


Abstract
The expression of flavin-containing monooxygenases (FMOs) in dog liver microsomes was suggested by a high methimazoleS-oxidase activity. When the reaction was catalyzed by dog liver microsomes, apparent Vmaxand Km values were 6.3 nmol/min/mg and 14 μM, respectively. This reaction was highly inhibited (73%) in the presence of imipramine, but it was also weakly affected by trimethylamine, suggesting the involvement of different isoforms. The sequences of dog FMO1 and FMO3 were obtained by reverse transcription-polymerase chain reaction and 5′/3′ terminal extension. The cDNAs of dog FMO1 and dog FMO3 encode proteins of 532 amino acids, which contain the NADPH- and FAD-binding sites. The dog FMO1 amino acid sequence is 88, 86, and 89% identical to sequences of human, rabbit, and pig FMO1, respectively. The dog FMO3 amino acid sequence is 83, 84, and 82% identical to sequences of human, rabbit, and rat FMO3, respectively. Dog FMO1 and dog FMO3 exhibited only 56% identities. The FMO1 and FMO3 recombinant proteins and the FMO1 and FMO3 microsomal proteins migrated with the same mobility (56 kDa), as determined in SDS-polyacrylamide gel electrophoresis and immunoblotting. By Western blotting, dog FMO1 and dog FMO3 were detected in microsomes from liver and lung but not in kidney microsomes. By Northern blotting, the probe for FMO1 specifically hybridized a 2.6-kilobase ( kb ) transcript in liver and lung samples only. The probe for FMO3 hybridized two transcripts of approximately 3 and 4.2 kb in the liver and lung samples.
The dog is widely used in pharmacotoxicological studies. Very limited information is available about drug metabolism enzymes in this animal species and particularly about the flavin-containing monooxygenases. Microsomal flavin-containing monooxygenase (FMO1; EC 1.14.13.8) catalyzes the FAD-, NADPH-, and O2-dependent oxidation of a large number of xenobiotics containing soft nucleophiles, including alkaloids, pesticides, and pharmaceutical substances (Ziegler, 1988, 1990). Since the initial purification of an FMO form from pig liver microsomes byZiegler and Mitchell (1972), different FMOs have been isolated, cloned, and sequenced from tissues of different animal species, including humans. To date, a maximum of five distinct isoforms of FMO have been identified. In the same species, the deduced amino acid sequences are 52 to 57% identical, whereas orthologous sequences share at least 80% of identity. Based on the cDNA sequence, the FMOs were classified into five subfamilies (FMO1 to FMO5) (Lawton et al., 1994). The isoforms differ in tissue distribution or substrate specificity.
FMO1 is the main isoform of the FMO family expressed in the liver of the pig (Sabourin et al., 1984; Gasser et al., 1990), rat (Itoh et al., 1993), rabbit (Tynes et al., 1985), male mouse (Sabourin et al., 1984), and fetal human (Dolphin et al., 1991). This isoform is also present in other tissues, such as kidney, brain, and lung (Itoh et al., 1993). FMO3 is also mainly expressed in the liver. In the adult human (Lomri et al., 1992), the female mouse (Falls et al., 1997a,b), and the sheep (Longin-Sauvageon et al., 1998), FMO3 is the predominant isoform expressed in liver. As for FMO1, this protein is also found in other tissues, such as the kidney. FMO2 was purified from rabbit lung (Williams et al., 1984) and was found principally in this tissue in all species studied. FMO4 has been cloned and sequenced from different animal species (Burnett et al., 1994), but the corresponding protein has not yet been isolated from any tissue. FMO5 seems to be expressed at a very low level in all the tissues studied (Code et al., 1998).
The evidence for the expression of FMO in dog liver was first reported by Dannan and Guengerich (1982), using rabbit anti-hog liver flavin-containing monooxygenase (FMO1) antisera. The involvement of FMO in drug metabolism in the dog has been confirmed by the FMO-dependentN-oxidation of ABT-418 (Rodrigues et al., 1995), ranitidine (Cross et al., 1990), and L-775,606 (Prueksaritanont et al., 2000) as being the main pathway of metabolism for these substrates in the dog liver. Furthermore, dog hepatocytes were shown to metabolize benzidamine, a substrate for FMO1, very efficiently (Ubeaud et al., 1999). Based on the methionine S-oxidation and immunoreactivity studies, Ripp et al. (1999) suggested a high expression of an FMO3 in dog liver but a minor expression in kidney microsomes. Data about the distribution of the predominant isoforms FMO1 and FMO3 in the dog are important because this animal is used in research as a model of human metabolism and toxicity. The purpose of this article was to characterize FMOs from dog liver microsomes. This is the first publication of the complete sequence of the cDNAs encoding for dog FMO1 and FMO3 and the tissue distribution of the corresponding mRNAs and proteins.
Experimental Procedures
Chemicals.
DNA polymerases and cDNA synthesis components were purchased from Promega (Charbonnieres, France), and all restriction enzymes were obtained from Roche Diagnostics (Meylan, France). The glutathioneS-transferase gene fusion vector pGEX-6P3 was obtained from Amersham Biosciences AB (Uppsala, Sweden). Oligonucleotides were manufactured by OligoExpress (Paris, France). Sequencing was performed by GenomeExpress (Meylan, France). The QIAquick PCR purification, QIAquick gel extraction, and QIAprep Spin Miniprep kits were purchased from QIAGEN (Courtaboeuf, France). All other chemicals were of the highest reagent or electrophoresis grade commercially available.
Biological Materials.
Dog livers, kidneys, lungs, and muscles were obtained from control animals in experiments carried out at the School of Veterinary Medicine and stored at −80°C. Microsomes were prepared from liver, kidney, and lung samples by differential centrifugation, as previously described (Moroni et al., 1995). Protein concentrations were determined by the Bradford method using serum bovine albumin as a standard. Poly(A) RNA was purified from dog liver (100 mg) with streptavidin-coated paramagnetic particles (PolyATract mRNA isolation system; Promega). Total RNA was isolated from dog liver, kidney, lung, and muscle by a single extraction with an acid guanidinium thiocyanate-phenol-chloroform solution, according to the method used byChomczynski and Sacchi (1987). Total RNA concentrations were evaluated spectrophotometrically from absorbance at 260 nm.
