective concentration required for PXR activation. To confirm our assumption, biotransformation of LK-935 (1 and 10 μM) was determined in suspension of primary human hepatocytes isolated from three donors. Extensive metabolism was observed in human liver cells. After 25 min, more than 80% of the initial concentration of 1 μM was metabolized (Fig. 5), whereas less than 50% of the starting concentration of 10 μM LK-935 remained unchanged. The elimination half-life was estimated to be 6.06 and 23.6 min at the concentrations of 1 and 10 μM, respectively (Table 2). On the basis of the pharmacokinetic parameters estimated in the in vitro hepatocyte system, LK-935 was considered to be a biologically unstable drug. It should be noted that LK-935 was proved to be chemically stable, because no loss of the substrate was observed in the medium in the absence of hepatocytes.
Kinetic parameters for LK-935 metabolism by primary human hepatocytes
The LC-MS/MS analysis of the incubation mixture with LK-935 revealed six metabolites (M1, M2, M3a, M3b, M4a, and M4b) produced by primary human hepatocytes (Fig. 6). All metabolites were eluted earlier than LK-935, indicating that the LK-935 derivatives had more hydrophilic properties than the parent compound. The MS/MS spectrum of LK-935 obtained by the EPI scan technique displayed an abundant peak of protonated molecular ion (M + H+) at m/z 353 and main fragment peaks at m/z 335 (water loss), at m/z 293 (loss of the propyl group), and at m/z 244 (cleavage of the C–C bond at the hydroxyl group). Further characteristic fragments of LK-935 were at m/z 173 formed by the cleavage of the N–C bond containing the chloro-substituted phenyl ring and at m/z 137 and 102 formed by the consecutive loss of HCl and Cl from the ion 173 (Fig. 7).
The MS/MS analysis revealed the structure of M1 formed by cleavage of the propyl group from the central nitrogen of LK-935 (Fig. 7). The mass shift of M1 with m/z 311 (M + H+) was –42 Da compared with LK-935. The EPI spectrum of M1 was similar to that of the parent compound (fragments at m/z 293, 173, 137, and 102). The minor peak at m/z 202 was considered to be the analog of the ion 244 in the EPI spectrum of the parent drug with the mass difference of –42 Da originating from the lack of propyl chain. The derivative with m/z 232 (M + H+) displayed a mass shift of –121 Da, corresponding to M2 formed by the cleavage of hydroxyethyl-pyridine moiety from the central nitrogen of LK-935. The EPI spectrum of M2 showed the ions at m/z 173, 137, and 102. During screening for m/z 369 (M + H+) corresponding to the mass shift of +16 Da, two metabolites (M3a and M3b) were detected, indicating monohydroxy derivatives of LK-935. It was supported by the fragment ions at m/z 351 and 333, representing consecutive losses of two H2O. The EPI spectrum of M3a showed a fragment ion at m/z 260, whereas that of M3b displayed an intense ion at m/z 244. Further characteristic fragments of M3 metabolites were at m/z 173, 137, and 102, indicating that the dichloro-phenyl-ethyl moiety of both monohydroxy metabolites was intact. We assumed that the ion at m/z 244 in the EPI spectrum of LK-935 was formed after the loss of the hydroxyethyl-pyridine part; thus, the M3a metabolite producing the fragment ion at m/z 260 (with mass shift of +16 Da comparing with m/z 244) contained the hydroxyl group at the propyl group. From these data, it can be concluded that the hydroxylation of LK-935 producing M3b took place at the pyridine ring. M4a and M4b as minor metabolites were detected at m/z 248 (M + H+), presenting the mass shift of +16 Da compared with M2. The water loss of 18 Da in the EPI spectrum proposed that the structure of M4 metabolites was the hydroxylated derivatives of M2. The presence of the fragment ion series 173, 137, and 102 suggested that the dichloro-phenyl-ethyl moiety was intact and the hydroxy-group was likely to locate at one of the carbon of the propyl group. For M4a, the additional ions at m/z 175, 161, and 139 derived from an isoxazolidine loss suggested the position of the hydroxy group at the terminal carbon atom.

Activation of hPXR and hCAR in response to statins, LK-935, and the PXR agonist rifampicin. HepG2 cells were transiently transfected with expression plasmid (pSG5-hPXR or pSG5-hCAR) together with luciferase reporter construct pGL3(CYP3A4/XREM[–7800/–7200]/–263/+11)LUC expression plasmid. The cells were treated for 24 h with rifampicin (3 μM), atorvastatin (10 μM), rosuvastatin (10 μM), and LK-935 (10 μM). Luciferase activities were determined as described under Materials and Methods.

In vitro kinetic analysis of LK-935 in primary human hepatocytes. LK-935 at the concentration of 1 μM was incubated in hepatocytes isolated from three donors. The consumption of LK-935 as a function of time was determined by LC-MS/MS as described under Materials and Methods.
