


Abstract
Bergamottin, a furanocoumarin isolated from grapefruit juice, was investigated for the ability to increase diazepam bioavailability and for its effect on cytochrome P450 (P450) enzymes in the beagle dog liver and intestine. To study the effect of bergamottin on diazepam pharmacokinetics, male beagle dogs were dosed with bergamottin (1 mg/kg) p.o. 0 or 2 h before p.o. diazepam (10 mg). In a second experiment, bergamottin (0.1 mg/kg) was dosed i.v. or p.o. 1 h before p.o. diazepam (10 mg). Plasma samples were collected over 24 h postdose, analyzed by liquid chromatography/mass tandem spectrometry, and diazepam pharmacokinetic parameters were determined. To study the effect of bergamottin on P450 enzymes, beagle dog liver and jejunum was harvested after a 10-day dosing regimen of bergamottin (1 mg/kg) p.o. per day; microsomes were prepared and analyzed for CYP3A12, CYP2B11, CYP1A1/2, and tolbutamide hydroxylase activity. Bergamottin predosing increased the plasma levels of diazepam as observed by Cmax (278.75 ng/ml versus 5.49 ng/ml) and the area under the curve [AUC(0-TLDC)] (247.69 versus 2.79 ng · hr/ml) in bergamottin versus placebo groups, respectively, indicating P450 enzyme inhibition. Diazepam plasma concentrations were increased to a similar level in the presence of i.v. and p.o. administered bergamottin. In hepatic microsomes, bergamottin treatment for 10 days reduced the activity of CYP3A12 by 50% and CYP1A1/2 by 75%. Tolbutamide hydroxylase activity did not change, and CYP2B11 activity was moderately induced. In jejunal microsomes, CYP3A12 activity doubled with bergamottin treatment. CYP2B11, CYP1A1/2 activity and tolbutamide hydroxylation was not detected. In conclusion, bergamottin is both an inhibitor and an inducer of P450 enzymes.
Grapefruit juice increases the plasma concentrations of drugs that are substrates for CYP3A4 (e.g., felodipine, triazolam, midazolam, terfenadine, cyclosporine, nifedipine, and diazepam) (Bailey et al., 1998b; Lundahl et al., 1998;Edwards et al., 1999; Mohri et al., 2000), CYP1A2 (e.g., caffeine) (Fuhr et al., 1993), and CYP2A6 (e.g., coumarin) (Merkel et al., 1994). In humans, grapefruit juice exposure decreases concentrations of intestinal CYP3A4 (Lown et al., 1997; Schmiedlin-Ren et al., 1997), does not effect CYP3A5, CYP1A1, CYP2D6 protein, or CYP3A4 mRNA (Lown et al., 1997), and inhibits P-glycoprotein activity (Sawada et al., 1998;Eagling et al., 1999; Edwards et al., 1999). Grapefruit juice consumption significantly increases plasma levels of orally administered cyclosporine and has no effect on the disposition of intravenously administered cyclosporine in human volunteers (Ducharme et al., 1995). Furthermore, hepatic CYP3A4 activity is not affected when measured by the erythromycin breath test (Lown et al., 1997), indicating that the grapefruit juice effect is primarily at the level of the intestine. In contrast to the results described above, long-term grapefruit juice treatment in rats has been shown to increase nifedipine clearance (Mohri et al., 2000). In mice, a single dose of grapefruit juice inhibits hepatic oxidative enzyme activity, whereas recurrent dosing increases activity (Dakovic et al., 1999). The results of these rodent studies imply that grapefruit juice is both an inhibitor and an inducer. To elucidate these effects of grapefruit juice, it is necessary to investigate the activity of the individual components.