Enzyme Assays.
The FMO-dependent methimazole S-oxidase activity was determined according to the method of Dixit and Roche (1984). Briefly, the S-oxidation of methimazole was assessed at 37°C in the presence of an NADPH-regenerating system; a typical reaction mixture contained Tris-HCl buffer (100 mM), pH 8.3, 100 μg of microsomal protein, 0.1 mM EDTA, 0.06 mM 5,5′-dithiobis(nitrobenzoic acid), 0.020 mM dithiothreitol, 0.1% Triton X-100, and 1 mM methimazole. The difference in absorbance between identical assay mixtures with and without methimazole was monitored at 412 nm for 3 min. Kinetic parameters of methimazole metabolism were determined from results obtained by the addition of decreasing amounts of methimazole (250, 125, 62.5, 50, 40, 30, 20, 10, 5, and 2.5 μM final concentration) to the sample cuvette. To examine the effects of varying pH on FMO activity, the pH of the reaction mixture (0.1 M tricine/KOH, 0.1 mM EDTA, 0.06 mM 5,5′-dithiobis(nitrobenzoic acid), 0.02 mM dithiothreitol, and 0.1 mM NADPH) was adjusted accordingly with KOH. To assess temperature stability, enzymatic fractions were first heated for 1 min at 50°C and then transferred to an ice-cold tube before determining the FMO activity. The effect of n-octylamine (1, 2.5, and 5 mM final concentration) on methimazole oxidation was observed using liver microsomes. The inhibitory effect of specific substrates, such as trimethylamine (1 mM) and imipramine (0.7 mM), on the methimazole oxidation activity of FMOs was assessed by adding these substrates to the sample and reference cuvette after the addition of methimazole (i.e., methimazole oxidation was monitored for 2 min first). Ethoxycoumarin-O-deethylase activity was assessed fluorometrically (Riviere et al., 1985).
RT-PCR.
The first-strand cDNA template was synthesized from dog liver mRNA (50 ng) in the presence of oligo(dT)15 (100 pmol) and 200 units of Moloney murine leukemia virus-reverse transcriptase (MMLV-RT) RNase H minus in 20 μl of standard reverse transcription buffer (50 mM Tris-HCl, pH 8.3, 3 mM MgCl2, 75 mM KCl, 10 mM dithiothreitol, and 200 μM each deoxynucleotide triphosphate) at 37°C for 1 h and then at 65°C for 10 min. The reaction product was purified on a silica gel column.
Two microliters of the resulting cDNA were amplified by PCR using primers designed from the most conserved regions of the sequences in humans (GenBank accession number 4503754), rats (GenBank accession number M84719), and rabbits (GenBank accession number M32030) FMO1 cDNA. The sequence of the sense primer FMO1-S1 and the antisense primer FMO1-AS1 were (5′-ATGGCCAAGCGAGTTGCAATTGTG-3′) and (5′-GTTGGCTTTTGCAGATGTGCAGGGAAG-3′), respectively. The PCR was performed using FMO1-S1 and FMO1-AS1 primers (20 pmol) andTaq DNA polymerase (1 unit) (Promega) in 50 μl of PCR buffer (10 mM Tris-HCl, pH 9.0, 1.5 mM MgCl2, 50 mM KCl, 0.1% Triton X-100, and 200 μM each deoxynucleotide triphosphate). The amplification was performed at 94°C for 3 min, then 35 cycles of 94°C for 30 s, 50°C for 30 s, and 72°C for 90 s, followed by a final extension at 72°C for 10 min. The PCR products were subjected to electrophoresis on agarose gel (1%). The principal product (1110 bp) was gel-purified, subcloned into pSTBlue-1 vector (perfectly blunt cloning kit; Novagen, Madison, WI), and sequenced.
For FMO3, the same strategy was performed with primers designed from the most conserved regions among the human (GenBank accession numberM83772), rabbit (GenBank accession number L10391), and mouse (GenBank accession number U87147) FMO3 cDNA sequences. The sequence of the sense primer FMO3-S1 and the antisense primer FMO3-AS1 were (5′-AAAGTGGCCATCATTGGA-3′) and (5′-CCAATGAAGGAGGMSAGTTCA-3′), respectively. The PCR was performed in similar conditions. The PCR products were analyzed on an ethidium bromide-stained 1% agarose gel. The principal product (1300 bp) was gel-purified, subcloned into pSTBlue-1 vector, and sequenced.
Amplification of cDNA Ends.
To obtain the cDNA ends, the SMART RACE cDNA amplification kit (CLONTECH, Palo Alto, CA) was used according to the manufacturer's recommendations. The specific primers used were designed from sequences of clones obtained after RT-PCR.
The 5′ first-strand cDNA template was synthesized from dog liver mRNA (50 ng) in the presence of a modified oligo(dT)30primer and a SMART II oligonucleotide in a standard reverse transcription buffer containing 200 units of MMLV-RT at 42°C for 1.5 h. The PCR was performed with anchor primer and a mix of polymerases (Advantage cDNA PCR kit; CLONTECH), using antisense primer FMO1-AS2 (25 pmol; nucleotide 168–198) or FMO3-AS2 (25 pmol; nucleotide 64–90) to generate the 5′ ends of dog FMO1 or FMO3, respectively. The PCR products were gel-purified, subcloned into pSTBlue-1 vector, and sequenced.
The 3′ first-strand cDNA template was obtained from dog liver mRNA (50 ng) in the presence of a (dT)30-anchor primer in a standard reverse transcription buffer containing 200 units of MMLV-RT at 42°C for 1.5 h. The PCR was performed in similar conditions, using sense primer FMO1-S3 (25 pmol; nucleotide 737–765) or FMO3-S3 (25 pmol; nucleotide 940–966) to generate the 3′ ends of dog FMO1 or FMO3, respectively. The PCR products were gel-purified, subcloned into pSTBlue-1 vector, and sequenced.
Isolation of Full-Length cDNA.