Involvement of P450 Forms in LK-935 Metabolism. Various P450 inhibitors (ketoconazole for CYP3A4, sulfaphenazole for CYP2C9, tranylcypromine for CYP2C19, and pentoxyresorufin for CYP2B6) were used to identify the individual isozymes participating in the formation of LK-935 metabolites (Table 3). The contributions of P450 isoforms to the production of LK-935 derivatives were characterized by inhibition constants (Ki values) determined at various concentrations of LK-935 (5, 10, and 20 μM) and of chemical inhibitors. The production of M1 was strongly reduced in the presence of ketoconazole and sulfaphenazole, suggesting contributions of CYP3A4 and CYP2C9 to the cleavage of the propyl group from the central nitrogen of LK-935. Dixon plots displayed the competitive inhibition of M1 formation both by ketoconazole and by sulfaphenazole. Ketoconazole was also found to be a potent competitive inhibitor of M2 formation with a submicromolar Ki value, providing evidence for the role of CYP3A4. Chemical inhibition of CYP2C9, CYP2C19, and CYP2B6 did not decrease the rate of cleavage of the hydroxyethyl-pyridine moiety from LK-935. Sulfaphenazole selective for CYP2C9 seemed to be a potent competitive inhibitor of LK-935 hydroxylation at the propyl-group (M3a formation) and at the pyridine ring (M3b formation). Ketoconazole also seemed to decrease M3b production; however, with a Ki value one magnitude higher than that of sulfaphenazole. In conclusion, the results of chemical inhibition studies indicated a possible involvement of CYP2C9 and CYP3A4 in the metabolism of LK-935, whereas the contributions of CYP2B6 and CYP2C19 were not confirmed (Fig. 7).
Inhibition of LK-935 metabolism by chemical inhibitors

LC-MS/MS chromatograms of LK-935 and the corresponding metabolites. LK-935 at the concentration of 10 μM was incubated in hepatocytes suspension for 1 h. LK-935 and its metabolites were determined by LC-MS/MS as described under Materials and Methods. The protonated molecular ion and the main fragment ion for quantification are indicated in each chromatogram.

Proposed metabolic scheme for LK-935. Characteristic fragmentation pathways determined by MS/MS is shown for LK-935. P450 enzymes playing significant role in formation of LK-935 metabolites are also displayed.
Discussion
Because of the increasing instances of hyperlipidemias and cardiovascular diseases in the developed world and the risk of severe adverse drug reactions (myopathies, rhabdomyolysis, renal toxicity, and hepatotoxicity) with statin-based therapy, development of novel strategies in cholesterol-lowering therapy is greatly needed (Tiwari et al., 2006; Alsheikh-Ali et al., 2007; Armitage, 2007; Martin and Krum, 2007; Rallidis et al., 2007). Searching for potential hypolipidemic drugs with new molecular targets is an important task for the pharmaceutical industry. The novel cholesterol-lowering drug candidates block cholesterol biosynthesis after the farnesyl pyrophosphate branchpoint, leaving the isoprene pathways untouched to avoid statin side effects (Fig. 1A) (Korosec et al., 2008). LK-935, a pyridylethanol(phenylethyl)amine derivative targeting human lanosterol 14α-demethylase (CYP51), was demonstrated to efficiently inhibit post-squalene cholesterol biosynthesis. The aim of our study was to reveal the drug interaction potential of LK-935 and to predict the P450-mediated interactions with coadministered drugs. We, therefore, investigated the induction of P450s by LK-935 in primary cultures of human hepatocytes and compared its P450-inducing potential with that of two frequently used statins, atorvastatin and rosuvastatin.
Atorvastatin, being an activator of hPXR and hCAR, has been reported to induce CYP3A4 and CYP2B6 (El-Sankary et al., 2001; Kocarek et al., 2002; Kobayashi et al., 2005). The results of the current studies provided unequivocal evidence that, in addition to these enzymes, the transcriptional expression and tolbutamide hydroxylation activity of CYP2C9 also increased in atorvastatin-treated hepatocytes. Rosuvastatin treatment also elevated the expression and activities of CYP3A4, CYP2C9, and CYP2B6; however, rosuvastatin did not seem to be as potent a P450 inducer as atorvastatin. In investigation of the role of nuclear receptors in rosuvastatin action, hCAR, but not hPXR, was supposed to mediate the increase in P450 expression (Fig. 4). LK-935, though, seemed to be a weak inducer in comparison with the statins or rifampicin, and the treatment of human hepatocytes with LK-935 led to up-regulation of CYP3A4, suggesting the involvement of PXR. In HepG2 cell-based assays, LK-935 proved to strongly activate hPXR; moreover, it seemed to be as potent a PXR activator as the well known PXR agonist rifampicin (Luo et al., 2002). The fact that LK-935 is an efficient agonist of hPXR is inconsistent with the findings of P450 induction studies, demonstrating a mild increase in CYP3A4 expression in primary hepatocytes as a consequence of LK-935 treatment. This inconsistency was assumed to be explained by the differences in the drug-metabolizing capacity between HepG2 cells and primary human hepatocytes. The activities and the expression of drug-metabolizing P450s have been demonstrated to be extremely low in HepG2 cells (Wilkening et al., 2003). In contrast, primary human hepatocytes were proved to extensively metabolize LK-935, rapidly decreasing the effective concentration presumably required for the maximal PXR activation. CYP3A4 and CYP2C9 were found to play a major role in the biotransformation of LK-935. CYP3A4, the most abundant isozyme of all P450s in human liver, is not detectable in HepG2 cells, and the expression of CYP2C9 is also negligible in this cell line (Wilkening et al., 2003).