Some of the principal components of grapefruit juice have been identified as the furanocoumarin bergamottin and its metabolite 6′,7′-dihydroxybergamottin, the flavonoids naringenin, naringin, quercetin, and kaempferol (Kane and Lipsky, 2000). All are reported to inhibit CYP3A activity in in vitro systems (Miniscalco et al., 1992;Cai et al., 1993; Ghosal et al., 1996; Schmiedlin-Ren et al., 1997; He et al., 1998; Eagling et al., 1999). It has been demonstrated that 6′,7′-dihydroxybergamottin, naringin (Bailey et al., 1998a), and quercetin (Rashid et al., 1993) are not the major components that inhibit CYP3A4 activity in humans. Bergamottin is a mechanism-based inhibitor of CYP3A4, with an IC50 of 1.04 μM in Caco-2 cells (Schmiedlin-Ren et al., 1997) and aKi of 7.7 μM in hepatic microsomes (He et al., 1998). In addition, bergamottin (10–100 μM) inhibits activities of CYP1A2, 2A6, 2C9, 2C19, 2D6, and 3A4 in human liver microsomes (He et al., 1998) and the activity of CYP1A1/2 and CYP2B in murine liver microsomes (Cai et al., 1993). To date, there are no reports on the in vivo or long-term effects of bergamottin on P4501 enzymes.
In an earlier article from this laboratory (He et al., 1998), it was speculated that, since bergamottin is a potent inactivator of P450 enzymes and is found in relatively high concentrations in grapefruit juice, it is most likely the principal active ingredient contributing to the “grapefruit juice effect” in vivo. Since all prior studies with bergamottin have been conducted in vitro and do not address in vivo disposition, we have tested this hypothesis by studying the effect of bergamottin on the plasma pharmacokinetics of the known CYP3A and CYP2C substrate diazepam. Diazepam was chosen because it is absorbed well, metabolized rapidly, and the interaction of diazepam and grapefruit juice has been previously characterized in humans (Özdemir et al., 1998). Preliminary experiments in Wistar rats (data not shown) revealed that bergamottin (1 mg/kg) did not affect diazepam PK, indicating that the rat was not an appropriate model. Trial experiments with beagle dogs showed an increase in diazepam PK values after bergamottin treatment, and this species was chosen for further PK studies. In addition, the effect of acute (10 days) bergamottin treatment on P450 activity in the liver and intestinal mucosa of beagle dogs was examined to clarify the major site of action of bergamottin.
Materials and Methods
This study was conducted in accordance with the NRC Guide for the Care and Use of Laboratory Animals. All animal use protocols were approved by the Institutional Animal Care and Use Committee of Pfizer Global Research and Development. Male beagle dogs (∼10 kg) were obtained from Marshall Farms, Inc. (Northrose, NY) and were fed no. 5006 canine lab diet (PMI Nutrition International, Brentwood OH). The dogs were housed in customized stainless steel suspended cages (Hoeltge, Inc., Cincinnati OH), one animal per cage in a temperature controlled environment (70–74°F; monitored by an Environmental Watchdog system, Waterford, WI) with a 12-h light/dark cycle. Bergamottin was obtained from Indofine Chemical Co. (Somerville, NJ); diazepam (Valium) tablets were purchased from Roche Products, Inc. (Manati, Puerto Rico); stable label diazepam-d5, 11β-hydroxy-testosterone, NADPH, testosterone, aprotinin, leupeptin, pepstatin, and phenylmethylsulfonyl fluoride were obtained from Sigma-Aldrich (St. Louis, MO). Resorufin and 7-ethoxyresorufin were purchased from Molecular Probes (Junction City, OR), and 6β-hydroxytestosterone was obtained from Steraloids, Inc. (Wilton, NH). BCA protein assay reagents and bovine serum albumin were obtained from Pierce Chemical Co. (Rockford, IL). Intralipid was purchased from Baxter (Deerfield, IL). Propylene glycol, lactose monohydrate, microcrystalline cellulose NF, sodium croscarmellose, sodium lauryl sulfate (SLS), magnesium stearate, and gray/gray gelatin capsules (size 3) were obtained from Warner Lambert CRM Centralized Raw Materials (Morris Plains, NJ). The i.v. catheters were obtained from Critikon (Tampa, FL). All solvents and other chemicals used were of HPLC grade or the highest purity available.
Drug Preparation.