A (dT)15 primed first-strand cDNA template was synthesized in the same conditions as described above. Sense FMO1-S4 [25 pmol; nucleotide (−)9–9] and antisense FMO1-AS4 (25 pmol; nucleotide 1797–1819) primers designed from the 5′ and 3′ ends of nonencoding sequences of dog FMO1, respectively, were used to perform the PCR amplification of the 1850-base pair fragment containing the complete coding region. After an initial denaturation step at 94°C for 2 min, Pfu DNA polymerase (1.25 units) (Promega) was added, and the reaction was incubated for 35 cycles at 94°C for 30 s, 50°C for 30 s, and 72°C for 4 min, and then for an additional 10 min at 72°C. The resulting product was gel-purified and cloned into pSTBlue-1. Inserts from four clones (clone dFMO1-1 to 4) were sequenced.
The same strategy was performed with primers designed from the 5′ and 3′ ends of nonencoding sequences of dog FMO3. The primers used corresponded to nucleotides (−)60 to (−)33 and 1737 to 1760 for sense (FMO3-S4)and antisense primer (FMO3-AS4), respectively. The resulting product (1800 bp) was gel-purified and cloned into pSTBlue-1. Inserts from four clones (clone dFMO3-1 to 4) were sequenced.
Construction of Escherichia coli Expression Vectors.
Dog FMO1 and FMO3 were expressed in the glutathioneS-transferase gene fusion vector pGEX-6P3 (Amersham Biosciences AB). The coding sequence corresponding to the dog FMO1 was amplified by PCR from clone dFMO1–2 using Pfu DNA polymerase and specific primers corresponding to nucleotides 1 to 13 (sense primer) and 1576 to 1599 (antisense primer), includingBamHI and SalI restriction sites in the extremities, respectively. After an initial denaturation step at 94°C for 2 min, Pfu DNA polymerase (1.25 units) was added, and the reaction incubated for 5 cycles at 94°C for 30 s, 55°C for 30 s, and 72°C for 4 min, for 20 cycles at 94°C for 30 s, 65°C for 30 s, and 72°C for 4 min, and then for an additional 10 min at 72°C. The gel-purified PCR product was digested by 10 units of BamHI and SalI restriction enzymes (Roche Diagnostics, Meylan, France) and ligated into the expression vector pGEX-6P3 linearized by the same enzymes. A clone named pGEX-dFMO1 was obtained and verified by sequencing.
The same strategy was performed for dog FMO3 from clone dFMO3–1, with specific primers corresponding to nucleotides 1 to 19 and 1575 to 1598, including BamHI and SalI restriction sites in the extremities, for sense and antisense primer, respectively. A plasmid, pGEX-dFMO3, was obtained in similar conditions and verified by sequencing.
Expression in E. coli of Recombinant Dog FMO1 and FMO3.
E. coli BL21 cells were transformed with pGEX -dFMO1 or pGEX-dFMO3 and grown at 37°C in Terrific Broth (Sigma, St. Louis, MO; 1.2% bacto tryptone, 2.4% yeast extract, 0.4% glycerol, and 90 mM potassium phosphate, pH 7.8) supplemented with ampicillin (100 μg/ml) to an absorbance of 0.6 to 0.8 at 600 nm. Isopropyl β-d-thio-galactopyranoside was then added to a final concentration of 0.2 mM, and the cells were further incubated overnight at 37°C. Cells were harvested by centrifugation at 10,000g for 10 min and resuspended in buffer A (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4, pH 7.3) containing 1 mM EDTA, 5 mM dithiothreitol, 500 μM phenylmethylsulfonyl fluoride, and 100 μg/ml lysozyme. After an incubation for 30 min at 4°C, the resuspended cells were disrupted by sonication (five 30-s pulses separated by a 30-s period of cooling). The solutions dFMO1 or dFMO3, treated with 1.5% lauroyl-sarcosine, were gently stirred for 1 h at 4°C and then centrifuged at 100,000g for 1 h at 4°C. The resulting supernatants were purified onto a Glutathione Sepharose 4B (Sigma) column.
Purification and Characterization of Recombinant Dog FMO1 and FMO3.
All purification procedures were carried out according to the manufacturer's recommendations for the purification of insoluble fusion protein. The supernatants that contained either recombinant FMO1 or FMO3 were treated with Triton X-100 to a final concentration of 2% and then applied to a Glutathione Sepharose 4B (Sigma) column (5 ml) equilibrated with buffer A containing 0.2% Triton X-100. After application of the FMO preparation, the column was washed with 5 column volumes of the same buffer and then with 5 column volumes of buffer B (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol, and 0.2% Triton X-100, pH 8.0). PreScission Protease (Amersham Biosciences AB) (50 units) was added. After incubation for 24 h at 4°C under gentle agitation, the cleavage result was eluted.
SDS-polyacrylamide gel electrophoresis and Western blotting were performed as described by Laemmli (1970) and Towbin et al. (1979), respectively. Purified recombinant dog FMO1 (1 μg) and dog FMO3 (1 μg) were detected by electrophoresis on a 10% polyacrylamide gel under denaturing conditions and stained with Coomassie blue. A mix of native dog FMO1 and dog FMO3, previously partially copurified in the laboratory from dog liver microsomes according to the method of Moroni et al. (1995), were used as a control. Immunoblots were incubated for 4 h with rabbit polyclonal anti-rat FMO1 (1:1000) or anti-rat FMO-3 IgG (1:1000) (13). Following a washing sequence in phosphate-buffered saline, alkaline phosphatase-linked antibodies anti-rabbit IgG were used as secondary antibodies, and blots were visualized with an alkaline phosphatase conjugate substrate kit (Bio-Rad, Hercules, CA).
Analysis of Proteins Distribution.
Western blotting of microsomal proteins were performed as previously described. Microsomes were prepared from the liver, kidney, and lung of two separate animals. Fifty micrograms of microsomal proteins were loaded, and immunoblottings were performed as described above. Each immunoblot contained, as standard, 1 μg of purified recombinant dog FMO1 or dog FMO3, according to the antibodies used.
Analysis of RNA Distribution.