Primary hepatocytes offer a useful model for estimation of in vitro pharmacokinetic properties of a drug and for prediction of in vivo pharmacokinetic parameters for most drugs. The predictability of in vivo Clint from in vitro data is considered to be good, even for drugs with in vivo Clint values ranging over 4 orders of magnitude. For LK-935 at the concentrations of both 1 and 10 μM, extremely high Clint values were estimated in primary human hepatocytes (198.7 and 47.96 ml/min/kg, respectively). The model (ClH) accounting for the influence of the metabolic (in vitro Clint) and nonenzymatic [binding to plasma proteins and hepatic blood flow (QH): 19 and 10.8 ml of plasma/min/kg, respectively] factors estimated LK-935 to be a high-extraction drug with low bioavailability (extraction ratios of 0.944 and 0.815 for 1 and 10 μM, respectively).
The in vitro pharmacokinetic studies using primary human hepatocytes also generated samples for the identification of metabolites. During the biotransformation, LK-935 underwent predominantly N-dealkylation and hydroxylation to a lesser extent. The cleavage of the propyl group from the tertiary amine of LK-935 resulted in the formation of M1, whereas the cleavage of the hydroxyethyl-pyridine part produced the major M2 metabolite. LC-MS/MS analysis identified two monohydroxy derivatives of LK-935 (M3 metabolites). One of them contained the hydroxy group at the propyl group and the other on the pyridine ring. The metabolite profile assumed that the main M2 metabolite underwent further hydroxylation at the propyl group, leading to the formation of two minor M4 metabolites (M4a and M4b). In vitro inhibition studies implicated CYP3A4 and CYP2C9 in the oxidative metabolism of LK-935. CYP3A4 seemed to have a significant role in the formation of the main metabolite M2. Clear evidence was provided for the participation of CYP2C9 in the hydroxylation of LK-935, whereas CYP3A4 and CYP2C9 seemed to account for equal contributions to the cleavage of the propyl group from the central nitrogen of LK-935.
In conclusion, we provide evidence for the induction of CYP3A4 by the novel cholesterol-lowering drug candidate LK-935. LK-935 has also been proved to be a potent hPXR activator as prerequisite for the transcriptional activation of the CYP3A4 gene; however, the rapid metabolism of LK-935 in primary hepatocytes decreases the efficient concentration required for maximal CYP3A4 induction. Therefore, the induction of CYP3A4 by LK-935 would be taken into account as the source of a mild or negligible drug-drug interaction. However, because CYP3A4 and CYP2C9 play a significant role in LK-935 metabolism, the inhibition of these P450s by coadministered drugs may lead to some increase in the LK-935 concentration required for the potent induction of CYP3A4. Our results also suggested the activation of hCAR by rosuvastatin and consequently for the induction of CYP3A4, CYP2C9, and CYP2B6 genes. The inductive response to rosuvastatin treatment can be the source of drug-drug interactions. Activation of hCAR and hPXR by atorvastatin and the subsequent induction of not only CYP2B6 and CYP3A4 but also CYP2C9 present additional targets by which atorvastatin, a widely used cholesterol-lowering drug, can modify the kinetics of numerous drugs.
Acknowledgments
We are indebted to Maria Grenyi for her skillful assistance in this study.
Footnotes
-
This study was supported by a János Bolyai Research Scholarship of the Hungarian Academy of Sciences [Grant BO/00413/05]; the Slovenian-Hungarian Intergovernmental S&T Cooperation Programme [Grant SLO-2/04]; and the European Community [Grant LSHG-CT-2005-512096, Steroltalk].
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.023887.
-
ABBREVIATIONS: P450, cytochrome P450; hCAR, human constitutive androstane receptor; PXR, pregnane X receptor; hPXR, human PXR; FF-MAS, follicular fluid meiosis activating sterol; LK-935, 2-((3,4-dichlorophenethyl)(propyl)amino)-1-(pyridin-3-yl)ethanol; DMEM, Dulbecco's modified Eagle's medium; RT, reverse transcriptase; PCR, polymerase chain reaction; TBS, Tris-buffered solution; DMSO, dimethyl sulfoxide; LC, liquid chromatography; MS/MS, tandem mass spectroscopy; EPI, enhanced product ion; MRM, multiple reaction monitoring; CAR, constitutive androstane receptor.
- Received August 13, 2008.
- Accepted October 23, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}