Capsules were prepared with bergamottin that was milled by a mortar and pestle in the presence of 10% SLS (w/w). Milled product was observed by optical microscopy to ensure uniform particle size and analyzed by reverse-phase HPLC for possible degradants (data not shown). Excipients were combined via geometric dilution to make a dry blend (5 g) as described: lactose (2.16 g) and microcrystalline cellulose (2.16 g) were mixed with a spatula on weigh paper. Sodium croscarmellose (0.1 g) and SLS (0.1 g; placebo blend only) were added in geometric dilution to the blend with magnesium stearate (0.025 g), incorporated last with only 1 min of mixing. Size 3 capsules were hand-filled with 1 or 10 mg of bergamottin and sufficient excipient blend for a total capsule weight of 100 mg, followed by thorough in-capsule mixing. Placebo capsules (size 3) contained only the excipient blend. Bergamottin i.v. formulation (0.2 mg/ml) was achieved by solubilizing 7 mg of bergamottin in 7.3 mg of propylene glycol by sonication, followed by the dropwise addition of this mixture into 28 ml of intralipid while vortexing. The formulation was administered immediately. Placebo i.v. formulation was prepared similarly without bergamottin.
Animal Dosing for Pharmacokinetic Studies.
Study 1
Male Beagle dogs (8–10 kg; n = 4–5) were treated with 10 mg of bergamottin or placebo capsules and 10-mg diazepam tablets (Valium; Roche Molecular Biochemicals). To evaluate when bergamottin dosing would have the greatest effect on diazepam plasma concentrations, beagle dogs were orally administered the following treatments: 1) diazepam + vehicle (placebo); 2) diazepam with bergamottin coadministered; and 3) diazepam dosed 2 h after bergamottin.
Study 2.
This study used a crossover design for the p.o. and i.v. arms of the investigation. Male beagle dogs (8–10 kg; n = 4–5) were fed 1 h before receiving 1 mg of bergamottin or placebo capsules as a pretreatment, and 1 h later, the dogs received 10 mg of diazepam p.o. After a 2-week wash-out period, dogs were administered an i.v. dose of 1 mg of bergamottin or placebo, followed immediately by p.o. diazepam (10 mg). The i.v. dosing was by a 2-ml i.v. infusion into the cephalic vein over a 2-min period.
Blood Sampling.
Serial blood collections were made from the jugular vein using a 20-gauge, 1.5-inch needle attached to a heparinized 3-ml syringe (100 μl of 1000 units/ml sodium heparin). Systemic blood (2 ml) was collected at 0, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 h for determination of plasma diazepam and bergamottin concentrations by LC/MS/MS. Blood was transferred to microfuge tubes, centrifuged at 10,000 rpm for 3 min, and plasma was separated and stored at −20°C until analysis.
LC/MS/MS Analysis of Dog Plasma.
All determinations were made by LC/MS/MS with a Micromass Quatro II tandem mass spectrometer (Manchester, UK), set to the electrospray positive-ionization mode, with Masslynx version 3.3 operating software. Parent to daughter transitions were established through direct infusion of each of the analytes into the mass spectrometer. The liquid chromatography system consisted of a PerkinElmer Series 200 autosampler and pump (flow rate, 0.2 ml/min) (Norwalk, CT). The analytes were separated on a C8 column (Zorbax XDB, 5 μm, 2.1 × 150 mm; MAC-MOD, Chadds, PA) eluted with 0.1% formic acid and acetonitrile by a gradient varying from acetonitrile/0.1% formic acid (35:65) to acetonitrile/0.1% formic acid (95:5) in 4 min. A liquid-liquid extraction with methyl t-butyl ether was conducted by adding to a 100-μl plasma sample 25 μl of diazepam-d5 as internal standard, 100 μl of 0.5 M K2HPO4 buffer, pH 12.0, and 20 μl of acetonitrile. The tubes were vortexed for 30 s before the addition of 650 μl of methyl t-butyl ether. After shaking for 15 min and centrifugation at 4000 rpm for 10 min, the organic layer was transferred to a second set of tubes and evaporated to dryness under nitrogen. Finally, the samples were reconstituted with 200 μl of acetonitrile/water (60:40, v/v), vortexed, and 5 μl was injected into the mass spectrometer. A 10-point standard curve (10 to 10,000 ng/ml) was prepared by spiking 100-μl blank plasma samples with 20 μl of standard working solutions containing bergamottin and diazepam in acetonitrile.
Animal Dosing for Enzyme Activity Studies.
Male beagle dogs (8–10 kg) were fed 1 h before receiving capsules containing placebo (n = 3) or 10 mg of bergamottin (n = 5) prepared as described above. Animals were dosed daily for 10 days, and livers were removed 24 h after the last dose. The animals used for this study were prescheduled for termination.