Twenty micrograms of total RNA isolated from dog liver, kidney, lung, and muscle were separated by electrophoresis on agarose gel (1.2%) containing formaldehyde (2 M) and then transferred to nylon membrane (Hybond N; Amersham Biosciences AB). The blot was washed in 5× SSC for 15 min at room temperature and baked at 80°C for 2 h. The membrane was prehybridized for 4 h at 42°C and then hybridized with a dog FMO1 coding region probe (EcoRI/EcoRI, base 1–1095) at 42°C for 24 h. The probe was labeled by a random primer method (Prime-A-Gene labeling system; Promega). The hybridized membrane was washed for two cycles of 10 min in 2× SSC (0.1% SDS), then two cycles of 15 min in 1× SSC (0.1% SDS) at room temperature, and at 55°C for two cycles of 15 min in 0.1× SSC (0.1% SDS). The membrane was subjected to autoradiography for 48 h at −80°C. The membrane was stripped with a 0.01× SSC (0.5% SDS) at 100°C. The removal of the probe was verified by autoradiography at −80°C. The blot was subsequently hybridized with the dog FMO3 coding region probe (EcoRI/KpnI, bases 572-1325) and finally a β-actin cDNA probe.
Data Analysis.
The estimation of the kinetic parameters was achieved by the incubation of 10 different concentrations of the substrate. Incubations were performed in triplicate. The ordinary least-squares criterion was used to fit the Michaelis-Menten model to the data, taking velocity as the dependent variable. The minimum values of the sum of squared residuals were computed using NAG foundation library routine EO4FDF (Numerical Algorithms Group Ltd., Natick, MA), a combined Gauss-Newton and modified Newton algorithm, using function values only.
Results
Enzyme Assays.
Methimazole S-oxidation activity was observed for liver and lung microsomes but not for kidney microsomes. The reaction was linear as a function of incubation time (more than 5 min) and protein concentration up to 500 μg of microsomal protein (results not shown). The apparent kinetic parameters for the methimazoleS-oxidation by liver microsomes were 14 μM and 6.3 nmol/min/mg for Km andVmax, respectively. The optimum pH for the reaction was found to be near 8.3. Heat treatment of liver microsomes (50°C for 1 min in the absence of NADPH) inhibited the FMO activity by 92 ± 2% (n = 6), whereas the cytochrome P450-dependent ethoxycoumarin-O-dealkylation activity was not significantly modified (Fig. 1).

Effect of elevated temperature (50°C for 1 min) on the activities of dog liver microsomes, as determined by methimazole S-oxidation and ethoxycoumarin-O-dealkylation.
Data presented represents the averages ± S.D. of three determinations. Samples were heated at 50°C for 1 min and then stored on ice until assayed at 37°C.
Methimazole S-oxidation was doubled in the presence ofn-octylamine (Table 1). Imipramine (0.7 mM) inhibited the methimazole S-oxidation catalyzed by liver or lung microsomes by 73 and 35%, respectively. Trimethylamine (1 mM) inhibited the methimazole S-oxidation catalyzed by liver or lung microsomes by 11 and 16%, respectively (Table 2).
Inhibition of methimazole S-oxidation in dog microsomes by FMO substrates
Enhancement of methimazole S-oxidation catalyzed by dog liver microsomes by several concentrations of n-octylamine
Sequencing of cDNA Encoding Dog FMO1.
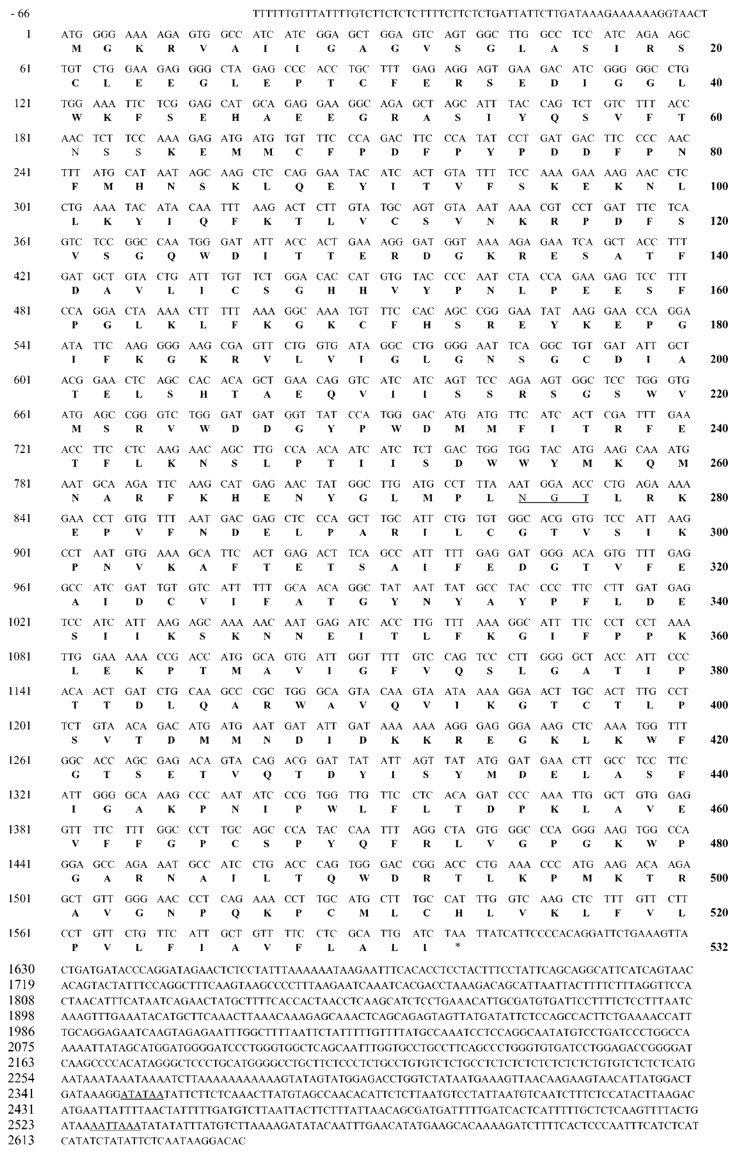
The cloning of dog FMO1 cDNA was performed by RT-PCR with sense primer FMO1-S1 and antisense primer FMO1-AS1, as described underExperimental Procedures. The RT-PCR generated a 1110-base pair fragment, which was sequenced and compared with other FMO1 cDNAs. The nucleotidic sequence obtained showed a 85% homology with human and mouse (GenBank accession number 6753889) FMO1 cDNAs and 84, 89, and 91% homology with rat, rabbit, and pig (M32031) FMO1 cDNAs, respectively. To obtain the dog FMO1 cDNA ends, the SMART RACE cDNA amplification kit (CLONTECH, Palo Alto, CA) was used as described underExperimental Procedures. 3′- and 5′-PCR products were obtained and sequenced. The 3′-PCR product contained the remainder of the protein-coding region and a 365-base pair of an apparent 3′-untranslated region. The 5′-PCR product contained a putative ATG start codon and 9 bp of the 5′-untranslated region. The flanking sequence to the proposed translational initiation codon, AACATGG, has a purine in position −3 and a G in position +4, conforming to the consensus Kozak sequence, for efficient initiation of translation of vertebrate mRNAs (Kozak, 1987).