Tissue Harvest and Microsomal Preparation.
Dogs were euthanized with a 10-ml sodium pentobarbital (65 mg/ml) injection into the jugular vein. Livers were rapidly perfused with cold saline, removed and flash frozen in liquid nitrogen, and stored at −80°C. Hepatic tissue (approximately 5 g) was homogenized in buffer containing 50 mM potassium phosphate, 150 mM potassium chloride, and 1 mM EDTA, pH 7.4. For intestinal microsomal isolation, the first 20 cm of the jejunum was removed, opened longitudinally, washed thoroughly with ice-cold saline containing 1 mM DTT and 1 mM EDTA, and the mucosal layer was blotted dry and scraped off with a glass microslide before flash freezing. In addition, all intestinal buffers contained 2 mM magnesium chloride, 5 mM 2-mercaptoethanol, 5 μg/ml aprotinin, 10 μg/ml leupeptin, 1 μg/ml pepstatin, and 40 μg/ml phenylmethylsulfonyl fluoride. Tissue homogenates were centrifuged at 10,000g for 25 min at 4°C using a Beckman XL-70 ultracentrifuge (Beckman Coulter, Inc, Fullerton, CA). Supernatants were collected and centrifuged at 105,000g for 65 min at 4°C in the same centrifuge. The supernatant was washed with 50 mM potassium phosphate, pH 7.7, 0.1 M sodium pyrophosphate, and 1 mM EDTA and centrifuged at 105,000g for 65 min at 4°C. The final microsomal pellets were resuspended in 3 to 5 ml of 50 mM potassium phosphate containing 20% glycerol and 1 mM EDTA, pH 7.4, with a Potter-E glass homogenizer from Kontes Glass Co. (Vineland, NJ) fitted with a Teflon pestle. A small aliquot was taken for protein determination, and all samples were stored at −80°C.
Protein and Enzyme Assays.
Protein analysis was conducted using the DC Microplate Protein Assay (Bio-Rad Laboratories, Hercules, CA). CYP3A12 activity was determined using testosterone 6β-hydroxylation with 6β-hydroxytestosterone quantitated by reverse-phase HPLC (Wood et al., 1983). CYP2C activity was measured by quantitating tolbutamide hydroxylation (Kunze et al., 1996). The O-dealkylation of 7-ethoxyresorufin was measured by a fluorometric method (Burke et al., 1985).
Statistical Analysis.
Results are expressed as mean ± S.E. Within each metabolism experiment, assays were performed in triplicate. Activity was calculated using an Excel spreadsheet (Microsoft Office 1997; Microsoft, Redmond, WA). Noncompartmental pharmacokinetic parameters were calculated using WinNonlin PK (3.0) (Pharsight, Mountain View, CA) or Watson software (6.2.0.02) from Innaphase Corporation (Philadelphia, PA). Statistical significance of treatment versus control was determined by Student's t test. Differences between treatment groups were analyzed by one-way analysis of variance, followed by the least significant difference multiple range tests.
Results
Pharmacokinetic Studies in Beagle Dogs.
Effect of bergamottin on diazepam kinetics with coadministration or predosing
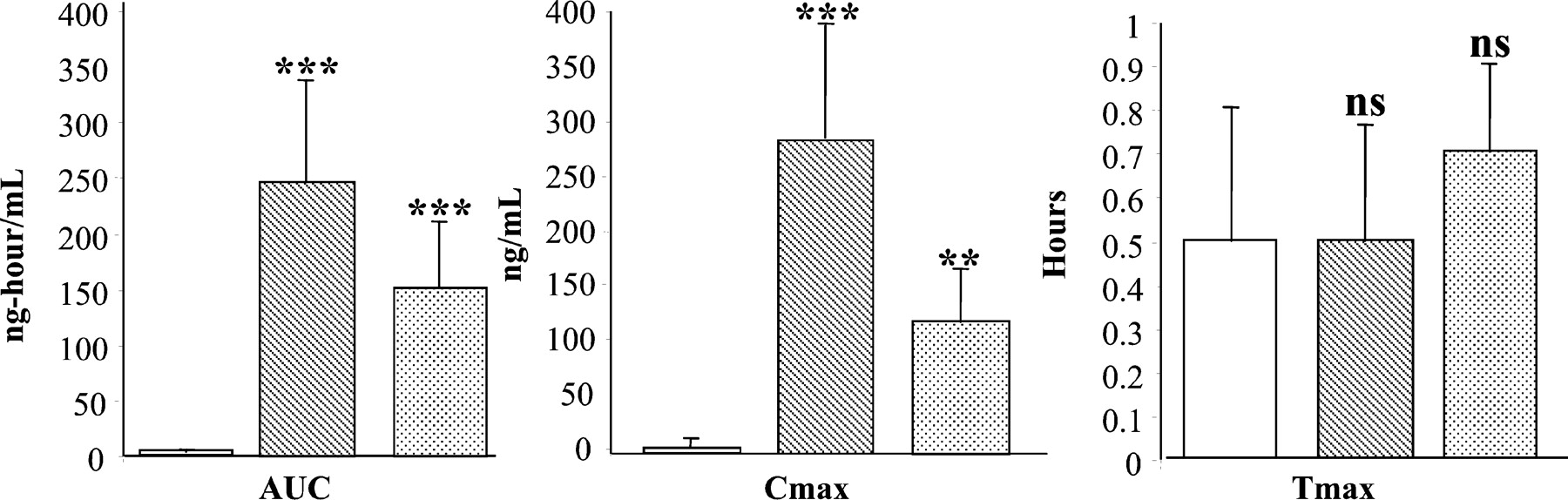
As shown in Fig. 1, bergamottin (1 mg/kg) significantly increased (p < 0.01) diazepam AUC(0-TLDC) after both pre- and concomitant dosing. Mean AUC(0-TLDC) for diazepam in the placebo group was 2.79 ± 3.24 ng · hr/ml compared with 247.69 ± 90.46 ng · hr/ml with bergamottin pretreatment and 150.46 ± 60.57 ng · hr/ml with coadministration, representing 89- and 54-fold increases in diazepam AUC(0-TLDC), respectively. Similarly, diazepamCmax (278.75 ± 109.2 and 103.10 ± 56.1 ng/ml for predosed and coadministration of bergamottin, respectively) increased by 51- and 19-fold over placebo (5.49 ± 7.47 ng/ml). There was no significant change in thetmax for diazepam with bergamottin administration (0.5 to 0.7 h). Plasma bergamottin levels were also monitored, and a mean tmax of 0.3 h was observed. Low concentrations of bergamottin preclude pharmacokinetic parameters from being calculated.

Effect of coadministration and predosing of bergamottin on diazepam pharmacokinetics.
Male beagle dogs were administered the following p.o. dosing regimens. Control: placebo capsules (open bars) along with diazepam; predosing: bergamottin (1 mg/kg) (hatched bars) administered 2 h before diazepam (10-mg tablets); coadministration: bergamottin (1 mg/kg) (stippled bars) administered along with diazepam (10-mg tablets). Systemic blood was collected over 24 h, diazepam plasma levels were measured by LC/MS/MS as described under Materials and Methods, and pharmacokinetic parameters were calculated using WinNonlin software. ∗∗, p < 0.01; ∗∗∗,p < 0.001; ns, not significant compared with control.
Comparative effect of i.v. and p.o. administration of bergamottin on diazepam kinetics.
These studies were conducted to test the effect of bergamottin (0.1 mg/kg) on diazepam (10 mg) plasma concentrations at a lower dose and after i.v. or p.o. administration (Fig. 2; Table1). PK analysis revealed no significant difference in diazepam plasma AUC when bergamottin was administered i.v. versus p.o.. With both routes of administration, AUC was significantly higher when compared with control, p.o. dosing resulting in a 3.6-fold increase over control and i.v. dosing a 3-fold increase. Similarly, Cmax was increased 2.6- and 2.3-fold over control for p.o. and i.v. bergamottin treatment, respectively. Both values were significantly higher than control (Table1). Diazepam half-life was similar when bergamottin was administered i.v. or p.o. Half-life determination could not be made for the control group due to low diazepam concentrations and insufficient data points. These studies also indicate that bergamottin is effective at increasing diazepam plasma levels at doses as low as 0.1 mg/kg. Due to the lower bergamottin dose in this study (0.1 mg/kg), plasma bergamottin concentrations were below the limit of quantitation by LC/MS/MS and pharmacokinetic analysis and could not be conducted on bergamottin.