A product encoding the entire open reading frame of dog FMO1 cDNA (from bp −9 to bp 1819 of the nucleotide sequence) was obtained by PCR using specific primers, and the PCR product was subcloned. Complete sequence analysis of the four clones confirmed that the inserts of all four clones contained sequences that encoded dog FMO1. A consensus nucleotide sequence was obtained by alignment of these sequences. The consensus nucleotide sequence for the dog FMO1 cDNA contains 1995 bases with a 9-base 5′-flanking region followed by an open reading frame of 1596 bases terminated with a TAA stop codon and a 3′-flanking region of 365 bases (Fig. 2). A polyadenylation signal (AATAAA) was found 60 bases upstream of a polyadenylated tail. The high degree of sequence identity with FMO1 cDNAs in the human (89%), pig (90%), rabbit (87%), mouse (84%), and rat (83%) identified the sequence as a dog FMO1 cDNA. All other heterologous FMO exhibited 55 to 60% identities.

Nucleotide sequence of dog FMO1 cDNA and deduced amino acid sequence of dog FMO1.
The open reading frame starts at nucleotide 1 and goes to nucleotide 1599. Asparagine-like glycosylation sites, stop codon, and the putative polyadenylation signal are underlined. The dog FMO1 sequence has been entered in the GenBank libraries under the provided accession numberAF384053.
Sequencing of cDNA Encoding Dog FMO3.
To obtain the dog FMO3 cDNA, a similar strategy was used. A 1300 base-pair fragment was obtained by RT-PCR with specific primers FMO3-S1 and FMO3-AS1 deduced from the most conserved regions among human, rabbit, and mouse FMO3 cDNAs. The sequence of this 1300-base pair fragment showed a high homology (from 83–87%) with the nucleotide sequences of FMO3s already known. The 5′ and 3′ ends of dog FMO3 cDNA were obtained by using the SMART RACE cDNA amplification kit. The 3′-PCR product contained the remainder of the protein-coding region and 1042 bp of an apparent 3′-untranslated region. The 5′-PCR product contained a putative ATG start codon and 66 bp of the 5′-untranslated region.
The entire sequence of the dog FMO3 cDNA (from nucleotide −60 to nucleotide 1760) was obtained by PCR with specific primers designed from 5′ and 3′ ends sequences. The 1700 bp fragment was subcloned. Complete sequence analysis of the four clones confirmed that the inserts of all four clones contained sequences that encode the dog FMO3. A consensus nucleotide sequence was obtained by alignment of these sequences. The consensus nucleotide sequence for the dog FMO3 cDNA contains 2704 bases with a 66 base 5′-flanking region followed by an open reading frame of 1596 bases terminated with a TAA stop codon and a 3′-flanking region of 1042 bases (Fig.3). FMO3 cDNA sequence contains multiple polyadenylation signals (AATAAA or other similar sequences). The high degree of sequence identity with FMO3 cDNAs in the human (86%), rabbit (84%), mouse (81%), and rat (82%), confirmed the identity of the dog cDNA obtained. The other heterologous FMO exhibited 55 to 60% identities.

Nucleotide sequence of dog FMO3 cDNA and deduced amino acid sequence of dog FMO3.
The open reading frame starts at nucleotide 1 and goes to nucleotide 1599. Asparagine-like glycosylation sites, stop codon, and the putative polyadenylation signal are underlined. The dog FMO3 sequence has been entered in the GenBank libraries under the provided accession numberAF384054.
Amino Acid Sequences Derived for Dog FMO1 and Dog FMO3 cDNAs.
The amino acid sequences derived from the consensus nucleotide sequence are shown in Fig. 2 and 3. The sequences of dog FMO1 (Fig. 2) and dog FMO3 (Fig. 3) encoded two proteins of 532 amino acid residues, with predicted molecular weights of 60,054 Da and 60,064 Da and theoretical pI of 9.28 and 7.94 for dog FMO1 and dog FMO3, respectively. The dog FMO1 amino acid sequence showed 88, 86, 89, 84, and 83% identity with the sequences of FMO1 from the human, rabbit, pig, mouse, and rat, respectively (Fig. 4). The dog FMO3 amino acid sequence showed 83, 84, 80, and 82% identity with the sequences of FMO3 from human, rabbit, mouse, and rat, respectively (Fig.5). Dog FMO1 and dog FMO3 exhibited only 56% identities. Putative FAD- and NADPH-binding domains were located in the dog FMO1 and FMO3 amino acid sequences between residues 9 and 14 and between residues 191 and 196, respectively. The putative FAD-binding site, G-A-G-V-S-G, was fully conserved between dog FMO1 and dog FMO3. The putative NADPH-binding site was different between dog FMO1 (G-M-G-N-S-G) and dog FMO3 (G-L-G-N-S-G).

Comparison of dog FMO1 deduced amino acid sequence with human, rabbit, pig, mouse and rat FMO1, and dog FMO3.
FAD-binding sites (residues 9 to 14), NADPH-binding sites (residues 191 to 196), and the FMO-characteristic pentapeptides –EGLEP- (residues 24 to 28) and –FATGY- (residues 331 to 335) are in boxes.

Comparison of dog FMO3 deduced amino acid sequence with human, rabbit, mouse and rat FMO3, and dog FMO1.
FAD-binding sites (residues 9 to 14), NADPH-binding sites (residues 191 to 196), and the FMO-characteristic pentapeptides –EGLEP- (residues 24 to 28) and –FATGY- (residues 327 to 331) are in boxes.
Expression of Recombinant Dog FMO1 and FMO3 in E.coli.
cDNAs encoding dog FMO1 and dog FMO3 were cloned into E. coli expression vectors (pGEX-6P3). Transformed cells were grown to mid-log phase and then induced with isopropyl β-d-thio-galactopyranoside (0.2 mM). The isopropyl β-d-thio-galactopyranoside-dependent expression of pGEX-dFMO1 or pGEX-dFMO3 in the E. coli BL21 produced a high glutathioneS-transferase activity but no detectable methimazoleS-oxidase activity. The glutathione S-transferase activity was used to trace steps in the purification of glutathioneS-transferase proteins. The proteins were solubilized from inclusion bodies using N-lauroyl-sarcosine (1.5%) because the induced fusion proteins containing dFMO1 or dFMO3 were insoluble in Triton X-100 (2%). This step compromised the ability to recover functional dog FMO1 or dog FMO3. Solubilized fusion proteins containing dFMO1 or dFMO3 were bound on a Glutathione Sepharose 4B. The dog FMO1 or dog FMO3 proteins were eluted by cleavage with PreScission Protease while the glutathione S-transferase remained on the column.
The dog FMO1 eluate (lane 3) was resolved into two bands on SDS-PAGE (Fig. 6). The apparent molecular weights of the two proteins were ∼56 and ∼59 kDa, respectively (lane 3). The SDS-PAGE of native dog FMOs (a mix of dFMO1 and dFMO3, previously partially copurified) (lane 2 and 5) revealed a band migrating with an apparent molecular weight of ∼56 kDa. Immunoblot analysis (Fig.7A) of purified FMO1 (lane 2) using rabbit polyclonal anti-rat FMO1 revealed the presence of a single immunoreactive protein migrating at ∼56 kDa. The microsomal enzyme was recognized at the same molecular weight (lane 1). The purified dog FMO1 was weakly recognized by rabbit polyclonal anti-rat FMO3 (lane 3).

Analysis of recombinant dFMO1 and dFMO3 products by SDS-PAGE.
Samples were electrophoresed on polyacrylamide gel in the presence of SDS and stained with Coomassie blue. Lane 1 shows the position of the molecular size standards. Lane 3, analysis of the purified recombinant dog FMO1; lane 4, the purified recombinant dog FMO3; lane 2 and 5, the native dog FMOs (mix of dFMO1 and dFMO3, partially copurified).

Analysis of recombinant dFMO1 and dFMO3 products by immunoblotting.
Fifty micrograms of dog liver microsomal protein (lane 1), purified recombinant dog FMO1 (lane 2), and purified recombinant dog FMO3 (lane 3) were electrophoresed on polyacrylamide gel in the presence of SDS, transferred to nitrocellulose, and revealed as described underExperimental Procedures with rabbit polyclonal anti-rat FMO1 (A) or rabbit polyclonal anti-rat FMO3 (B). dFMO1 and dFMO3 were detected at ∼56 kDa.
The purified protein dog FMO3 migrated as a single band on SDS-PAGE (Fig. 6, lane 4). The molecular weight was determined to be ∼56 kDa, which was similar to the SDS-PAGE mobility of native dog FMOs (Fig. 6, lane 2). By immunoblot analysis, the purified FMO3 (Fig. 7B, lane 3) and the microsomal enzyme (Fig. 7B, lane 1) cross-reacted with rabbit polyclonal anti-rat FMO3 at the same molecular weight. The recombinant dog FMO3 was not recognized by rabbit polyclonal anti-rat FMO1 (Fig.7B, lane 2).
Protein Distribution.
Figure 8 depicts representative immunoblots in which antibodies to the FMO1 and FMO3 forms were reacted with dog microsomes from liver, kidney, and lung. In Fig. 8A, a 56-kDa protein corresponding to expressed dog FMO1 was detected with antibodies against rat FMO1 in microsomes from liver (lane 2) and lung (lane 4) but not in kidney microsomes (lane 3), suggesting tissue-dependent expression of FMO1.

Analysis of FMO1 and FMO3 protein distribution by immunoblotting.
Microsomal samples (lanes 2 to 4; 50 μg of protein) were loaded as follows: lane 2, dog liver; lane 3, dog kidney; lane 4, dog lung. Purified recombinant dog FMO1 (lane 1A) and dog FMO3 (lane 1B) were loaded. Immunoblotting was performed as described underExperimental Procedures with rabbit polyclonal anti-rat FMO1 (A) or rabbit polyclonal anti-rat FMO3 (B). The proteins were detected at ∼56 kDa.
Immunoblot examination of FMO3 distribution (Fig. 8B) detected a band corresponding to the expressed dog FMO3 (∼56 kDa) in the liver (lane 2) and lung (lane 4) samples only but not in the kidney samples, suggesting a tissue-specific expression of FMO3. A minor cross-reacting protein of higher molecular weight (∼61 kDa) was seen with microsomes from the liver and kidney.
mRNA Distribution.