Effect of i.v. or p.o. bergamottin treatment on p.o. diazepam pharmacokinetics.
Male beagle dogs were administered one of two dosing regimens. p.o.: bergamottin (0.1 mg/kg) (square symbols) or placebo (triangular symbol) capsules were administered 1 h before 10-mg diazepam tablets; i.v.: bergamottin (0.1 mg/kg) (round symbol) was administered at the same time as diazepam (10 mg), as described under Materials and Methods. Plasma diazepam levels were measured by LC/MS/MS, and pharmacokinetic parameters were calculated using WinNonlin software.
Effect of p.o. or i.v. administration of bergamottin (0.1 mg/kg) on diazepam (10 mg) pharmacokinetics in beagle dogs
Effect of bergamottin on drug-metabolizing enzymes.
To characterize the effect of bergamottin on P450 enzymes, male beagle dogs were administered 10 mg of bergamottin once daily for 10 days before harvesting liver and intestinal tissue for enzyme activity determinations (Fig. 3). CYP3A12 activity significantly decreased (p < 0.01) in the liver with bergamottin treatment (control, 905 ± 0.16 nM/min/mg of protein; treated, 445 ± 0.07 nM/min/mg of protein). The jejunal mucosa showed the opposite trend, with bergamottin treatment doubling CYP3A12 activity from 250 ± 0.01 nM/min/mg of protein to 507 ± 0.07 nM/min/mg of protein (p < 0.001). There was no change in tolbutamide 4-hydroxylase activity (usually associated with CYP2C in other species) after bergamottin treatment (control, 5.76 ± 1.31 nM/min/mg of protein; treated, 5.27 ± 0.75 nM/min/mg of protein), and no activity was detected in intestinal microsomes. Basal CYP2B11 activity was below the limits of detection by LC/MS/MS in both hepatic and intestinal microsomes. After bergamottin treatment, hepatic microsomal CYP2B11 activity was determined to be 0.885 ± 0.48 nM/min/mg of protein, suggesting moderate induction by bergamottin in the liver. Bergamottin treatment reduced hepatic CYP1A1/2 activity to one-third of control levels [control, 9.430 ± 1.83 nM/min/mg of protein; bergamottin, 2.290 ± 0.60 nM/min/mg of protein (p < 001)]. No CYP1A1/2 activity was detected in the control or treated dog jejunal mucosa by the ethoxyresorufin assay.

Effect of chronic bergamottin treatment on hepatic and intestinal P450 enzymes in Beagle dogs.
Male beagle dogs were administered bergamottin (10 mg) (filled bars) or placebo as control (open bars) each day for 10 consecutive days. Hepatic and intestinal (first 20 cm of jejunum) tissues were harvested on day 11, and microsomes were prepared as described underMaterials and Methods. Microsomal analysis was conducted for CYP3A12 (A) (testosterone 6β-hydroxylation), CYP2C (B) (tolbutamide hydroxylation), CYP2B11 (C) [formation of 7-pentoxyresorufin O-dealkylase (PROD)], and CYP1A1/2 (D) [formation of ethoxyresorufin O-dealkylase (EROD)]. Results are expressed as mean ± S.E. ∗∗,p < 0.01; ∗∗∗, p < 0.001; ns, not significant compared with control.
Discussion
Changes in P450 activity represent the basis for many cases of clinical drug-drug interactions. These changes may be a result of up-regulation or down-regulation of the associated genes, inhibition of enzyme activity, or decreased degradation of protein. Although typically a drug-drug interaction is regarded as an adverse event, if managed effectively, this interaction could be used to enhance plasma concentrations of high-clearance compounds, leading to increased bioavailability and, presumably, efficacy. The effect of grapefruit juice on plasma pharmacokinetics of CYP3A4 substrates is well characterized in humans. Grapefruit juice has been reported to increase plasma concentrations of the CYP3A4 substrates nifedipine, lovastatin, and atorvastatin (Bailey et al., 1998b; Mohri et al., 2000) and the CYP1A2 substrate caffeine (Fuhr et al., 1993). Furanocoumarins, including those present in grapefruit juice, have been shown to inactivate P450 enzymes in human liver microsomes (He et al., 1998) and increase the bioavailability of CYP3A4 substrates (Bailey et al., 2000;Malhotra et al., 2001). Bergamottin is a major component of grapefruit juice, with concentrations as high as 10 μg/ml (Bailey et al., 1998a). The present studies were conducted to test the hypothesis that bergamottin, a potent P450 inactivator, effects an increase in plasma levels of compounds rapidly metabolized by P450 enzymes (e.g., diazepam).