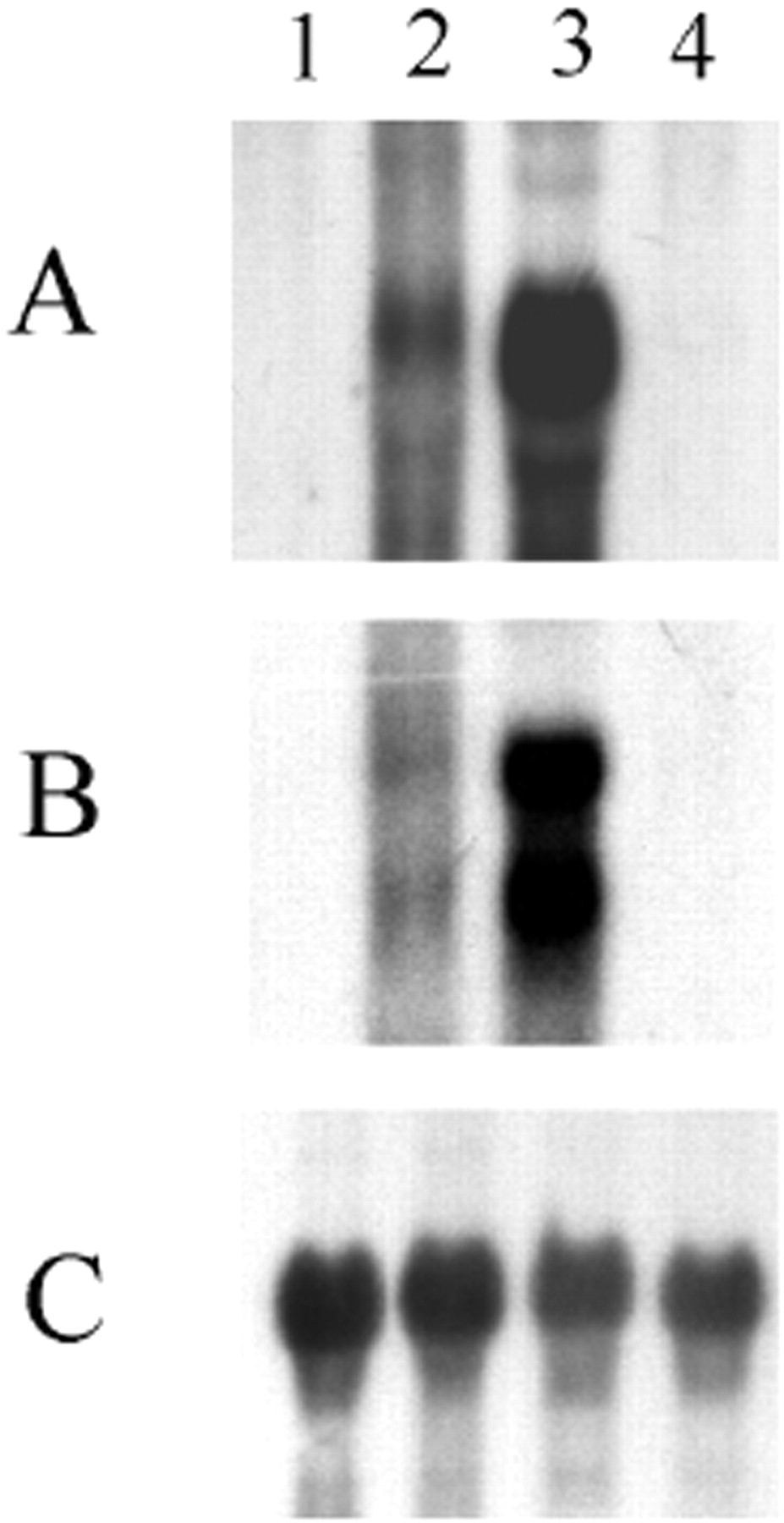
To determine whether the tissue distribution observed at the protein level was evident at the level of transcription, mRNA expression was examined by Northern blot. Expression of mRNAs encoding FMO1 and FMO3 in dog liver (lane 3), kidney (lane 4), lung (lane 2), and muscle (lane 1) is shown in Fig. 9. The probe for FMO1 (fragment containing bases 1 to 1095) strongly and specifically hybridized a 2.6-kb transcript in the liver and lung samples (Fig. 9A). In contrast, the FMO1 probe did not hybridize with any mRNA from the kidney and muscle samples (Fig. 9A). Hybridization with the FMO3 cDNA probe (fragment containing bases 572 to 1325) revealed two transcripts of approximately 3 and 4.2 kb in the liver and lung samples (Fig. 9B). No signal was detected in the kidney and muscle samples. FMO1 and FMO3 mRNA levels were higher in the liver compared with the lung. Blots stripped and probed with actin cDNA demonstrated equal loading of mRNA (Fig. 9C).

Analysis for FMO1 and FMO3 expression of mRNA isolated from dog muscle, lung, liver, and kidney.
Samples of mRNA (20 μg) isolated from muscle (lane 1), lung (lane 2), liver (lane 3), and kidney (lane 4) were electrophoresed in agarose (1.2%) containing formaldehyde (2 M), transferred to nylon membranes, and hybridized with a 32P-labeledEcoRI/EcoRI-fragment (bases 1–1095) of the cDNA for dog FMO1 (A) or a 32P-labeledEcoRI/KpnI-fragment (bases 572-1325) of the cDNA for dog FMO3 (B). Relative abundance of β-actin cDNA is presented in C.
Discussion
Although dogs are widely used in pharmacotoxicological studies, very limited information is available about drug metabolism enzymes in this animal and particularly about flavin containing-monooxygenases. We present in this study some data about catalytic properties of dog liver microsomes using methimazole, one of the most specific substrate of FMOs. Methimazole S-oxidation activity in dog liver microsomes (6.3 ± 1.8 nmol/min/mg) is similar to that in other species used in drug metabolism studies, such as the rat or the mouse. The methimazole S-oxidation activity present in dog liver microsomes possesses all the specific properties of the FMOs: a high optimal pH (8.3), an inhibition of the activity by thermal treatment (1 min at 50°C in the absence of NADPH) with no modification of the cytochrome P450-dependent ethoxycoumarin-O-dealkylation activity, a lack of sensitivity of activity to nonionic detergents, and a relatively high affinity toward methimazole (Km, 14 μM). Furthermore, methimazoleS-oxidase is present in lung microsomes (3.4 ± 0.3 nmol/min/mg) but not in kidney microsomes.
Imipramine, a substrate for FMO1, was used as an FMO1-selective inhibitor for the methimazole S-oxidation activity. Imipramine inhibited the methimazole S-oxidation activity by 73 and 35%, respectively, when dog microsomes from the liver or the lung were used. These results argue for a major involvement of dog FMO1 in the liver. The enhancement of the methimazole activity in the presence of n-octylamine is also frequently reported as a property of the FMO1 orthologs. When trimethylamine, a substrate for FMO3, was used, the methimazole S-oxidation catalyzed by liver or lung microsomes was inhibited by 11 and 16%, respectively. These results argue for a minor expression of an FMO3 ortholog in the liver and in the lung. The inhibition rates observed when selective substrates of FMO1 and FMO3 were used to inhibit the methimazoleS-oxidation were high (73 and 11%) when the reaction was catalyzed by liver microsomes. However these inhibition rates were more limited (35 and 16%) when lung microsomes were used, arguing for the expression of another FMO, probably an FMO2 ortholog, in the lung.
When dog liver microsomes were subjected to Western blot analysis using either antibodies against rat FMO1 or antibodies against rat FMO3, a clear band was observed at a mol. wt. close to 56 kDa. An attempt to purify FMOs from the liver of a dog was carried out. A chromatographic method, which allowed us to separate rat FMO1 from rat FMO3 (Moroni et al., 1995), did not succeed in the separation of the two antigens in the dog. The semipurified preparation obtained is recognized by these two antibodies.
The catalytic activities and the immunological properties of liver microsomes suggest the involvement of two FMOs isoforms, FMO1 and FMO3. To examine tissue-dependent expression of dog FMO1 and FMO3, we attempted to obtain the cDNA sequences of dog FMO1 and dog FMO3 to use cDNA recombinant expression system to estimate the specificity of antibodies anti rat-FMO1 and anti-rat FMO3.
A strategy of reverse transcription-polymerase chain reaction and 5′/3′ terminal extension was used to obtain the corresponding cDNAs sequences. Specific FMO1 or FMO3 oligonucleotides were designed from the known sequences of different FMO1s or FMO3, respectively. Two different cDNA sequences encoding two different proteins of 532 amino acids were obtained from dog liver. These two deduced amino acid sequences exhibited only 56% identities. The homologous FMOs are 50 to 60% identical. One of them revealed more than 80% identities with other FMO1 amino acid sequences. The designation of “dog FMO1” to name this cDNA is in agreement with the actual nomenclature based on the comparison of the amino acid sequence of the mammalian FMOs. The other protein, which exhibits more than 80% identities with known FMO3 amino acid sequences, is named dog FMO3. As for the previously described forms of FMO, the sequences exhibit the presence of characteristic FAD- and NADPH-binding sites, and the absolute conservation of two pentapeptides (–EGLEP- beginning at residue 24 and –FATGY- beginning at residue 327).
Recombinant proteins were produced as inclusion bodies only. An attempt to solubilize these proteins with nonionic detergent was not successful. Recombinant proteins were solubilized with 1.5% lauroyl-sarcosine. Given that this detergent fully abolished the methimazole S-oxidase activity from even the liver microsomes, it was not surprising that these recombinant proteins were not catalytically active. As a consequence it was not possible to study the catalytic properties of the recombinant isoforms. The recombinant dog FMO1, using Western blotting, was recognized by antibodies raised against rat FMO1 and weakly by antibodies raised against rat FMO3. However, the recombinant dog FMO3 was recognized by the antibodies raised against rat FMO3 only. The high recognition of a 56 kDa protein by these two antibodies in liver and lung microsomes or in the semipurified fraction demonstrates that in dog liver and lung microsomes, dog FMO1 and dog FMO3 are both expressed. Neither dFMO1 nor dFMO3 was detected using Western blot with kidney microsomes.
Northern blot analysis using specific probes for dFMO1 or dFMO3 confirm the expression and distribution of the corresponding transcripts of these two FMOs in the liver and in the lung but not in the kidney. Using our FMO1 probe, the size of the recognized transcript (2.6 kb) is slightly longer than the sequence (1995-base pair) reported above. This difference could be due to the use of the 5′/3′ terminal extension methods. The size of the FMO1 mRNA is different from those of the transcripts detected using the FMO3 probe. This confirms the specificity of the probes used despite the 60% identity between dog FMO1 and FMO3 nucleotide sequences. Two transcripts (3 kb and 4.2 kb) were detected with the FMO3 probe in the liver and lung. This is probably due to alternative 3′ processing at multiple polyadenylation signals (AATAAA or other similar sequences); the FMO3 cDNA sequence contains multiple polyadenylation signals (Fig. 3).
All these results are consistent with the expression of FMO1 and FMO3 in the liver and in the lung but not in the kidney, FMO1 being predominant in the liver. In the liver of the pig (Gasser et al., 1990), rat (Itoh et al., 1993), male mouse (Sabourin et al., 1984), rabbit (Tynes et al., 1985), and dog, the predominant FMO belongs to the FMO1 subfamily. On the contrary, FMO3 is predominant in the liver of female mouse (Falls et al., 1997a,b) and sheep (Longin-Sauvageon et al., 1998). In human beings, FMO1 is found in the fetal liver only, and FMO3 is predominant in the adult liver.
Neither FMO1 nor FMO3 is present in the kidney of dogs. In the kidney of other animal species, such as rat (Itoh et al., 1993), mouse (Itoh et al., 1997), and human (Phillips et al., 1995; Dolphin et al., 1996), either FMO1 or FMO3 (or both) is highly expressed. The expression of these enzymes is, on the contrary, very limited in the kidney of rabbits, but in this last species, the expression of FMO4 is high (Burnett et al., 1994). It would be interesting to evaluate the expression of the three other isoforms of FMO, particularly FMO2 in the lung and FMO4 in the kidney.
Acknowledgments
We thank L. Olivier and C. Moulin for technical assistance.
Footnotes
-
This study was funded in part by a grant from Direction Générale de l'Enseignement et de la Recherche (DGER) Formation par la Recherche, Ministère de l'Agriculture, France. The nucleotide sequences reported in this article have been submitted to the GenBank under the provided accession number AF384053 for dFMO1 and AF384054 for dFMO3.
- Abbreviations used are::
- FMO
- flavin-containing monooxygenase
- ABT-418
- (S)-3-methyl-5-(1-methyl-2-pyrrolidinyl) isoxazole
- L-775,606
- 3-(3-{4-[2-(3-fluorophenyl)ethyl]piperazin-1-yl}propyl)-5-[1,2,4]triazol-4-yl-1H-indol
- RT-PCR
- reverse transcription-polymerase chain reaction
- MMLV-RT
- Moloney murine leukemia virus-reverse transcriptase
- bp
- base pair
- SSC
- standard sodium citrate
- PAGE
- polyacrylamide gel electrophoresis
- kb
- kilobases
- Received July 12, 2001.
- Accepted October 25, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}