Diazepam, despite being well absorbed from the gastrointestinal tract, has low oral bioavailability in humans due to high first-pass metabolism by CYP3A4 and CYP2C9 (Nemeroff et al., 1996). In human volunteers, a single dose of grapefruit juice has been shown to increase diazepam plasma AUC by 3.2-fold andCmax by 1.5-fold (Özdemir et al., 1998). Studies have suggested that grapefruit juice acts primarily on intestinal CYP3A4 activity since short-term treatment does not affect the i.v. clearance of coadministered CYP3A4 substrates (Ducharme et al., 1995). The primary observed effect of grapefruit juice on oral medication is an increase in Cmax, with no change in the rate of elimination (Bailey et al., 1995) since hepatic CYP3A is presumably not changed.
We chose the beagle dog as our model after preliminary experiments (data not shown) revealed increased diazepam levels (Cmax, 10.2-fold; AUC, 31-fold over control) in the plasma of beagle dogs administered two doses of 1 mg/kg bergamottin each; 24 and 0.5 h before diazepam dosing. This unexpectedly large increase in AUC suggested that the primary metabolic pathway(s) for diazepam (CYP3A12 and/or CYP2C21) was inhibited and similar to grapefruit juice; bergamottin could enhance bioavailability of orally administered P450 substrates. In subsequent experiments in beagle dogs, coadministration of bergamottin with diazepam revealed no obvious difference in tmax for bergamottin (0.80 ± 0.11 h) and diazepam (0.70 ± 0.21 h) demonstrating also that both compounds are rapidly absorbed. Although diazepam AUC and Cmax increased significantly in both experimental groups, predosing resulted in higher diazepam plasma concentration (Fig. 1), indicating that time is required for bergamottin to distribute and achieve effective tissue concentrations for a maximal effect on P450 inhibition in vivo. Earlier studies with grapefruit juice (Lown et al., 1997) showed that the effect on CYP3A4 was intestinal, and there was no hepatic component. To see whether this was true with bergamottin, we administered beagle dogs bergamottin i.v. or p.o.. The results revealed no apparent difference in the diazepam plasma AUC(0-TLDC) andCmax between the i.v. and p.o. groups, indicating that there was probably a hepatic component as well. Given the study design used, we could not discern whether the P450 enzymes inhibited by bergamottin were predominantly in the intestine or the liver.
The effect of bergamottin on hepatic and intestinal P450 enzymes in the beagle dog was evaluated by treating dogs with bergamottin p.o. for 10 days and harvesting livers and jejunal mucosa. There was a 50% decrease in CYP3A12 activity and a 75% decrease in CYP1A1/2 activity in bergamottin treated livers compared with controls. In vitro studies using human (He et al., 1998) and murine hepatic microsomes (Cai et al., 1993) reveal the same trend. In our investigation, there was no change in 4-hydroxytolbutamide activity in the hepatic microsomes after bergamottin treatment. This activity is associated with CYP2C in other species (Sharer et al., 1995) and presumably of CYP2C21 in the dog. No CYP2B11 activity was detected in the control dog livers, and there was measurable activity after bergamottin treatment, albeit to low levels, implying CYP2B11 induction. Although there are no earlier reports of the effect of bergamottin on CYP2B in intact cell systems, in murine microsomes bergamottin is a potent inhibitor of CYP2B11 activity (Cai et al., 1993). This is the first time that induction of CYP2B activity by a component of grapefruit juice has been reported.
Surprisingly, in intestinal microsomes, CYP3A12 activity doubled with repeated bergamottin administration. This is in contrast to the inhibitory effect of bergamottin on the dog liver, implying that there is more than one mechanism by which bergamottin acts on P450 enzymes. Inhibitory and inductive effects of grapefruit juice on activity and concentrations of total P450 have previously been observed in rats in which chronic grapefruit juice administration reduced intestinal total P450 and increased hepatic total P450 (Mohri et al., 2000). In addition, nifedipine (a CYP3A substrate) was cleared significantly faster in these rats after chronic grapefruit juice administration (Mohri et al., 2000). Since CYP3A is a major inducible hepatic P450 in rats, it can be presumed that this increase in hepatic total P450 levels is primarily due to an increase in CYP3A. The observed difference between this rat study (increased hepatic CYP3A) and ours (decreased hepatic CYP3A) could be due to interspecies variability and/or other components in grapefruit juice. The concentration of bergamottin in the grapefruit juice used in that study is not known. Bergamottin concentrations vary significantly in different preparations of grapefruit juice (Vanakoski et al., 1996), and this variability could contribute to the differences observed. Both inhibition and induction of hepatic oxidative enzymes by grapefruit juice have been reported in mice in which a single dose of grapefruit juice decreased and multiple dosing increased xanthine oxidase, glutathione peroxidase, lipid peroxidase, and liver glutathione content (Dakovic et al., 1999).
It is well established that long-term administration of potent CYP3A inhibitors can result in up-regulation of CYP3A mRNA and protein because inhibitors can also be inducers. Some examples of drugs that inhibit CYP3A catalytic activity with short-term treatment and up-regulate CYP3A mRNA and protein content with recurrent administration are protease inhibitors (Gass et al., 1998), macrolide antibiotics (Wrighton et al., 1985), and imidazole antimycotic drugs (Hostetler et al., 1989; Schmiedlin-Ren et al., 1993). Similarly, our results indicate that bergamottin is both an inhibitor and an inducer of CYP3A. The difference in the response of intestinal and hepatic CYP3A12 to recurrent bergamottin treatment in beagle dogs may be caused by reduced concentrations reaching the liver compared with the intestine after oral dosing, thereby reducing the likelihood of an induction effect in the liver. Another explanation is that the effect on the liver could be due to a metabolite of bergamottin. It is also possible that CYP3A12 transcription is increased in the liver by bergamottin, and the enzyme, once formed, is inactivated by bergamottin. In recent experiments conducted in our laboratory using primary human hepatocytes, bergamottin inhibited CYP3A4 activity, slightly increased CYP3A4 protein (Kostrubsky et al., 2000), and elevated mRNA levels 8-fold (data not shown). These results imply that bergamottin increases transcription of the CYP3A4 protein, and once formed, it is inactivated by bergamottin, which is a mechanism-based inhibitor of CYP3A4. These results corroborate our findings with the dog, namely that bergamottin inhibits and induces CYP3A. Considering the above findings, long-term coadministration of bergamottin with a high-clearance drug would not be expected to increase bioavailability of CYP3A substrates carte blanche.
In conclusion, the results presented here demonstrate that bergamottin increases the oral bioavailability of diazepam in beagle dogs, indicating that bergamottin inhibits P450 enzyme activity. There is no significant difference in diazepam p.o. pharmacokinetics after acute oral or i.v. administration of bergamottin. Bergamottin is both an inhibitor and an inducer of CYP3A12. Recurrent treatment with bergamottin inhibits hepatic CYP1A1/2 activity and moderately induces CYP2B11 activity. We have demonstrated for the first time that repeated administration of bergamottin inhibits CYP1A1/2 and CYP3A12 activity in the liver and induces intestinal CYP3A12 and hepatic CYP2B11 activity.
Acknowledgments
We thank Dr. Thomas Woolf for intellectual support and for reviewing the manuscript, Dr. Michael Sinz for help with the study design, Dr. Louis Radulovic for help with the interpretation of PK data, and Betsy Mitchell and Kelly Rose for technical support.
Footnotes
- Abbreviations used are::
- P450
- cytochrome P450
- PK
- pharmacokinetic
- SLS
- sodium lauryl sulfate
- HPLC
- high-performance liquid chromatography
- LC/MS/MS
- liquid chromatography/tandem mass spectrometry
- AUC
- area under the curve
- TLDC
- time of last detectable concentration
- Received August 10, 2001.
- Accepted November 1, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}